

Isochores and Heat Capacity of Liquid Water in Terms of the Ion–Molecular Model

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
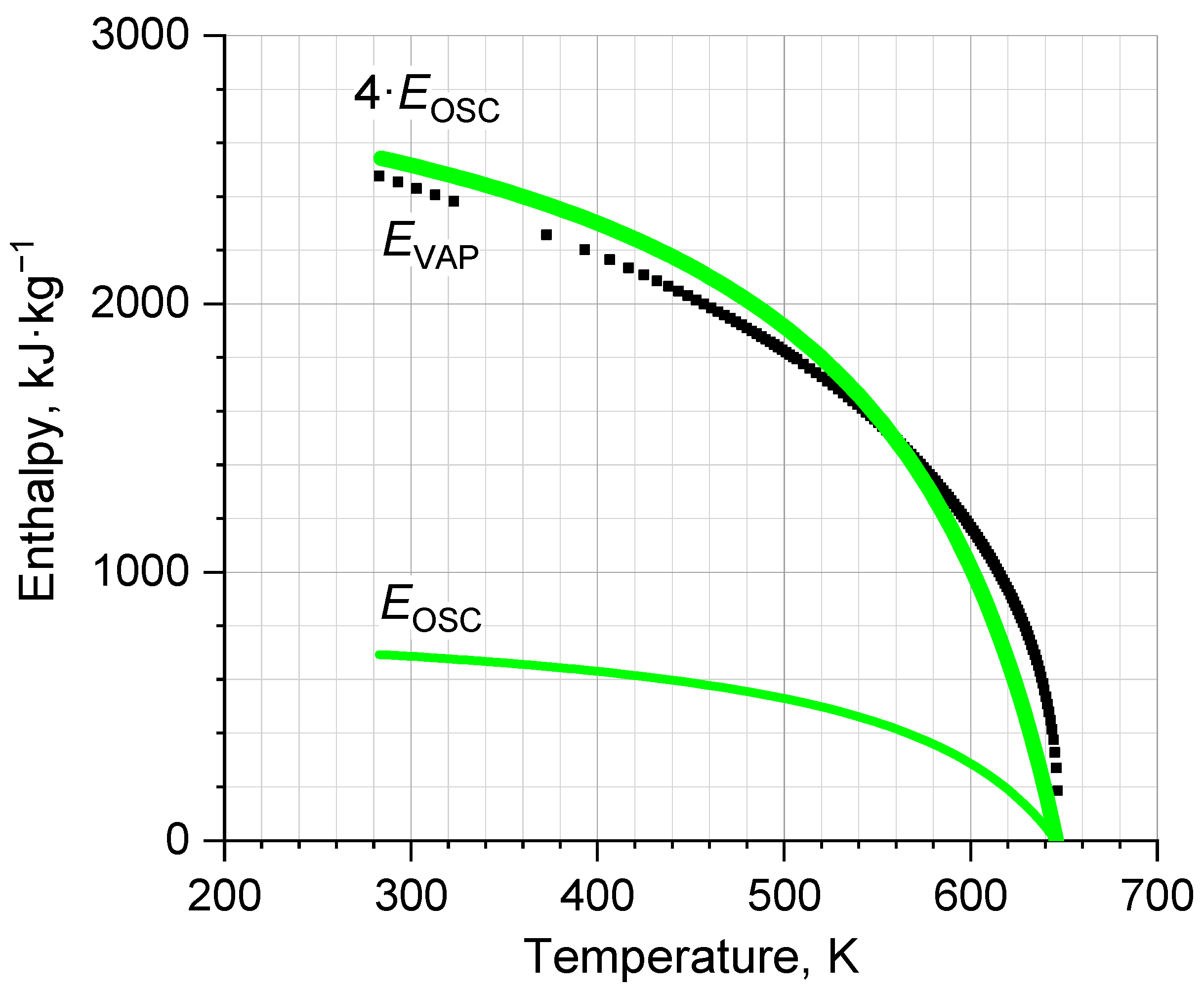{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results and Discussion
2.1. The IM Model of Liquid Water
2.2. The Basic Postulate
2.3. The 5-THz Molecular Oscillator
2.4. Thermodynamic Outline
2.5. Isochores and Heat Capacity
2.6. The Ion Concentration
2.7. The Ion Concentration Issue
3. Materials and Methods
3.1. General Strategy
3.2. Resolution of the 180 cm−1 (5-THz) Peak Issue
3.3. Striving for Simplicity and Clarity
4. Conclusions
Author Contributions
Funding
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| HB | hydrogen bond |
| IM | Ion–molecular |
| DS | dielectric spectroscopy |
| IR | infrared |
References
- Schilling, H. Statistische Physik in Beispielen; Fachbuch: Leipzig, Germany, 1972. [Google Scholar]
- Stillinger, F.H. Water revisited. Science 1980, 209, 451–457. [Google Scholar] [CrossRef] [PubMed]
- Henchman, R.H. Water’s dual nature and its continuously changing hydrogen bonds. J. Phys. Condens. Matter 2016, 28, 384001. [Google Scholar] [CrossRef] [PubMed]
- Brini, E.; Fennell, C.J.; Fernandez-Serra, M.; Hribar-Lee, B.; Lukšič, M.; Dill, K.A. How water’s properties are encoded in its molecular structure and energies. Chem. Rev. 2017, 117, 12385–12414. [Google Scholar] [CrossRef] [PubMed]
- Pettersson, L.G.M.; Henchman, R.H.; Nilsson, A. Water—The most anomalous liquid. Chem. Rev. 2016, 116, 7459–7462. [Google Scholar] [CrossRef]
- Volkov, A.A.; Artemov, V.G.; Pronin, A.V. A radically new suggestion about the electrodynamics of water: Can the pH index and the Debye relaxation be of a common origin? EPL 2014, 106, 46004. [Google Scholar] [CrossRef]
- Volkov, A.A.; Artemov, V.G.; Volkov, A.A., Jr.; Sysoev, N.N. Possible mechanism of molecular motion in liquid water from dielectric spectroscopy data. J. Mol. Liq. 2017, 248, 564–568. [Google Scholar] [CrossRef]
- Volkov, A.A.; Vasin, A.A.; Volkov, A.A., Jr. Cohesion and heat capacity of liquid water from the viewpoint of an electrostatic model. Bull. Russ. Acad. Sci. Phys. 2020, 84, 48–52. [Google Scholar] [CrossRef]
- Volkov, A.A.; Vasin, A.A.; Volkov, A.A., Jr. Dipole-ion model medium with properties of liquid water. Ferroelectrics 2020, 561, 57–64. [Google Scholar] [CrossRef]
- Chaplin, M. Water Structure and Science. Anomalous Properties of Water. Available online: https://water.lsbu.ac.uk/water/water_anomalies.html (accessed on 31 January 2023).
- Del Valle, J.C.; Aragó, C.; Marqués, M.I.; Gonzalo, J.A. Paraelectric response of water in the range 0–100 °C. Ferroelectrics 2014, 466, 166–180. [Google Scholar] [CrossRef]
- Abascal, J.L.F.; Vega, C. A general purpose model for the condensed phases of water: TIP4P/2005. J. Chem. Phys. 2005, 123, 234505. [Google Scholar] [CrossRef]
