
Diapause-Linked Gene Expression Pattern and Related Candidate Duplicated Genes of the Mountain Butterfly Parnassius glacialis (Lepidoptera: Papilionidae) Revealed by Comprehensive Transcriptome Profiling

Abstract
:1. Introduction
2. Results
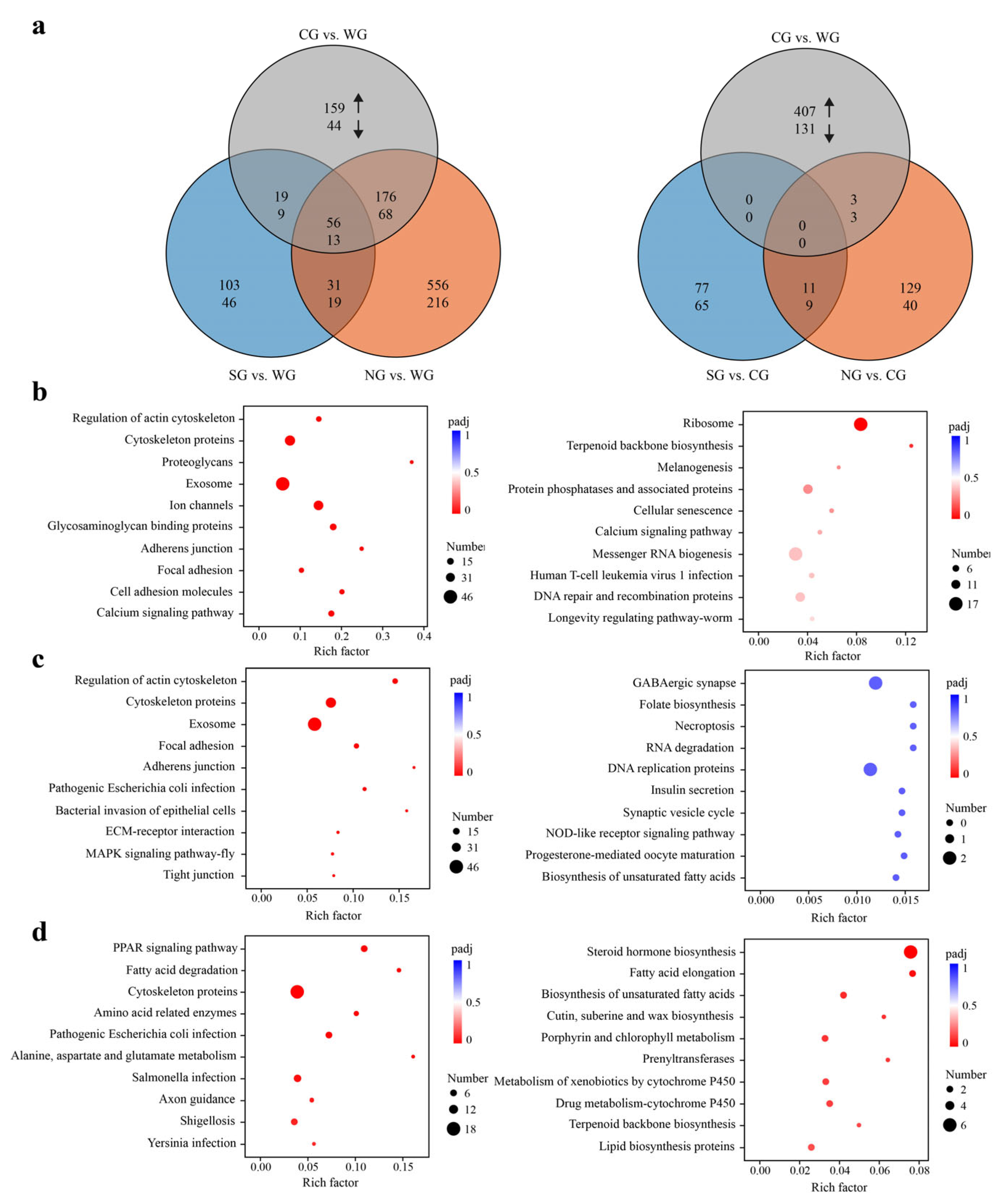
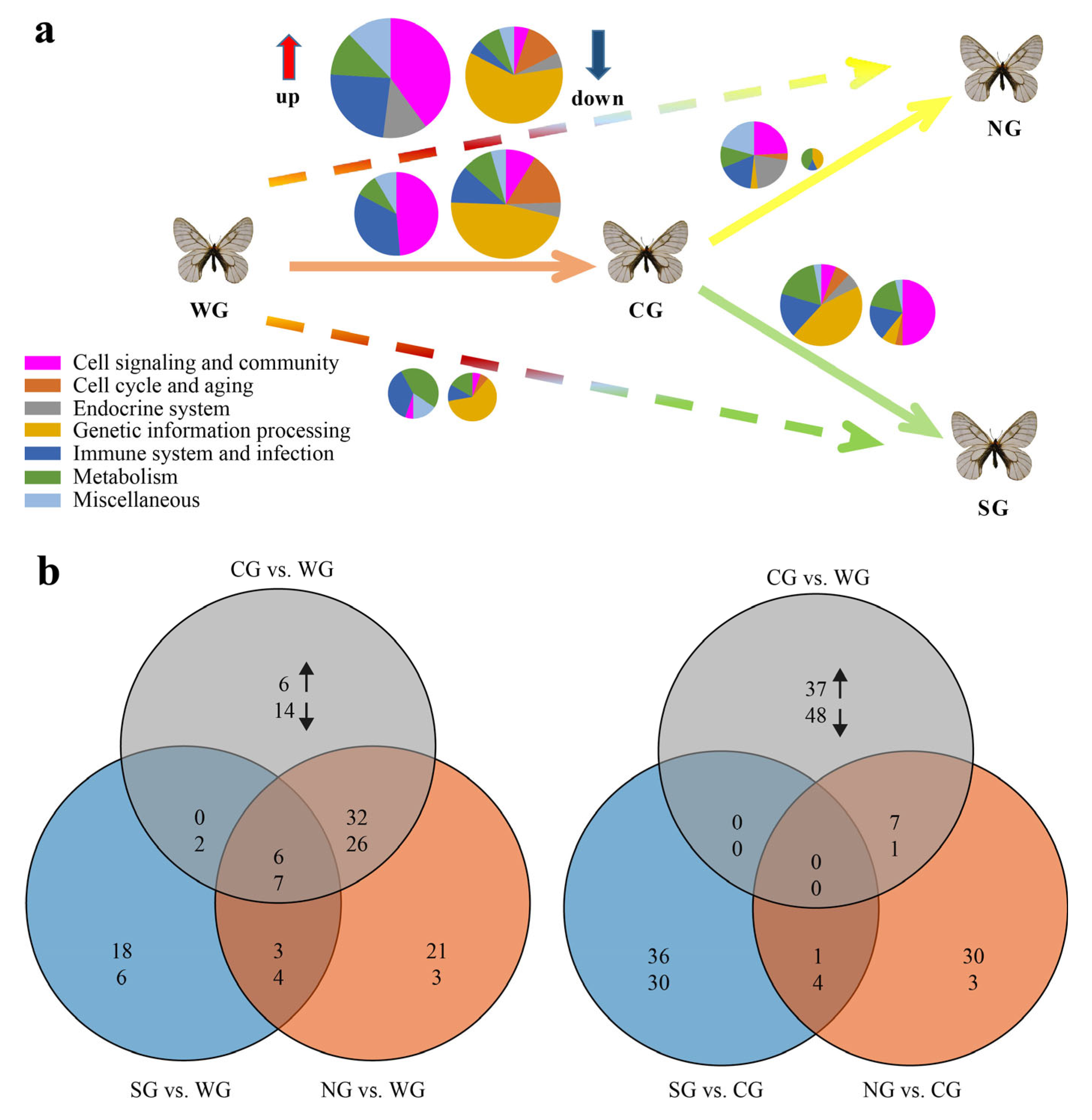
2.1. Statistics of Differentially Expressed Genes (DEGs) and Functional Enrichment Analysis
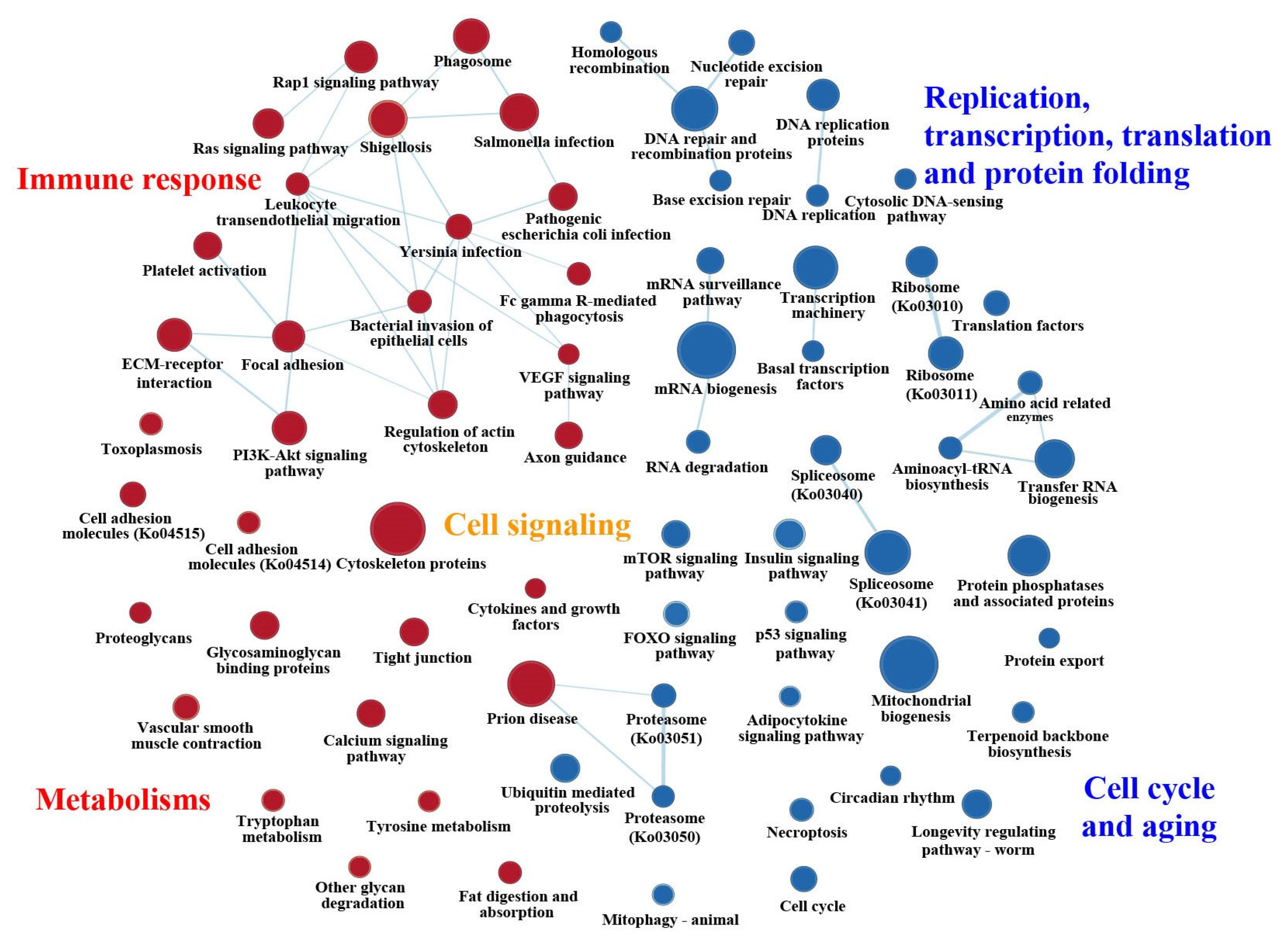
2.2. General Statistics of Enriched KEGG Pathways Based on GSEAs
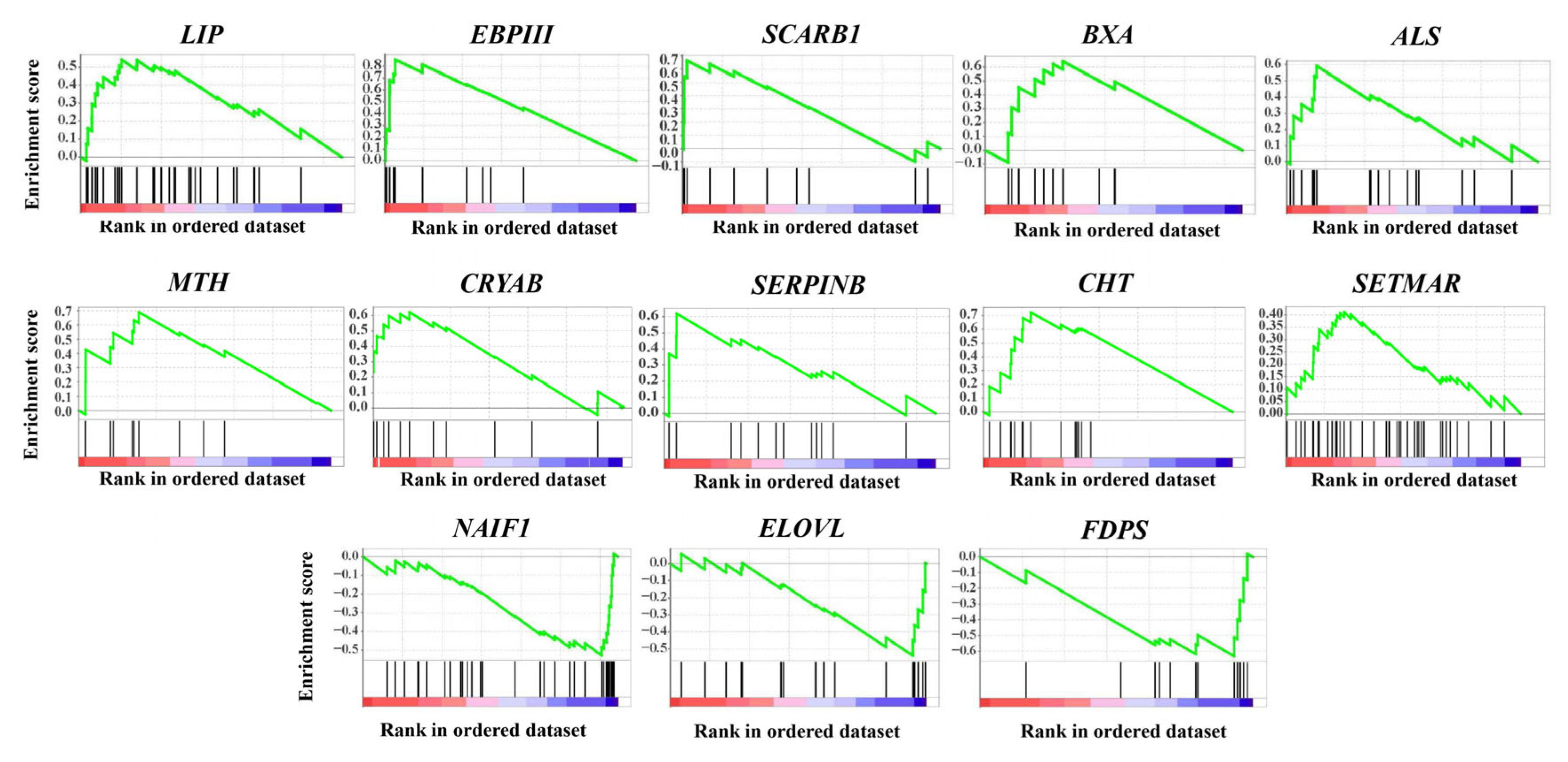
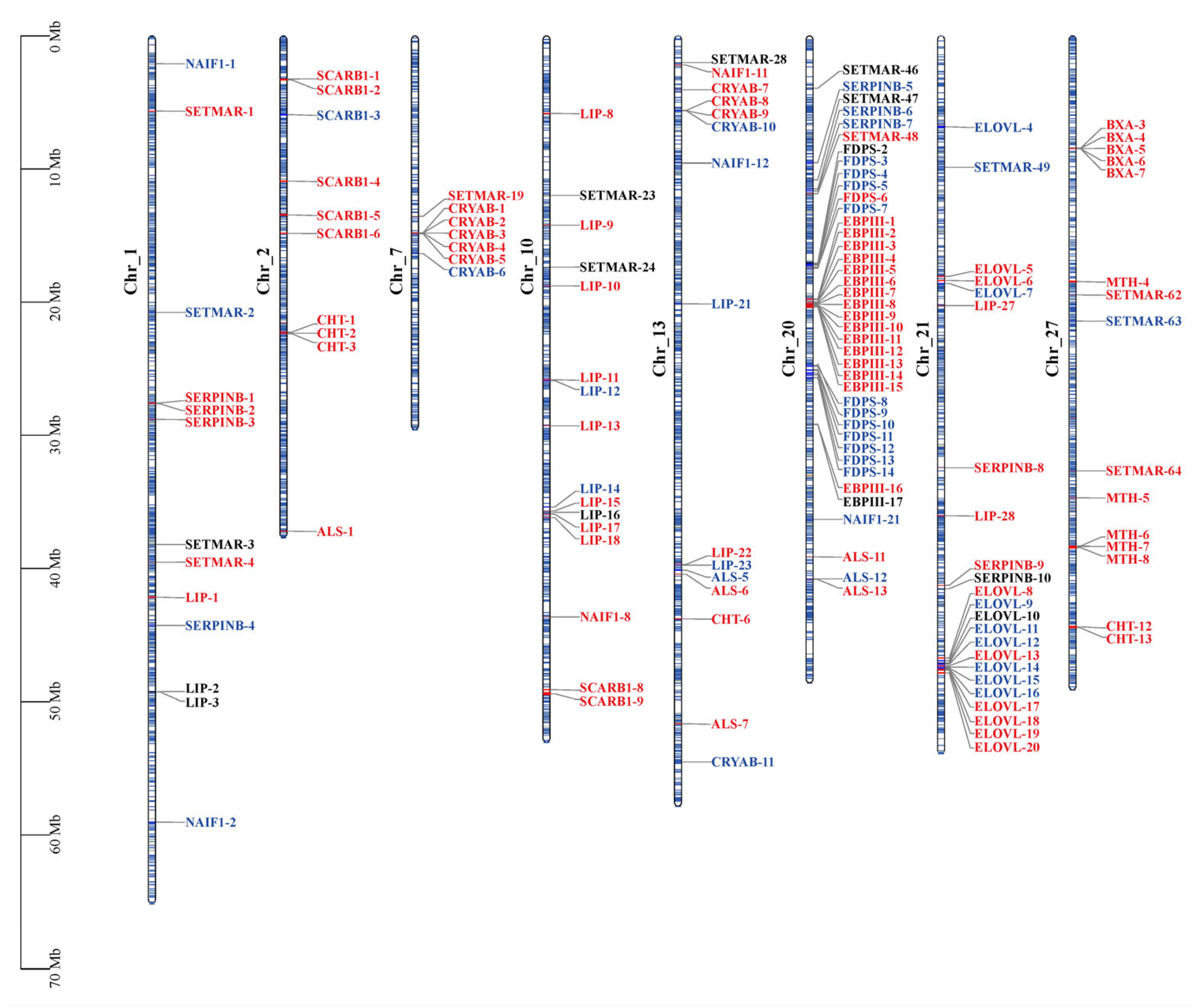
2.3. Featured Gene Sets Based on GSEAs
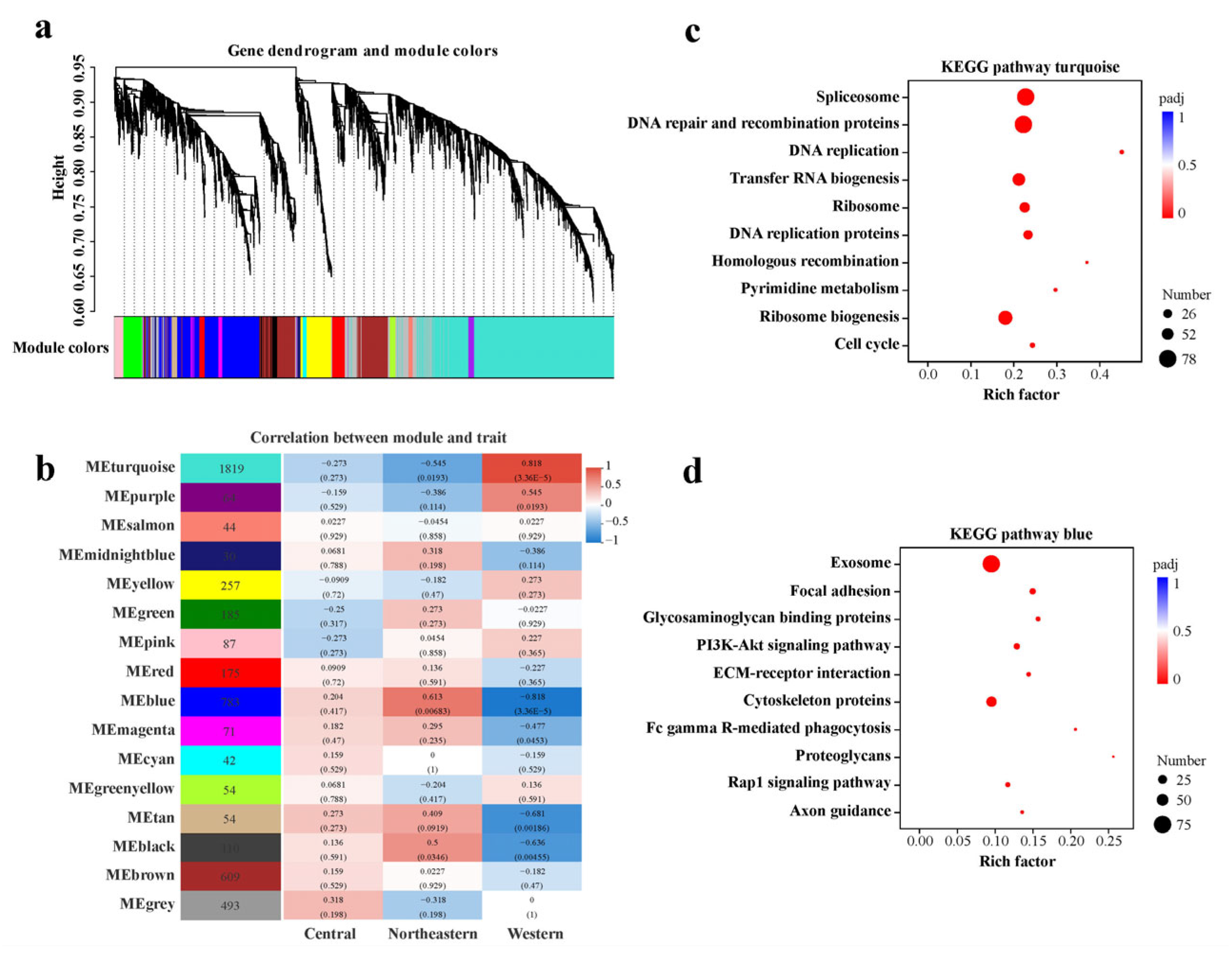
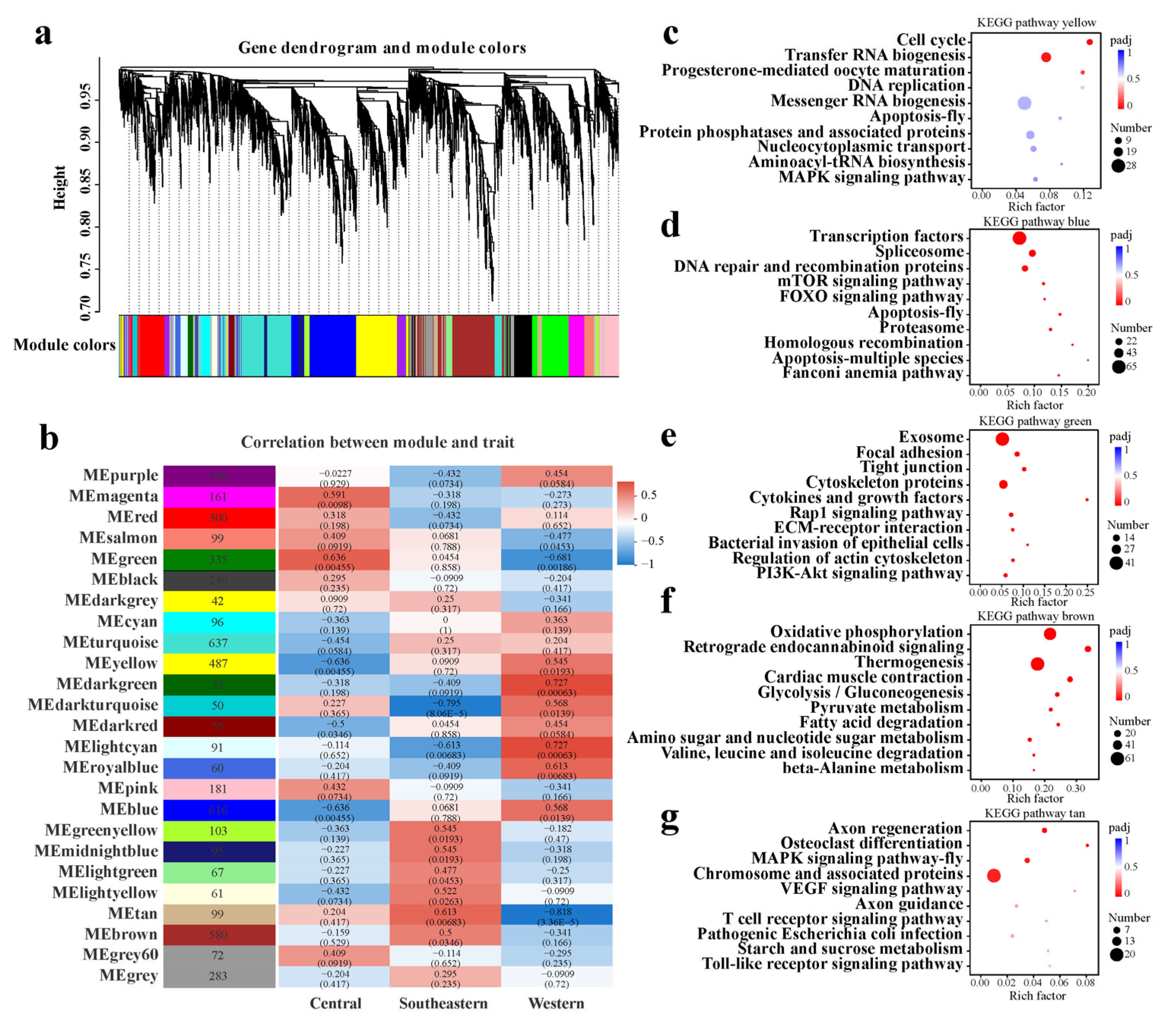
2.4. Featured Modules Based on WGCNA
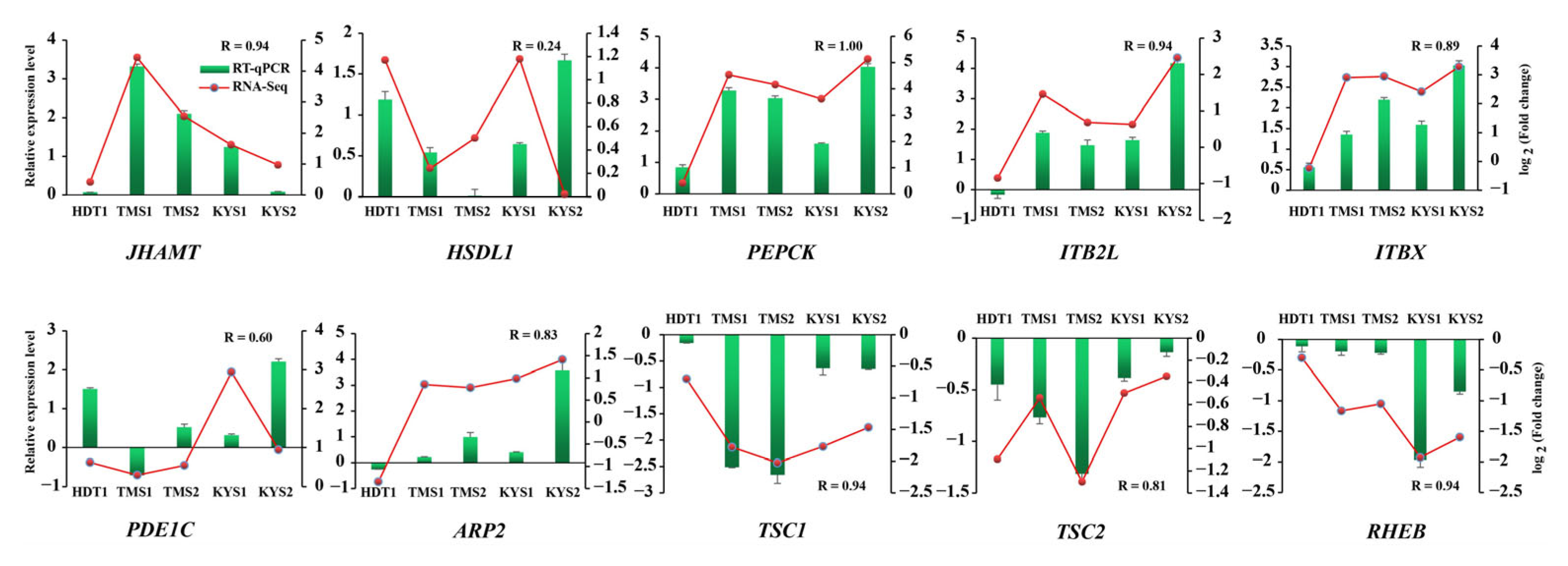
2.5. RNA-Seq Validation Using RT-qPCR
3. Discussion
4. Materials and Methods
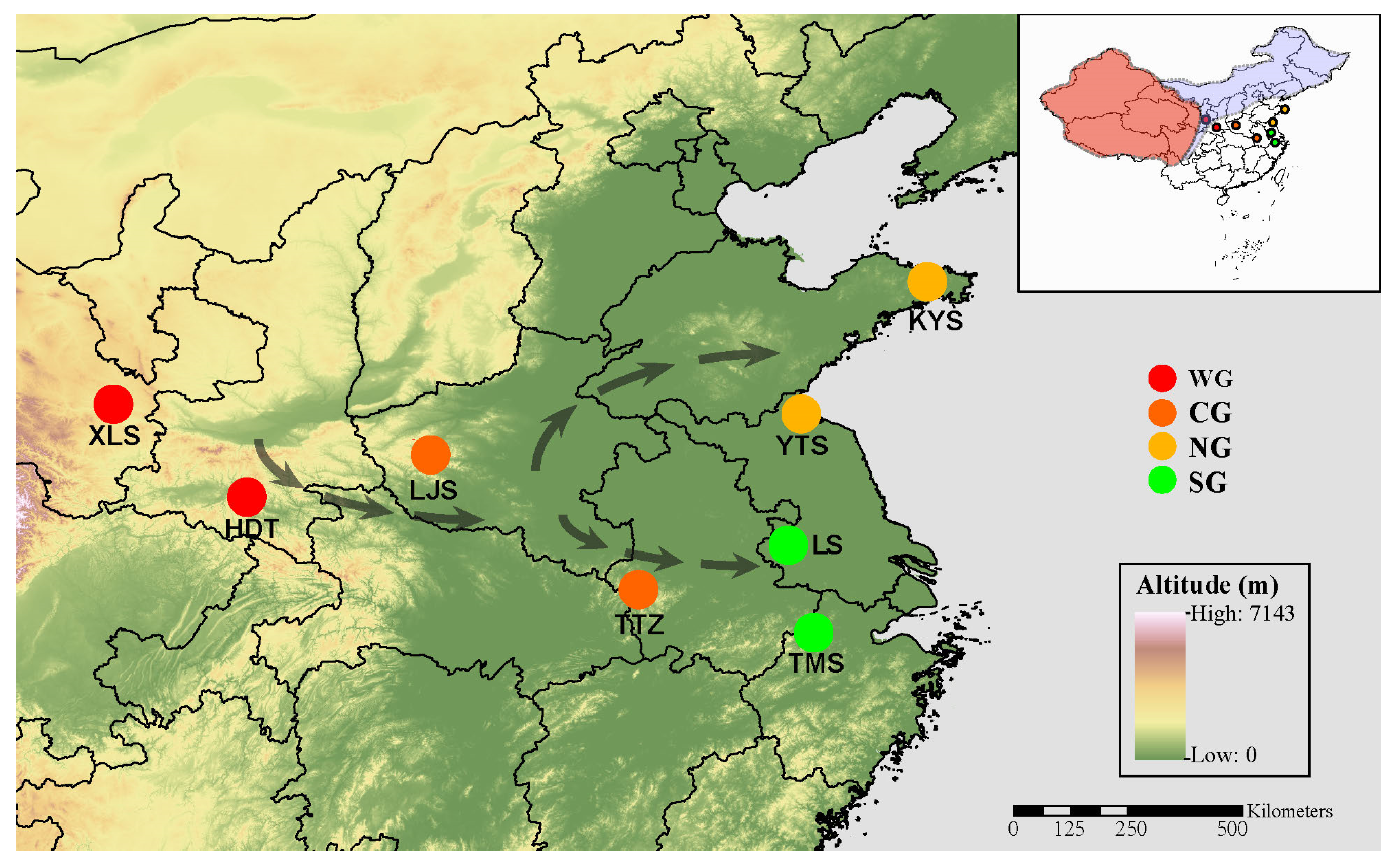
4.1. Sample Collection
4.2. mRNA-Seq Library Construction and Illumina Sequencing
4.3. Mapping, Transcript- and Gene-Level Abundance Estimation
4.4. Expression Level Normalization and Differential Expression Analysis
4.5. Gene Set Enrichment Analysis (GSEA)
4.6. Weighted Gene Co-Expression Network Analysis (WGCNA)
4.7. Validation of Gene Expression by Real-Time RT-qPCR
4.8. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Zhang, J.; Marshall, K.E.; Westwood, J.T.; Clark, M.S.; Sinclair, B.J. Divergent transcriptomic responses to repeated and single cold exposures in Drosophila melanogaster. J. Exp. Biol. 2011, 214, 4021–4029. [Google Scholar] [CrossRef] [Green Version]
- Chown, S.L.; Gaston, K.J. Macrophysiology for a changing world. Proc. R. Soc. B-Biol. Sci. 2008, 275, 1469–1478. [Google Scholar] [CrossRef] [Green Version]
- Sorensen, J.G.; Schou, M.F.; Kristensen, T.N.; Loeschcke, V. Thermal fluctuations affect the transcriptome through mechanisms independent of average temperature. Sci. Rep. 2016, 6, 30975. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lindestad, O.; Wheat, C.W.; Nylin, S.; Gotthard, K. Local adaptation of photoperiodic plasticity maintains life cycle variation within latitudes in a butterfly. Ecology 2019, 100, e02550. [Google Scholar] [CrossRef] [Green Version]
- McBryan, T.L.; Anttila, K.; Healy, T.M.; Schulte, P.M. Responses to temperature and hypoxia as interacting stressors in fish: Implications for adaptation to environmental change. Integr. Comp. Biol. 2013, 53, 648–659. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sorensen, J.G.; Schou, M.F.; Loeschcke, V. Evolutionary adaptation to environmental stressors: A common response at the proteomic level. Evolution 2017, 71, 1627–1642. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boardman, L.; Mitchell, K.A.; Terblanche, J.S.; Sorensen, J.G. A transcriptomics assessment of oxygen-temperature interactions reveals novel candidate genes underlying variation in thermal tolerance and survival. J. Insect Physiol. 2018, 106, 179–188. [Google Scholar] [CrossRef] [PubMed]
