
Activation of TLRs Triggers GLP-1 Secretion in Mice
 ,
,
Abstract
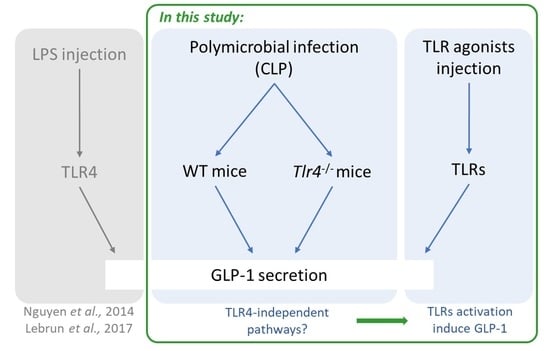
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
2.1. CLP Induces Systemic Inflammation and Modulates Inflammatory Gene Expression in the Gut
2.2. TLR Agonists Increase Cytokines and Expression of TLRs
2.3. GLP-1 Secretion Is Mediated through Multiple TLRs
3. Discussion
4. Materials and Methods
4.1. Experimental Animals and Samplings
4.2. Cecal Ligation Puncture (CLP) Treatment
4.3. Drugs Administration in Mice
4.4. Real-Time Quantitative PCR
4.5. Plasma and Tissues Biochemical Analyses
4.6. Statistics
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Jandhyala, S.M.; Talukdar, R.; Subramanyam, C.; Vuyyuru, H.; Sasikala, M.; Reddy, D.N. Role of the Normal Gut Microbiota. World J. Gastroenterol. 2015, 21, 8787–8803. [Google Scholar] [CrossRef]
- Mörbe, U.M.; Jørgensen, P.B.; Fenton, T.M.; von Burg, N.; Riis, L.B.; Spencer, J.; Agace, W.W. Human Gut-Associated Lymphoid Tissues (GALT); Diversity, Structure, and Function. Mucosal Immunol. 2021, 14, 793–802. [Google Scholar] [CrossRef] [PubMed]
- Abreu, M.T.; Fukata, M.; Arditi, M. TLR Signaling in the Gut in Health and Disease. J. Immunol. 2005, 174, 4453–4460. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Drucker, D.J.; Habener, J.F.; Holst, J.J. Discovery, Characterization, and Clinical Development of the Glucagon-like Peptides. J. Clin. Investig. 2017, 127, 4217–4227. [Google Scholar] [CrossRef] [Green Version]
- Rowlands, J.; Heng, J.; Newsholme, P.; Carlessi, R. Pleiotropic Effects of GLP-1 and Analogs on Cell Signaling, Metabolism, and Function. Front. Endocrinol. (Lausanne) 2018, 9, 672. [Google Scholar] [CrossRef] [Green Version]
- Iorga, R.A.; Bacalbasa, N.; Carsote, M.; Bratu, O.G.; Stanescu, A.M.A.; Bungau, S.; Pantis, C.; Diaconu, C.C. Metabolic and Cardiovascular Benefits of GLP-1 Agonists, besides the Hypoglycemic Effect (Review). Exp. Ther. Med. 2020, 20, 2396–2400. [Google Scholar] [CrossRef] [PubMed]
- Vandemark, C.; Nguyen, J.; Zhao, Z.-Q. Cardiovascular Protection with a Long-Acting GLP-1 Receptor Agonist Liraglutide: An Experimental Update. Molecules 2023, 28, 1369. [Google Scholar] [CrossRef]
- Merza, N.; Akram, M.; Mengal, A.; Mustafa Rashid, A.; Mahboob, A.; Faryad, M.; Fatima, Z.; Ahmed, M.; Ansari, S.A. The Safety and Efficacy of GLP-1 Receptor Agonists in Heart Failure Patients: A Systematic Review and Meta-Analysis. Curr. Probl. Cardiol. 2023, 48, 101602. [Google Scholar] [CrossRef]
- Perl, S.H.; Bloch, O.; Zelnic-Yuval, D.; Love, I.; Mendel-Cohen, L.; Flor, H.; Rapoport, M.J. Sepsis-Induced Activation of Endogenous GLP-1 System Is Enhanced in Type 2 Diabetes. Diabetes/Metab. Res. Rev. 2018, 34, e2982. [Google Scholar] [CrossRef]