- Kremer, F.; Schönhals, A. (Eds.) Broadband Dielectric Spectroscopy; Springer: Berlin/Heidelberg, Germany; New York, NY, USA, 2003; ISBN 978-3-642-62809-2. [Google Scholar]
- Kaatze, U.; Feldman, Y. Broadband dielectric spectrometry of liquids and biosystems. Meas. Sci. Technol. 2006, 17, R17–R35. [Google Scholar] [CrossRef]
- Dissado, L. Dielectric response. In Springer Handbook of Electronic and Photonic Materials; Kasap, S., Capper, P., Eds.; Springer International Publishing AG: Cham, Switzerland, 2017; pp. 219–245. ISBN 978-3-319-48933-9. [Google Scholar] [CrossRef]
- Woodward, W.H.H. Broadband dielectric spectroscopy—A practical guide. In Broadband Dielectric Spectroscopy: A Modern Analytical Technique; Woodward, W.H.H., Ed.; American Chemical Society: Washington, DC, USA, 2021; pp. 3–59. ISBN 9780841298484. [Google Scholar] [CrossRef]
- Buixaderas, E.; Kamba, S.; Petzelt, J. Lattice dynamics and central-mode phenomena in the dielectric response of ferroelectrics and related materials. Ferroelectrics 2004, 308, 131–192. [Google Scholar] [CrossRef]
- Hasted, J.B. Liquid water: Dielectric properties. In Water: A Comprehensive Treatise, Vol. 1: The Physics and Physical Chemistry of Water; Franks, F., Ed.; Plenum Press: New York, NY, USA, 1972; pp. 255–309. ISBN 978-1-4684-8336-9. [Google Scholar] [CrossRef]
- Von Hippel, A.R. The dielectric relaxation spectra of water, ice, and aqueous solutions, and their interpretation. I. Critical survey of the status-quo for water. IEEE Trans. Electr. Insul. 1988, 23, 801–816. [Google Scholar] [CrossRef]
- Buchner, R.; Barthel, J.; Stauber, J. The dielectric relaxation of water between 0 °C and 35 °C. Chem. Phys. Lett. 1999, 306, 57–63. [Google Scholar] [CrossRef]
- Fukasawa, T.; Sato, T.; Watanabe, J.; Hama, Y.; Kunz, W.; Buchner, R. Relation between dielectric and low-frequency Raman spectra of hydrogen-bond liquids. Phys. Rev. Lett. 2005, 95, 197802. [Google Scholar] [CrossRef] [PubMed]
- Yada, H.; Nagai, M.; Tanaka, K. The intermolecular stretching vibration mode in water isotopes investigated with broadband terahertz time-domain spectroscopy. Chem. Phys. Lett. 2009, 473, 279–283. [Google Scholar] [CrossRef]
- Elton, D.C. The origin of the Debye relaxation in liquid water and fitting the high frequency excess response. Phys. Chem. Chem. Phys. 2017, 19, 18739–18749. [Google Scholar] [CrossRef]
- Popov, I.; Ben Ishai, P.; Khamzin, A.; Feldman, Y. The mechanism of the dielectric relaxation in water. Phys. Chem. Chem. Phys. 2016, 18, 13941–13953. [Google Scholar] [CrossRef]
- Hansen, J.S.; Kisliuk, A.; Sokolov, A.P.; Gainaru, C. Identification of structural relaxation in the dielectric response of water. Phys. Rev. Lett. 2016, 116, 237601. [Google Scholar] [CrossRef]
- Shiraga, K.; Tanaka, K.; Arikawa, T.; Saito, S.; Ogawa, Y. Reconsideration of the relaxational and vibrational line shapes of liquid water based on ultrabroadband dielectric spectroscopy. Phys. Chem. Chem. Phys. 2018, 20, 26200–26209. [Google Scholar] [CrossRef]