- Kucerova, L.; Kubrak, O.I.; Bengtsson, J.M.; Strnad, H.; Nylin, S.; Theopold, U.; Nassel, D.R. Slowed aging during reproductive dormancy is reflected in genome-wide transcriptome changes in Drosophila melanogaster. BMC Genomics 2016, 17, 50. [Google Scholar] [CrossRef] [Green Version]
- Forrest, J.R. Complex responses of insect phenology to climate change. Curr. Opin. Insect Sci. 2016, 17, 49–54. [Google Scholar] [CrossRef]
- Dowle, E.J.; Powell, T.H.Q.; Doellman, M.M.; Meyers, P.J.; Calvert, M.B.; Walden, K.K.O.; Robertson, H.M.; Berlocher, S.H.; Feder, J.L.; Hahn, D.A.; et al. Genome-wide variation and transcriptional changes in diverse developmental processes underlie the rapid evolution of seasonal adaptation. Proc. Natl. Acad. Sci. USA 2020, 117, 23960–23969. [Google Scholar] [CrossRef]
- Kostal, V.; Stetina, T.; Poupardin, R.; Korbelova, J.; Bruce, A.W. Conceptual framework of the eco-physiological phases of insect diapause development justified by transcriptomic profiling. Proc. Natl. Acad. Sci. USA 2017, 114, 8532–8537. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Condamine, F.L.; Rolland, J.; Hohna, S.; Sperling, F.A.H.; Sanmartin, I. Testing the role of the Red Queen and Court Jester as drivers of the macroevolution of Apollo butterflies. Syst. Biol. 2018, 67, 940–964. [Google Scholar] [CrossRef]
- Su, C.; Xie, T.; Wang, Y.; Si, C.; Li, L.; Ma, J.; Li, C.; Sun, X.; Hao, J.; Yang, Q. Miocene diversification and high-altitude adaptation of Parnassius butterflies (Lepidoptera: Papilionidae) in Qinghai-Tibet Plateau revealed by large-scale transcriptomic data. Insects 2020, 11, 754. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; He, B.; Tao, R.; Su, C.; Ma, J.; Hao, J.; Yang, Q. Phylogeny and biogeographic history of Parnassius butterflies (Papilionidae: Parnassiinae) reveal their origin and deep diversification in West China. Insects 2022, 13, 406. [Google Scholar] [CrossRef]
- Tao, R.; Xu, C.; Wang, Y.; Sun, X.; Li, C.; Ma, J.; Hao, J.; Yang, Q. Spatiotemporal differentiation of alpine butterfly Parnassius glacialis (Papilionidae: Parnassiinae) in China: Evidence from mitochondrial DNA and nuclear single nucleotide polymorphisms. Genes 2020, 11, 188. [Google Scholar] [CrossRef] [Green Version]
- Zhang, W.; Chen, J.; Keyhani, N.O.; Jin, K.; Wei, Q.; Xia, Y. Central nervous system responses of the Oriental migratory, Locusta migratoria manilensis, to fungal infection. Sci. Rep. 2017, 7, 10340. [Google Scholar] [CrossRef] [Green Version]
- Veenstra, J.A. Arthropod IGF, relaxin and gonadulin, putative orthologs of Drosophila insulin-like peptides 6, 7 and 8, likely originated from an ancient gene triplication. PeerJ 2020, 8, e9534. [Google Scholar] [CrossRef] [PubMed]
- De Loof, A.; Lindemans, M.; Liu, F.; De Groef, B.; Schoofs, L. Endocrine archeology: Do insects retain ancestrally inherited counterparts of the vertebrate releasing hormones GnRH, GHRH, TRH, and CRF? Gen. Comp. Endocrinol. 2012, 177, 18–27. [Google Scholar] [CrossRef]