- Braun, J.-P.; Buhner, S.; Kastrup, M.; Dietz, E.; Langer, K.; Dohmen, P.; Lochs, H.; Spies, C. Barrier Function of the Gut and Multiple Organ Dysfunction after Cardiac Surgery. J. Int. Med. Res. 2007, 35, 72–83. [Google Scholar] [CrossRef]
- Nguyen, M.; Tavernier, A.; Gautier, T.; Aho, S.; Morgant, M.C.; Bouhemad, B.; Guinot, P.-G.; Grober, J. Glucagon-like Peptide-1 Is Associated with Poor Clinical Outcome, Lipopolysaccharide Translocation and Inflammation in Patients Undergoing Cardiac Surgery with Cardiopulmonary Bypass. Cytokine 2020, 133, 155182. [Google Scholar] [CrossRef] [PubMed]
- Yusta, B.; Baggio, L.L.; Koehler, J.; Holland, D.; Cao, X.; Pinnell, L.J.; Johnson-Henry, K.C.; Yeung, W.; Surette, M.G.; Bang, K.W.A.; et al. GLP-1R Agonists Modulate Enteric Immune Responses Through the Intestinal Intraepithelial Lymphocyte GLP-1R. Diabetes 2015, 64, 2537–2549. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zou, Z.; Wang, Z. Liraglutide Attenuates Intestinal Ischemia/Reperfusion Injury via NF-ΚB and PI3K/Akt Pathways in Mice. Life Sci. 2022, 309, 121045. [Google Scholar] [CrossRef] [PubMed]
- Nozu, T.; Miyagishi, S.; Kumei, S.; Nozu, R.; Takakusaki, K.; Okumura, T. Glucagon-like Peptide-1 Analog, Liraglutide, Improves Visceral Sensation and Gut Permeability in Rats. J. Gastroenterol. Hepatol. 2018, 33, 232–239. [Google Scholar] [CrossRef] [Green Version]
- Brubaker, P.L. The Molecular Determinants of Glucagon-like Peptide Secretion by the Intestinal L Cell. Endocrinology 2022, 163, bqac159. [Google Scholar] [CrossRef]
- Tolhurst, G.; Heffron, H.; Lam, Y.S.; Parker, H.E.; Habib, A.M.; Diakogiannaki, E.; Cameron, J.; Grosse, J.; Reimann, F.; Gribble, F.M. Short-Chain Fatty Acids Stimulate Glucagon-like Peptide-1 Secretion via the G-Protein-Coupled Receptor FFAR2. Diabetes 2012, 61, 364–371. [Google Scholar] [CrossRef] [Green Version]
- Parker, H.E.; Wallis, K.; le Roux, C.W.; Wong, K.Y.; Reimann, F.; Gribble, F.M. Molecular Mechanisms Underlying Bile Acid-Stimulated Glucagon-like Peptide-1 Secretion. Br. J. Pharm. 2012, 165, 414–423. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cani, P.D.; Knauf, C. A Newly Identified Protein from Akkermansia Muciniphila Stimulates GLP-1 Secretion. Cell Metab. 2021, 33, 1073–1075. [Google Scholar] [CrossRef]
- Tomaro-Duchesneau, C.; LeValley, S.L.; Roeth, D.; Sun, L.; Horrigan, F.T.; Kalkum, M.; Hyser, J.M.; Britton, R.A. Discovery of a Bacterial Peptide as a Modulator of GLP-1 and Metabolic Disease. Sci. Rep. 2020, 10, 4922. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, A.T.; Mandard, S.; Dray, C.; Deckert, V.; Valet, P.; Besnard, P.; Drucker, D.J.; Lagrost, L.; Grober, J. Lipopolysaccharides-Mediated Increase in Glucose-Stimulated Insulin Secretion: Involvement of the Glucagon-like Peptide 1 (GLP1) Pathway. Diabetes 2014, 63, 471. [Google Scholar] [CrossRef] [Green Version]