- Volkov, A.A.; Chuchupal, S.V. Dielectric spectra of liquid water: Ultrabroadband modeling and interpretation. J. Mol. Liq. 2022, 365, 120044. [Google Scholar] [CrossRef]
- Ellison, W.J. Permittivity of pure water, at standard atmospheric pressure, over the frequency range 0–25 THz and the temperature range 0–100 °C. J. Phys. Chem. Ref. Data 2007, 36, 1–18. [Google Scholar] [CrossRef]
- Jackson, J.D. Classical Electrodynamics, 3rd ed.; John Wiley & Sons: Hoboken, NJ, USA, 1998; ISBN 978-0-471-30932-1. [Google Scholar]
- Wagner, W.; Pruß, A. The IAPWS formulation 1995 for the thermodynamic properties of ordinary water substance for general and scientific use. J. Phys. Chem. Ref. Data 2002, 31, 387–535. [Google Scholar] [CrossRef]
- Slater, J.C. Quantum Theory of Molecules and Solids, Vol. 1: Electronic Structure of Molecules; McGraw-Hill: New York, NY, USA, 1963; ISBN 978-0-07-058035-0. [Google Scholar]
- Zelsmann, H.R. Temperature dependence of the optical constants for liquid H2O and D2O in the far IR region. J. Mol. Struct. 1995, 350, 95–114. [Google Scholar] [CrossRef]
- Reif, F. Berkeley Physics Course, Vol. 5: Statistical Physics; McGraw-Hill: New York, NY, USA, 1967; ISBN 978-0-07-004862-1. [Google Scholar]
- Eisenberg, D.; Kauzmann, W. The Structure and Properties of Water; Oxford University Press: New York, NY, USA, 2005; ISBN 978-0-19-857026-4. [Google Scholar]
- Becker, R. Theory of Heat, 2nd ed.; Springer: Berlin, Germany, 1967; ISBN 978-3-642-49257-0. [Google Scholar]
- Silbey, R.J.; Alberty, R.A.; Bawendi, M.G. Physical Chemistry, 4th ed.; John Wiley & Sons: Hoboken, NJ, USA, 2005; ISBN 978-0-471-21504-2. [Google Scholar]
- Atkins, P.; de Paula, J. Physical Chemistry, 8th ed.; Oxford University Press: New York, NY, USA, 2006; ISBN 978-0-7167-8759-4. [Google Scholar]
- Dougherty, R.C.; Howard, L.N. Equilibrium structural model of liquid water: Evidence from heat capacity, spectra, density, and other properties. J. Chem. Phys. 1998, 109, 7379–7393. [Google Scholar] [CrossRef]
- Holten, V.; Sengers, J.V.; Anisimov, M.A. Equation of state for supercooled water at pressures up to 400 MPa. J. Phys. Chem. Ref. Data 2014, 43, 043101. [Google Scholar] [CrossRef]
- Mallamace, F.; Corsaro, C.; Mallamace, D.; Fazio, E.; Chen, S.-H.; Cupane, A. Specific heat and transport functions of water. Int. J. Mol. Sci. 2020, 21, 622. [Google Scholar] [CrossRef]
- Kaatze, U. Water, the special liquid. J. Mol. Liq. 2018, 259, 304–318. [Google Scholar] [CrossRef]
- Bockris, J.O.; Reddy, A.K.N. Modern Electrochemistry, Vol. 1: Ionics, 2nd ed.; Kluwer Academic/Plenum Publishers: New York, NY, USA, 1998; ISBN 978-0-306-45554-4. [Google Scholar]
- Rogers, E.M. Physics for the Inquiring Mind; Princeton University Press: Princeton, NJ, USA, 1960; ISBN 978-0-691-08016-1. [Google Scholar]