- Nagare, M.; Ayachit, M.; Agnihotri, A.; Schwab, W.; Joshi, R. Glycosyltransferases: The multifaceted enzymatic regulator in insects. Insect Mol. Biol. 2021, 30, 123–137. [Google Scholar] [CrossRef] [PubMed]
- Soya, S.; Sahar, U.; Karacali, S. Monosaccharide profiling of silkworm (Bombyx mori L.) nervous system during development and aging. Invert. Neurosci. 2016, 16, 8. [Google Scholar] [CrossRef]
- Somvanshi, V.S.; Gahoi, S.; Banakar, P.; Thakur, P.K.; Kumar, M.; Sajnani, M.; Pandey, P.; Rao, U. A transcriptomic insight into the infective juvenile stage of the insect parasitic nematode, Heterorhabditis indica. BMC Genomics 2016, 17, 166. [Google Scholar] [CrossRef] [Green Version]
- Meldolesi, J. Exosomes and ectosomes in intercellular communication. Curr. Biol. 2018, 28, R435–R444. [Google Scholar] [CrossRef] [Green Version]
- Teets, N.M.; Yi, S.X.; Lee, R.E., Jr.; Denlinger, D.L. Calcium signaling mediates cold sensing in insect tissues. Proc. Natl. Acad. Sci. USA 2013, 110, 9154–9159. [Google Scholar] [CrossRef] [Green Version]
- Hao, Y.J.; Zhang, Y.J.; Si, F.L.; Fu, D.Y.; He, Z.B.; Chen, B. Insight into the possible mechanism of the summer diapause of Delia antiqua (Diptera: Anthomyiidae) through digital gene expression analysis. Insect Sci. 2016, 23, 438–451. [Google Scholar] [CrossRef]
- Green, D.A.; Kronforst, M.R. Monarch butterflies use an environmentally sensitive, internal timer to control overwintering dynamics. Mol. Ecol. 2019, 28, 3642–3655. [Google Scholar] [CrossRef] [PubMed]
- Roh, H.; Kim, N.; Lee, Y.; Park, J.; Kim, B.S.; Lee, M.K.; Park, C.I.; Kim, D.H. Dual-organ transcriptomic analysis of rainbow trout infected with Ichthyophthirius multifiliis through co-expression and machine learning. Front. Immunol. 2021, 12, 677730. [Google Scholar] [CrossRef] [PubMed]
- Sideri, M.; Tsakas, S.; Markoutsa, E.; Lampropoulou, M.; Marmaras, V.J. Innate immunity in insects: Surface-associated dopa decarboxylase-dependent pathways regulate phagocytosis, nodulation and melanization in medfly haemocytes. Immunology 2008, 123, 528–537. [Google Scholar] [CrossRef]
- Chen, X.D.; Neupane, S.; Gill, T.A.; Gossett, H.; Pelz-Stelinski, K.S.; Stelinski, L.L. Comparative transcriptome analysis of thiamethoxam susceptible and resistant Asian citrus psyllid, Diaphorina citri (Hemiptera: Liviidae), using RNA-sequencing. Insect Sci. 2021, 28, 1708–1720. [Google Scholar] [CrossRef]
- Yang, H.; Gao, X.; Li, X.; Zhang, H.; Chen, N.; Zhang, Y.; Liu, X.; Zhang, X. Comparative transcriptome analysis of red swamp crayfish (Procambarus clarkia) hepatopancreas in response to WSSV and Aeromonas hydrophila infection. Fish Shellfish Immunol. 2018, 83, 397–405. [Google Scholar] [CrossRef]
- De Gregorio, E.; Han, S.J.; Lee, W.J.; Baek, M.J.; Osaki, T.; Kawabata, S.; Lee, B.L.; Iwanaga, S.; Lemaitre, B.; Brey, P.T. An immune-responsive Serpin regulates the melanization cascade in Drosophila. Dev. Cell 2002, 3, 581–592. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mitsou, I.; Multhaupt, H.A.B.; Couchman, J.R. Proteoglycans, ion channels and cell-matrix adhesion. Biochem. J. 2017, 474, 1965–1979. [Google Scholar] [CrossRef] [PubMed]