- Kahles, F.; Meyer, C.; Möllmann, J.; Diebold, S.; Findeisen, H.M.; Lebherz, C.; Trautwein, C.; Koch, A.; Tacke, F.; Marx, N.; et al. GLP-1 Secretion Is Increased by Inflammatory Stimuli in an IL-6–Dependent Manner, Leading to Hyperinsulinemia and Blood Glucose Lowering. Diabetes 2014, 63, 3221–3229. [Google Scholar] [CrossRef] [Green Version]
- Lebrun, L.J.; Lenaerts, K.; Kiers, D.; Pais de Barros, J.-P.; Le Guern, N.; Plesnik, J.; Thomas, C.; Bourgeois, T.; Dejong, C.H.C.; Kox, M.; et al. Enteroendocrine L Cells Sense LPS after Gut Barrier Injury to Enhance GLP-1 Secretion. Cell Rep. 2017, 21, 1160–1168. [Google Scholar] [CrossRef] [Green Version]
- Worthington, J.J.; Reimann, F.; Gribble, F.M. Enteroendocrine Cells-Sensory Sentinels of the Intestinal Environment and Orchestrators of Mucosal Immunity. Mucosal Immunol. 2018, 11, 3–20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aravanis, C.V.; Kapelouzou, A.; Vagios, S.; Tsilimigras, D.I.; Katsimpoulas, M.; Moris, D.; Demesticha, T.D.; Schizas, D.; Kostakis, A.; Machairas, A.; et al. Toll-Like Receptors -2, -3, -4 and -7 Expression Patterns in the Liver of a CLP-Induced Sepsis Mouse Model. J. Investig. Surg 2020, 33, 109–117. [Google Scholar] [CrossRef] [PubMed]
- Krivan, S.; Kapelouzou, A.; Vagios, S.; Tsilimigras, D.I.; Katsimpoulas, M.; Moris, D.; Aravanis, C.V.; Demesticha, T.D.; Schizas, D.; Mavroidis, M.; et al. Increased Expression of Toll-like Receptors 2, 3, 4 and 7 MRNA in the Kidney and Intestine of a Septic Mouse Model. Sci. Rep. 2019, 9, 4010. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bakopoulos, A.; Kapelouzou, A.; Tsilimigras, D.I.; Katsimpoulas, M.; Schizas, D.; Aravanis, C.; Balafas, E.; Mavroidis, M.; Pavlakis, K.; Machairas, A.; et al. Expression of Toll-like Receptors (TLRs) in the Lungs of an Experimental Sepsis Mouse Model. PLoS ONE 2017, 12, e0188050. [Google Scholar] [CrossRef] [PubMed]
- Bogunovic, M.; Davé, S.H.; Tilstra, J.S.; Chang, D.T.W.; Harpaz, N.; Xiong, H.; Mayer, L.F.; Plevy, S.E. Enteroendocrine Cells Express Functional Toll-like Receptors. Am. J. Physiol. Gastrointest. Liver Physiol. 2007, 292, G1770–G1783. [Google Scholar] [CrossRef]
- Gribble, F.M.; Reimann, F. Function and Mechanisms of Enteroendocrine Cells and Gut Hormones in Metabolism. Nat. Rev. Endocrinol. 2019, 15, 226–237. [Google Scholar] [CrossRef]
- Harrison, E.; Lal, S.; McLaughlin, J.T. Enteroendocrine Cells in Gastrointestinal Pathophysiology. Curr. Opin. Pharmacol. 2013, 13, 941–945. [Google Scholar] [CrossRef]
- Genton, L.; Kudsk, K.A. Interactions between the Enteric Nervous System and the Immune System: Role of Neuropeptides and Nutrition. Am. J. Surg. 2003, 186, 253–258. [Google Scholar] [CrossRef]