- Elton, D.C.; Fernández-Serra, M.-V. Polar nanoregions in water: A study of the dielectric properties of TIP4P/2005, TIP4P/2005f and TTM3F. J. Chem. Phys. 2014, 140, 124504. [Google Scholar] [CrossRef]
- Heyden, M.; Sun, J.; Funkner, S.; Mathias, G.; Forbert, H.; Havenith, M.; Marx, D. Dissecting the THz spectrum of liquid water from first principles via correlations in time and space. Proc. Natl. Acad. Sci. USA 2010, 107, 12068–12073. [Google Scholar] [CrossRef]
- Torii, H. Intermolecular electron density modulations in water and their effects on the far-infrared spectral profiles at 6 THz. J. Phys. Chem. B 2011, 115, 6636–6643. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, O.F.; Bilde, M.; Frosch, M. Water activity. Spectrosc. Int. J. 2012, 27, 565–569. [Google Scholar] [CrossRef]
- Sega, M.; Schröder, C. Dielectric and terahertz spectroscopy of polarizable and nonpolarizable water models: A comparative study. J. Phys. Chem. A 2015, 119, 1539–1547. [Google Scholar] [CrossRef] [PubMed]
- Shi, L.; Ni, Y.; Drews, S.E.P.; Skinner, J.L. Dielectric constant and low-frequency infrared spectra for liquid water and ice Ih within the E3B model. J. Chem. Phys. 2014, 141, 084508. [Google Scholar] [CrossRef] [PubMed]
- Kittel, C. Introduction to Solid State Physics, 2nd ed.; John Wiley & Sons: New York, NY, USA, 1953. [Google Scholar]
- Kontogeorgis, G.M.; Holster, A.; Kottaki, N.; Tsochantaris, E.; Topsøe, F.; Poulsen, J.; Bache, M.; Liang, X.; Blom, N.S.; Kronholm, J. Water structure, properties and some applications—A review. Chem. Thermodyn. Therm. Anal. 2022, 6, 100053. [Google Scholar] [CrossRef]
- Feynman, R.; Leighton, R.; Sands, M. The Feynman Lectures on Physics, The New Millennium Edition. Available online: https://www.feynmanlectures.caltech.edu/info/ (accessed on 31 January 2023).
- Bernal, J.D.; Fowler, R.H. A theory of water and ionic solution, with particular reference to hydrogen and hydroxyl ions. J. Chem. Phys. 1933, 1, 515–548. [Google Scholar] [CrossRef]
- Dill, K.A.; Truskett, T.M.; Vlachy, V.; Hribar-Lee, B. Modeling water, the hydrophobic effect, and ion solvation. Annu. Rev. Biophys. Biophys. Biomol. Struct. 2005, 34, 173–199. [Google Scholar] [CrossRef]








Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Volkov, A.A.; Chuchupal, S.V. Isochores and Heat Capacity of Liquid Water in Terms of the Ion–Molecular Model. Int. J. Mol. Sci. 2023, 24, 5630. https://doi.org/10.3390/ijms24065630
Volkov AA, Chuchupal SV. Isochores and Heat Capacity of Liquid Water in Terms of the Ion–Molecular Model. International Journal of Molecular Sciences. 2023; 24(6):5630. https://doi.org/10.3390/ijms24065630
Chicago/Turabian StyleVolkov, Alexander A., and Sergey V. Chuchupal. 2023. "Isochores and Heat Capacity of Liquid Water in Terms of the Ion–Molecular Model" International Journal of Molecular Sciences 24, no. 6: 5630. https://doi.org/10.3390/ijms24065630