- Jiang, T.; Zhu, Y.; Peng, Y.; Zhang, W.; Xiao, H. Universal and differential transcriptional regulatory pathways involved in the preparation of summer and winter diapauses in Pieris melete. Bull. Entomol. Res. 2021, 111, 371–378. [Google Scholar] [CrossRef] [PubMed]
- Yocum, G.D.; Rinehart, J.P.; Horvath, D.P.; Kemp, W.P.; Bosch, J.; Alroobi, R.; Salem, S. Key molecular processes of the diapause to post-diapause quiescence transition in the alfalfa leafcutting bee Megachile rotundata identified by comparative transcriptome analysis. Physiol. Entomol. 2015, 40, 103–112. [Google Scholar] [CrossRef]
- Tu, X.; Wang, J.; Hao, K.; Whitman, D.W.; Fan, Y.; Cao, G.; Zhang, Z. Transcriptomic and proteomic analysis of pre-diapause and non-diapause eggs of migratory locust, Locusta migratoria L. (Orthoptera: Acridoidea). Sci. Rep. 2015, 5, 11402. [Google Scholar] [CrossRef] [Green Version]
- Deng, Y.; Li, F.; Rieske, L.K.; Sun, L.L.; Sun, S.H. Transcriptome sequencing for identification of diapause-associated genes in fall webworm, Hyphantria cunea Drury. Gene 2018, 668, 229–236. [Google Scholar] [CrossRef]
- Zhai, Y.; Dong, X.; Gao, H.; Chen, H.; Yang, P.; Li, P.; Yin, Z.; Zheng, L.; Yu, Y. Quantitative proteomic and transcriptomic analyses of metabolic regulation of adult reproductive diapause in Drosophila suzukii (Diptera: Drosophilidae) females. Front. Physiol. 2019, 10, 344. [Google Scholar] [CrossRef] [Green Version]
- Lin, X.; Smagghe, G. Roles of the insulin signaling pathway in insect development and organ growth. Peptides 2019, 122, 169923. [Google Scholar] [CrossRef]
- Kramer, J.M.; Slade, J.D.; Staveley, B.E. Foxo is required for resistance to amino acid starvation in Drosophila. Genome 2008, 51, 668–672. [Google Scholar] [CrossRef]
- Xiong, S.; Yu, K.; Ye, X.; Fang, Q.; Deng, Y.; Xiao, S.; Yang, L.; Wang, B.; Wang, F.; Yan, Z.; et al. Genes acting in longevity-related pathways in the endoparasitoid, Pteromalus puparum. Arch. Insect Biochem. Physiol. 2020, 103, e21635. [Google Scholar] [CrossRef]
- Hay, N. Interplay between FOXO, TOR, and Akt. BBA-Mol. Cell Res. 2011, 1813, 1965–1970. [Google Scholar] [CrossRef] [Green Version]
- Oldham, S. Obesity and nutrient sensing TOR pathway in flies and vertebrates: Functional conservation of genetic mechanisms. Trends Endocrinol. Met. 2011, 22, 45–52. [Google Scholar] [CrossRef] [Green Version]
- Fontana, L.; Partridge, L.; Longo, V.D. Extending healthy life span-from yeast to humans. Science 2010, 328, 321–326. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Poupardin, R.; Schottner, K.; Korbelova, J.; Provaznik, J.; Dolezel, D.; Pavlinic, D.; Benes, V.; Kostal, V. Early transcriptional events linked to induction of diapause revealed by RNAseq in larvae of drosophilid fly, Chymomyza costata. BMC Genom. 2015, 16, 720. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, S.Z.; Zhu, L.B.; You, L.L.; Wang, J.; Cao, H.H.; Liu, Y.X.; Toufeeq, S.; Wang, Y.L.; Kong, X.; Xu, J.P. A novel digestive proteinase Lipase member H-A in Bombyx mori contributes to digestive juice antiviral activity against B. mori nucleopolyhedrovirus. Insects 2020, 11, 154. [Google Scholar] [CrossRef] [Green Version]