- Lee, J.H.; Patel, K.; Tae, H.J.; Lustig, A.; Kim, J.W.; Mattson, M.P.; Taub, D.D. Ghrelin Augments Murine T-Cell Proliferation by Activation of the Phosphatidylinositol-3-Kinase, Extracellular Signal-Regulated Kinase and Protein Kinase C Signaling Pathways. FEBS Lett. 2014, 588, 4708–4719. [Google Scholar] [CrossRef] [Green Version]
- Reardon, C.; Duncan, G.S.; Brüstle, A.; Brenner, D.; Tusche, M.W.; Olofsson, P.S.; Rosas-Ballina, M.; Tracey, K.J.; Mak, T.W. Lymphocyte-Derived ACh Regulates Local Innate but Not Adaptive Immunity. Proc. Natl. Acad. Sci. USA 2013, 110, 1410–1415. [Google Scholar] [CrossRef] [Green Version]
- Palazzo, M.; Balsari, A.; Rossini, A.; Selleri, S.; Calcaterra, C.; Gariboldi, S.; Zanobbio, L.; Arnaboldi, F.; Shirai, Y.F.; Serrao, G.; et al. Activation of Enteroendocrine Cells via TLRs Induces Hormone, Chemokine, and Defensin Secretion. J. Immunol. 2007, 178, 4296–4303. [Google Scholar] [CrossRef] [Green Version]
- Drucker, D.J.; Yusta, B. Physiology and Pharmacology of the Enteroendocrine Hormone Glucagon-like Peptide-2. Annu. Rev. Physiol 2014, 76, 561–583. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Drucker, D.J. Mechanisms of Action and Therapeutic Application of Glucagon-like Peptide-1. Cell Metab. 2018, 27, 740–756. [Google Scholar] [CrossRef] [Green Version]
- Hunt, J.E.; Holst, J.J.; Jeppesen, P.B.; Kissow, H. GLP-1 and Intestinal Diseases. Biomedicines 2021, 9, 383. [Google Scholar] [CrossRef]
- Keller, J.; Binnewies, U.; Rösch, M.; Juul Holst, J.; Beglinger, C.; Andresen, V.; Layer, P. Gastric Emptying and Disease Activity in Inflammatory Bowel Disease. Eur. J. Clin. Investig. 2015, 45, 1234–1242. [Google Scholar] [CrossRef] [PubMed]
- Lucotti, P.; Lovati, E.; Lenti, M.V.; Valvo, B.; Sprio, E.; Aronico, N.; Giuffrida, P.; Dell’Aera, D.; Pasini, A.; Ubezio, C.; et al. Abnormal Post-Prandial Glucagon-like Peptide Release in Patients with Crohn’s Disease. Clin. Res. Hepatol. Gastroenterol. 2021, 45, 101533. [Google Scholar] [CrossRef]
- Brakenridge, S.C.; Moore, F.A.; Mercier, N.R.; Cox, M.; Wu, Q.; Moldawer, L.L.; Mohr, A.M.; Efron, P.A.; Smith, R.S. Persistently Elevated Glucagon-Like Peptide 1 Levels Among Critically-Ill Surgical Patients After Sepsis and Development of Chronic Critical Illness and Dismal Long-Term Outcomes. J. Am. Coll Surg. 2019, 229, 58–67.e1. [Google Scholar] [CrossRef] [PubMed]
- Lebherz, C.; Schlieper, G.; Möllmann, J.; Kahles, F.; Schwarz, M.; Brünsing, J.; Dimkovic, N.; Koch, A.; Trautwein, C.; Flöge, J.; et al. GLP-1 Levels Predict Mortality in Patients with Critical Illness as Well as End-Stage Renal Disease. Am. J. Med. 2017, 130, 833–841.e3. [Google Scholar] [CrossRef]
- Zanotti-Cavazzoni, S.L.; Guglielmi, M.; Parrillo, J.E.; Walker, T.; Dellinger, R.P.; Hollenberg, S.M. Fluid Resuscitation Influences Cardiovascular Performance and Mortality in a Murine Model of Sepsis. Intensive Care Med. 2009, 35, 748–754. [Google Scholar] [CrossRef] [PubMed]