- Zhang, S.D.; Li, X.; Bin, Z.; Du, M.F.; Yin, X.M.; An, S.H. Molecular identification of a pancreatic lipase-like gene involved in sex pheromone biosynthesis of Bombyx mori. Insect Sci. 2014, 21, 459–468. [Google Scholar] [CrossRef]
- Martin-Blazquez, R.; Chen, B.; Kang, L.; Bakkali, M. Evolution, expression and association of the chemosensory protein genes with the outbreak phase of the two main pest locusts. Sci. Rep. 2017, 7, 6653. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, F.; Venthur, H.; Wang, S.; Homem, R.A.; Zhou, J.J. Evidence for the involvement of the chemosensory protein AgosCSP5 in resistance to insecticides in the cotton aphid, Aphis gossypii. Insects 2021, 12, 335. [Google Scholar] [CrossRef]
- Herboso, L.; Talamillo, A.; Perez, C.; Barrio, R. Expression of the Scavenger Receptor class B type I (SR-BI) family in Drosophila melanogaster. Int. J. Dev. Biol. 2011, 55, 603–611. [Google Scholar] [CrossRef] [Green Version]
- Shen, W.J.; Hu, J.; Hu, Z.; Kraemer, F.B.; Azhar, S. Scavenger Receptor class B type I (SR-BI): A versatile receptor with multiple functions and actions. Metabolism 2014, 63, 875–886. [Google Scholar] [CrossRef]
- Mizoguchi, A.; Okamoto, N. Insulin-like and IGF-like peptides in the silkmoth Bombyx mori: Discovery, structure, secretion, and function. Front. Physiol. 2013, 4, 217. [Google Scholar] [CrossRef] [Green Version]
- Arquier, N.; Geminard, C.; Bourouis, M.; Jarretou, G.; Honegger, B.; Paix, A.; Leopold, P. Drosophila ALS regulates growth and metabolism through functional interaction with insulin-like peptides. Cell Metab. 2008, 7, 333–338. [Google Scholar] [CrossRef]
- Cao, C.; Sun, L.; Du, H.; Moural, T.W.; Bai, H.; Liu, P.; Zhu, F. Physiological functions of a methuselah-like G protein coupled receptor in Lymantria dispar Linnaeus. Pestic. Biochem. Physiol. 2019, 160, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Wojtowicz, I.; Jablonska, J.; Zmojdzian, M.; Taghli-Lamallem, O.; Renaud, Y.; Junion, G.; Daczewska, M.; Huelsmann, S.; Jagla, K.; Jagla, T. Drosophila small heat shock protein CryAB ensures structural integrity of developing muscles, and proper muscle and heart performance. Development 2015, 142, 994–1005. [Google Scholar] [CrossRef] [Green Version]
- Arakane, Y.; Muthukrishnan, S. Insect chitinase and chitinase-like proteins. Cell Mol. Life Sci. 2010, 67, 201–216. [Google Scholar] [CrossRef] [PubMed]
- Castillo-Gracia, M.; Couillaud, F. Molecular cloning and tissue expression of an insect farnesyl diphosphate synthase. Eur. J. Biochem. 1999, 262, 365–370. [Google Scholar] [CrossRef] [Green Version]
- Zhu, J.; Khalil, S.M.; Mitchell, R.D.; Bissinger, B.W.; Egekwu, N.; Sonenshine, D.E.; Roe, R.M. Mevalonate-farnesal biosynthesis in ticks: Comparative synganglion transcriptomics and a new perspective. PLoS ONE 2016, 11, e0141084. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cardoso-Moreira, M.; Arguello, J.R.; Gottipati, S.; Harshman, L.G.; Grenier, J.K.; Clark, A.G. Evidence for the fixation of gene duplications by positive selection in Drosophila. Genome Res. 2016, 26, 787–798. [Google Scholar] [CrossRef] [Green Version]