- Assimakopoulos, S.F.; Triantos, C.; Thomopoulos, K.; Fligou, F.; Maroulis, I.; Marangos, M.; Gogos, C.A. Gut-Origin Sepsis in the Critically Ill Patient: Pathophysiology and Treatment. Infection 2018, 46, 751–760. [Google Scholar] [CrossRef] [PubMed]
- Gibson, D.L.; Ma, C.; Rosenberger, C.M.; Bergstrom, K.S.B.; Valdez, Y.; Huang, J.T.; Khan, M.A.; Vallance, B.A. Toll-like Receptor 2 Plays a Critical Role in Maintaining Mucosal Integrity during Citrobacter Rodentium-Induced Colitis. Cell Microbiol. 2008, 10, 388–403. [Google Scholar] [CrossRef]
- Kamdar, K.; Johnson, A.M.F.; Chac, D.; Myers, K.; Kulur, V.; Truevillian, K.; DePaolo, R.W. Innate Recognition of the Microbiota by Toll-like Receptor-1 Promotes Epithelial Homeostasis and Prevents Chronic Inflammation. J. Immunol. 2018, 201, 230–242. [Google Scholar] [CrossRef] [Green Version]
- Cario, E.; Gerken, G.; Podolsky, D.K. Toll-like Receptor 2 Controls Mucosal Inflammation by Regulating Epithelial Barrier Function. Gastroenterology 2007, 132, 1359–1374. [Google Scholar] [CrossRef]
- Dheer, R.; Santaolalla, R.; Davies, J.M.; Lang, J.K.; Phillips, M.C.; Pastorini, C.; Vazquez-Pertejo, M.T.; Abreu, M.T. Intestinal Epithelial Toll-Like Receptor 4 Signaling Affects Epithelial Function and Colonic Microbiota and Promotes a Risk for Transmissible Colitis. Infect. Immun. 2016, 84, 798–810. [Google Scholar] [CrossRef] [Green Version]
- Guo, S.; Al-Sadi, R.; Said, H.M.; Ma, T.Y. Lipopolysaccharide Causes an Increase in Intestinal Tight Junction Permeability in Vitro and in Vivo by Inducing Enterocyte Membrane Expression and Localization of TLR-4 and CD14. Am. J. Pathol. 2013, 182, 375–387. [Google Scholar] [CrossRef] [Green Version]
- Nighot, M.; Al-Sadi, R.; Guo, S.; Rawat, M.; Nighot, P.; Watterson, M.D.; Ma, T.Y. Lipopolysaccharide-Induced Increase in Intestinal Epithelial Tight Permeability Is Mediated by Toll-Like Receptor 4/Myeloid Differentiation Primary Response 88 (MyD88) Activation of Myosin Light Chain Kinase Expression. Am. J. Pathol. 2017, 187, 2698–2710. [Google Scholar] [CrossRef] [Green Version]
- Tatum, P.M.; Harmon, C.M.; Lorenz, R.G.; Dimmitt, R.A. Toll-like Receptor 4 Is Protective against Neonatal Murine Ischemia-Reperfusion Intestinal Injury. J. Pediatr. Surg. 2010, 45, 1246–1255. [Google Scholar] [CrossRef] [Green Version]
- Chen, L.-W.; Chang, W.-J.; Chen, P.-H.; Liu, W.-C.; Hsu, C.-M. TLR Ligand Decreases Mesenteric Ischemia and Reperfusion Injury-Induced Gut Damage through TNF-α Signaling. Shock 2008, 30, 563. [Google Scholar] [CrossRef]
- Kitajima, S.; Takuma, S.; Morimoto, M. Changes in Colonic Mucosal Permeability in Mouse Colitis Induced with Dextran Sulfate Sodium. Exp. Anim. 1999, 48, 137–143. [Google Scholar] [CrossRef] [Green Version]