- Rogers, R.L.; Shao, L.; Thornton, K.R. Tandem duplications lead to novel expression patterns through exon shuffling in Drosophila yakuba. PLoS Genet. 2017, 13, e1006795. [Google Scholar] [CrossRef] [Green Version]
- Cordaux, R.; Udit, S.; Batzer, M.A.; Feschotte, C. Birth of a chimeric primate gene by capture of the transposase gene from a mobile element. Proc. Natl. Acad. Sci. USA 2006, 103, 8101–8106. [Google Scholar] [CrossRef] [Green Version]
- Sinzelle, L.; Izsvak, Z.; Ivics, Z. Molecular domestication of transposable elements: From detrimental parasites to useful host genes. Cell Mol. Life Sci. 2009, 66, 1073–1093. [Google Scholar] [CrossRef]
- Nicolau, M.; Picault, N.; Moissiard, G. The evolutionary volte-face of transposable elements: From harmful jumping genes to major drivers of genetic innovation. Cells 2021, 10, 2952. [Google Scholar] [CrossRef]
- Lv, B.; Shi, T.; Wang, X.; Song, Q.; Zhang, Y.; Shen, Y.; Ma, D.; Lou, Y. Overexpression of the novel human gene, nuclear apoptosis-inducing factor 1, induces apoptosis. Int. J. Biochem. Cell Biol. 2006, 38, 671–683. [Google Scholar] [CrossRef]
- Rech, G.E.; Radio, S.; Guirao-Rico, S.; Aguilera, L.; Horvath, V.; Green, L.; Lindstadt, H.; Jamilloux, V.; Quesneville, H.; Gonzalez, J. Population-scale long-read sequencing uncovers transposable elements associated with gene expression variation and adaptive signatures in Drosophila. Nat. Commun. 2022, 13, 1948. [Google Scholar] [CrossRef] [PubMed]
- Kankare, M.; Parker, D.J.; Merisalo, M.; Salminen, T.S.; Hoikkala, A. Transcriptional differences between diapausing and non-diapausing D. montana females reared under the same photoperiod and temperature. PLoS ONE 2016, 11, e0161852. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yocum, G.D.; Toutges, M.J.; Roehrdanz, R.L.; Dihle, P.J. Insertion of miniature subterminal inverted repeat-like elements in diapause-regulated genes in the Colorado potato beetle, Leptinotarsa decemlineata (Coleoptera: Chrysomelidae). Eur. J. Entomol. 2011, 108, 197–203. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.; Jiang, Z.; Fan, P.; Ericson, P.G.P.; Song, G.; Luo, X.; Lei, F.; Qu, Y. The combination of genomic offset and niche modelling provides insights into climate change-driven vulnerability. Nat. Commun. 2022, 13, 4821. [Google Scholar] [CrossRef]
- Guo, S.; Tian, Z.; Wu, Q.W.; King-Jones, K.; Liu, W.; Zhu, F.; Wang, X.P. Steroid hormone ecdysone deficiency stimulates preparation for photoperiodic reproductive diapause. PLoS Genet. 2021, 17, e1009352. [Google Scholar] [CrossRef]
- Herman, W.S.; Tatar, M. Juvenile hormone regulation of longevity in the migratory monarch butterfly. Proc. Biol. Sci. 2001, 268, 2509–2514. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, T.Y.; Sun, J.S.; Zhang, Q.R.; Xu, J.; Jiang, R.J.; Xu, W.H. The diapause hormone-pheromone biosynthesis activating neuropeptide gene of Helicoverpa armigera encodes multiple peptides that break, rather than induce, diapause. J. Insect Physiol. 2004, 50, 547–554. [Google Scholar] [CrossRef]