- Fukata, M.; Michelsen, K.S.; Eri, R.; Thomas, L.S.; Hu, B.; Lukasek, K.; Nast, C.C.; Lechago, J.; Xu, R.; Naiki, Y.; et al. Toll-like Receptor-4 Is Required for Intestinal Response to Epithelial Injury and Limiting Bacterial Translocation in a Murine Model of Acute Colitis. Am. J. Physiol.-Gastrointest. Liver Physiol. 2005, 288, G1055–G1065. [Google Scholar] [CrossRef] [Green Version]
- Rose, W.A.; Sakamoto, K.; Leifer, C.A. TLR9 Is Important for Protection against Intestinal Damage and for Intestinal Repair. Sci. Rep. 2012, 2, 574. [Google Scholar] [CrossRef] [Green Version]
- Chassaing, B.; Ley, R.E.; Gewirtz, A.T. Intestinal Epithelial Cell Toll-like Receptor 5 Regulates the Intestinal Microbiota to Prevent Low-Grade Inflammation and Metabolic Syndrome in Mice. Gastroenterology 2014, 147, 1363–1377.e17. [Google Scholar] [CrossRef] [Green Version]
- Del Olmo-Garcia, M.I.; Merino-Torres, J.F. GLP-1 Receptor Agonists and Cardiovascular Disease in Patients with Type 2 Diabetes. J. Diabetes Res. 2018, 2018, 4020492. [Google Scholar] [CrossRef] [Green Version]
- Bang-Berthelsen, C.H.; Holm, T.L.; Pyke, C.; Simonsen, L.; Søkilde, R.; Pociot, F.; Heller, R.S.; Folkersen, L.; Kvist, P.H.; Jackerott, M.; et al. GLP-1 Induces Barrier Protective Expression in Brunner’s Glands and Regulates Colonic Inflammation. Inflamm. Bowel Dis. 2016, 22, 2078–2097. [Google Scholar] [CrossRef] [Green Version]
- Zatorski, H.; Sałaga, M.; Fichna, J. Role of Glucagon-like Peptides in Inflammatory Bowel Diseases—Current Knowledge and Future Perspectives. Naunyn-Schmiedeberg’s Arch. Pharm. 2019, 392, 1321–1330. [Google Scholar] [CrossRef] [Green Version]
- Lourie, J. A Novel Use of Liraglutide: Induction of Partial Remission in Ulcerative Colitis and Ankylosing Spondylitis. Clin Med Rev Case Rep. 2019, 6, 281. [Google Scholar] [CrossRef]
- Villumsen, M.; Schelde, A.B.; Jimenez-Solem, E.; Jess, T.; Allin, K.H. GLP-1 Based Therapies and Disease Course of Inflammatory Bowel Disease. eClinicalMedicine 2021, 37, 100979. [Google Scholar] [CrossRef]
- Helmstädter, J.; Keppeler, K.; Aust, F.; Küster, L.; Frenis, K.; Filippou, K.; Vujacic-Mirski, K.; Tsohataridis, S.; Kalinovic, S.; Kröller-Schön, S.; et al. GLP-1 Analog Liraglutide Improves Vascular Function in Polymicrobial Sepsis by Reduction of Oxidative Stress and Inflammation. Antioxidants 2021, 10, 1175. [Google Scholar] [CrossRef]
- Hirasawa, H.; Oda, S.; Nakamura, M. Blood Glucose Control in Patients with Severe Sepsis and Septic Shock. World J. Gastroenterol. 2009, 15, 4132–4136. [Google Scholar] [CrossRef] [Green Version]
- Jafar, N.; Edriss, H.; Nugent, K. The Effect of Short-Term Hyperglycemia on the Innate Immune System. Am. J. Med. Sci. 2016, 351, 201–211. [Google Scholar] [CrossRef]