- Nylin, S. Induction of diapause and seasonal morphs in butterflies and other insects: Knowns, unknowns and the challenge of integration. Physiol. Entomol. 2013, 38, 96–104. [Google Scholar] [CrossRef] [Green Version]
- Poelchau, M.F.; Reynolds, J.A.; Elsik, C.G.; Denlinger, D.L.; Armbruster, P.A. Deep sequencing reveals complex mechanisms of diapause preparation in the invasive mosquito, Aedes albopictus. Proc. R. Soc. B-Biol. Sci. 2013, 280, 20130143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuzmin, E.; Taylor, J.S.; Boone, C. Retention of duplicated genes in evolution. Trends Genet. 2022, 38, 59–72. [Google Scholar] [CrossRef] [PubMed]
- Sun, B.J.; Huebner, C.; Treidel, L.A.; Clark, R.M.; Roberts, K.T.; Kenagy, G.J.; Williams, C.M. Nocturnal dispersal flight of crickets: Behavioural and physiological responses to cool environmental temperatures. Funct. Ecol. 2020, 34, 1907–1920. [Google Scholar] [CrossRef]
- Loli, D.; Bicudo, J.E. Control and regulatory mechanisms associated with thermogenesis in flying insects and birds. Biosci. Rep. 2005, 25, 149–180. [Google Scholar] [CrossRef]
- Kim, D.; Paggi, J.M.; Park, C.; Bennett, C.; Salzberg, S.L. Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nat. Biotechnol. 2019, 37, 907–915. [Google Scholar] [CrossRef]
- Pertea, M.; Pertea, G.M.; Antonescu, C.M.; Chang, T.C.; Mendell, J.T.; Salzberg, S.L. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat. Biotechnol. 2015, 33, 290–295. [Google Scholar] [CrossRef] [Green Version]
- Chen, C.; Chen, H.; Zhang, Y.; Thomas, H.R.; Frank, M.H.; He, Y.; Xia, R. TBtools: An integrative toolkit developed for interactive analyses of big biological data. Mol. Plant. 2020, 13, 1194–1202. [Google Scholar] [CrossRef]
- Ren, Y.; Yu, G.; Shi, C.; Liu, L.; Guo, Q.; Han, C.; Zhang, D.; Zhang, L.; Liu, B.; Gao, H.; et al. Majorbio Cloud: A one-stop, comprehensive bioinformatic platform for multiomics analyses. iMeta 2022, 1, e12. [Google Scholar] [CrossRef]
- Brawand, D.; Soumillon, M.; Necsulea, A.; Julien, P.; Csardi, G.; Harrigan, P.; Weier, M.; Liechti, A.; Aximu-Petri, A.; Kircher, M.; et al. The evolution of gene expression levels in mammalian organs. Nature 2011, 478, 343–348. [Google Scholar] [CrossRef]
- Darbellay, F.; Necsulea, A. Comparative transcriptomics analyses across species, organs, and developmental stages reveal functionally constrained lncRNAs. Mol. Biol. Evol. 2020, 37, 240–259. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. EdgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef] [Green Version]
- Merico, D.; Isserlin, R.; Stueker, O.; Emili, A.; Bader, G.D. Enrichment map: A network-based method for gene-set enrichment visualization and interpretation. PLoS ONE 2010, 5, e13984. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef]