- Leonidou, L.; Mouzaki, A.; Michalaki, M.; DeLastic, A.L.; Kyriazopoulou, V.; Bassaris, H.P.; Gogos, C.A. Cytokine Production and Hospital Mortality in Patients with Sepsis-Induced Stress Hyperglycemia. J. Infect. 2007, 55, 340–346. [Google Scholar] [CrossRef]
- Yang, F.; Zeng, F.; Luo, X.; Lei, Y.; Li, J.; Lu, S.; Huang, X.; Lan, Y.; Liu, R. GLP-1 Receptor: A New Target for Sepsis. Front. Pharm. 2021, 12, 706908. [Google Scholar] [CrossRef]
- Xiao, R.; Wang, R.; Li, S.; Kang, X.; Ren, Y.; Sun, E.; Wang, C.; He, J.; Zhan, J. Preliminary Evaluation of Potential Properties of Three Probiotics and Their Combination with Prebiotics on GLP-1 Secretion and Type 2 Diabetes Alleviation. J. Food Qual. 2022, 2022, e8586843. [Google Scholar] [CrossRef]
- Pegah, A.; Abbasi-Oshaghi, E.; Khodadadi, I.; Mirzaei, F.; Tayebinai, H. Probiotic and Resveratrol Normalize GLP-1 Levels and Oxidative Stress in the Intestine of Diabetic Rats. Metab. Open 2021, 10, 100093. [Google Scholar] [CrossRef]
- Wei, S.-H.; Chen, Y.-P.; Chen, M.-J. Selecting Probiotics with the Abilities of Enhancing GLP-1 to Mitigate the Progression of Type 1 Diabetes in Vitro and in Vivo. J. Funct. Foods 2015, 18, 473–486. [Google Scholar] [CrossRef]
- Yadav, H.; Lee, J.-H.; Lloyd, J.; Walter, P.; Rane, S.G. Beneficial Metabolic Effects of a Probiotic via Butyrate-Induced GLP-1 Hormone Secretion. J. Biol. Chem. 2013, 288, 25088–25097. [Google Scholar] [CrossRef] [Green Version]
- Williams, L.; Alshehri, A.; Robichaud, B.; Cudmore, A.; Gagnon, J. The Role of the Bacterial Muramyl Dipeptide in the Regulation of GLP-1 and Glycemia. Int. J. Mol. Sci. 2020, 21, 5252. [Google Scholar] [CrossRef]
- Klein, A.; Deckert, V.; Schneider, M.; Dutrillaux, F.; Hammann, A.; Athias, A.; Le Guern, N.; Pais de Barros, J.-P.; Desrumaux, C.; Masson, D.; et al. Alpha-Tocopherol Modulates Phosphatidylserine Externalization in Erythrocytes: Relevance in Phospholipid Transfer Protein-Deficient Mice. Arter. Thromb. Vasc. Biol. 2006, 26, 2160–2167. [Google Scholar] [CrossRef] [Green Version]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lebrun, L.J.; Dusuel, A.; Xolin, M.; Le Guern, N.; Grober, J. Activation of TLRs Triggers GLP-1 Secretion in Mice. Int. J. Mol. Sci. 2023, 24, 5333. https://doi.org/10.3390/ijms24065333
Lebrun LJ, Dusuel A, Xolin M, Le Guern N, Grober J. Activation of TLRs Triggers GLP-1 Secretion in Mice. International Journal of Molecular Sciences. 2023; 24(6):5333. https://doi.org/10.3390/ijms24065333
Chicago/Turabian StyleLebrun, Lorène J., Alois Dusuel, Marion Xolin, Naig Le Guern, and Jacques Grober. 2023. "Activation of TLRs Triggers GLP-1 Secretion in Mice" International Journal of Molecular Sciences 24, no. 6: 5333. https://doi.org/10.3390/ijms24065333