- Blum, M.; Chang, H.Y.; Chuguransky, S.; Grego, T.; Kandasaamy, S.; Mitchell, A.; Nuka, G.; Paysan-Lafosse, T.; Qureshi, M.; Raj, S.; et al. The InterPro protein families and domains database: 20 years on. Nucleic. Acids Res. 2021, 49, D344–D354. [Google Scholar] [CrossRef]
- Langfelder, P.; Horvath, S. WGCNA: An R package for weighted correlation network analysis. BMC Bioinform. 2008, 9, 559. [Google Scholar] [CrossRef] [Green Version]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- Pan, H.; Yang, X.; Bidne, K.; Hellmich, R.L.; Siegfried, B.D.; Zhou, X. Selection of reference genes for RT-qPCR analysis in the monarch butterfly, Danaus plexippus (L.), a migrating bio-indicator. PLoS ONE 2015, 10, e0129482. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Enriched Group | Gene Name | Description | Main Function |
|---|---|---|---|
| CG and NG | LIP | Lipase | Fat catabolism |
| CG and NG | EBPIII | Ejaculatory bulb-specific protein 3-like | Chemoreception and resistance to insecticides |
| CG and NG | BXA | Bombyxin | Metabolism, growth and longevity regulation |
| CG and NG | CHT | Chitinase | Digestion, molting and immune defense |
| NG | SCARB1 | Scavenger receptor class B member 1 | Steroid hormone production and immune defense |
| NG | CRYAB | Crystalline alpha B | Stress responses and extended lifespan |
| NG | SERPINB | Serpin B | Immune defense |
| NG | ALS | Insulin-like growth factor-binding protein complex acid labile subunit | Metabolism, growth and longevity regulation |
| CG | MTH | G protein-coupled receptor Mth | Chemoreception and extended lifespan |
| CG | SETMAR | Histone-lysine N-methyltransferase SETMAR | DNA repair and epigenetic modification |
| SG | NAIF1 | Nuclear apoptosis-inducing factor 1 | Apoptosis regulation |
| SG | ELOVL | Elongation of very long chain fatty acids protein | Long-chain fatty acids biosynthesis |
| SG | FDPS | Farnesyl diphosphate synthase | Juvenile hormone production |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Su, C.; Ding, C.; Zhao, Y.; He, B.; Nie, R.; Hao, J. Diapause-Linked Gene Expression Pattern and Related Candidate Duplicated Genes of the Mountain Butterfly Parnassius glacialis (Lepidoptera: Papilionidae) Revealed by Comprehensive Transcriptome Profiling. Int. J. Mol. Sci. 2023, 24, 5577. https://doi.org/10.3390/ijms24065577
Su C, Ding C, Zhao Y, He B, Nie R, Hao J. Diapause-Linked Gene Expression Pattern and Related Candidate Duplicated Genes of the Mountain Butterfly Parnassius glacialis (Lepidoptera: Papilionidae) Revealed by Comprehensive Transcriptome Profiling. International Journal of Molecular Sciences. 2023; 24(6):5577. https://doi.org/10.3390/ijms24065577
Chicago/Turabian StyleSu, Chengyong, Chen Ding, Youjie Zhao, Bo He, Ruie Nie, and Jiasheng Hao. 2023. "Diapause-Linked Gene Expression Pattern and Related Candidate Duplicated Genes of the Mountain Butterfly Parnassius glacialis (Lepidoptera: Papilionidae) Revealed by Comprehensive Transcriptome Profiling" International Journal of Molecular Sciences 24, no. 6: 5577. https://doi.org/10.3390/ijms24065577