1. Introduction
The inherited blistering skin disease epidermolysis bullosa is associated with severe debilitating conditions, which significantly impact patients’ quality of life [
1]. In general, the severity and heterogeneity of the disease justifies the ongoing development of therapeutic options at the DNA and RNA level, of which some are at the preclinical development stage, whereas others have already been applied clinically [
2]. Furthermore, cDNA replacement and splicing-modulating therapies, gene editing via designer nucleases, in particular, has become the main focus of research over the last years. Although clinical translation remains a big challenge, designer nucleases such as CRISPR/Cas9 are constantly improving in regards to efficiency and safety and therefore hold great promise for future clinical applications as therapeutics for genodermatoses [
3,
4,
5]. For the junctional form of EB (JEB), mutations in genes encoding integrin-α6β4, laminin-332, and type XVII collagen (C17) are the predominant cause of disease [
6]. We have recently developed patient-specific Cas9 nuclease and nickase-based targeting strategies to either reframe [
7] or repair [
8] the
COL17A1 gene using endogenous end joining (EJ) as well as homology-directed repair (HDR) pathways in JEB keratinocytes. Mutations in
COL17A1 result in reduced or absent expression of type XVII collagen (C17), a transmembrane protein that is necessary for the connection of the basal keratinocytes of the epidermis to the underlying lamina lucida [
9,
10]. Using a
COL17A1 reframing approach, we could successfully restore C17 expression in 46% of treated primary JEB keratinocytes. The reframed C17 protein variants expressed, encoded
COL17A1 transcripts predominantly carrying 25- and 37-nt deletions [
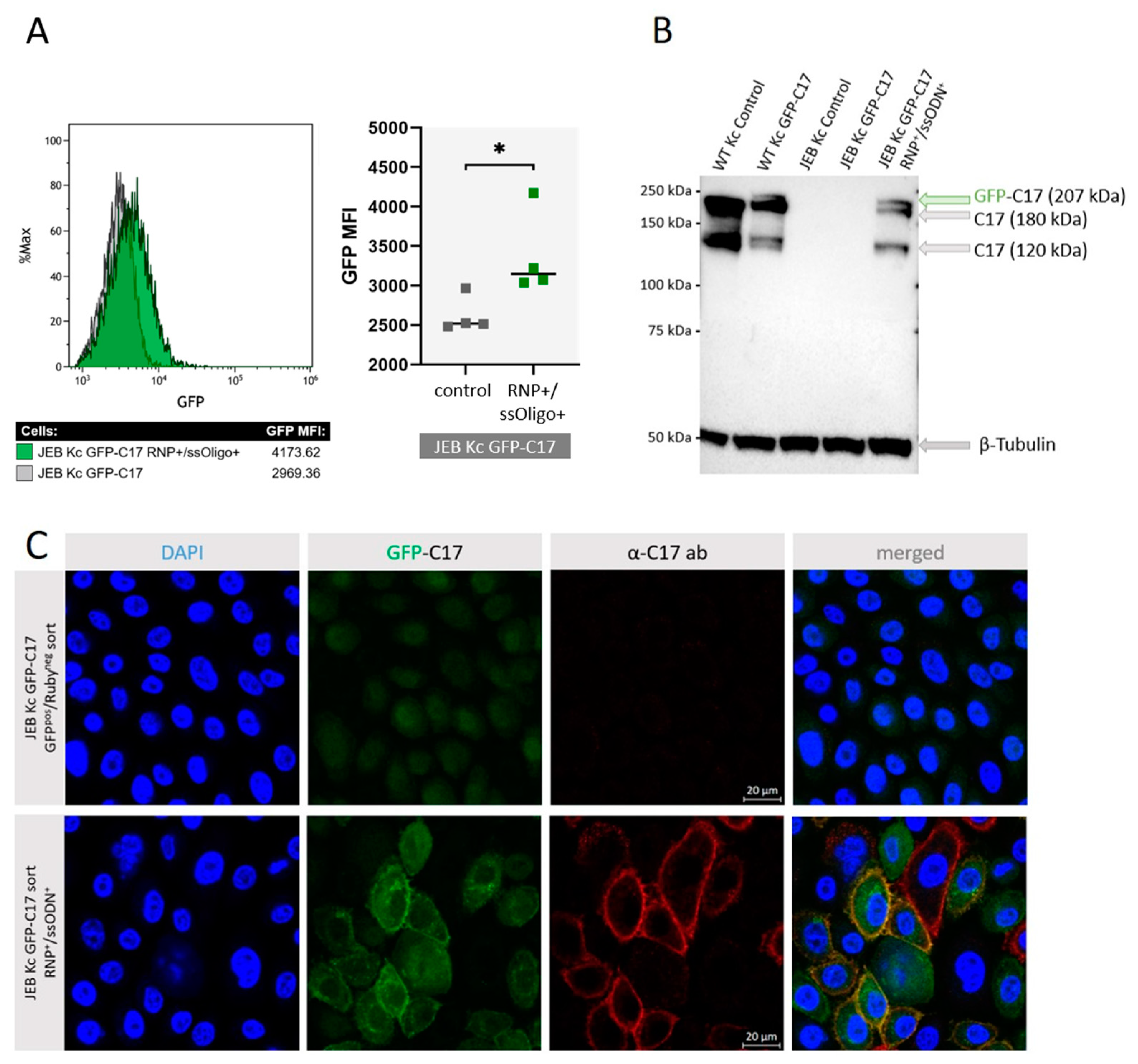
7]. Additional inclusion of a single-stranded oligonucleotide (ssODN) template for HDR increased C17 restoration levels up to 60% as assessed by flow cytometry [
8]. Here, the combined induction of EJ and HDR led to a significant boost of gene repair/reframing and, most importantly, to an increased adhesive strength of C17 to its binding partner laminin-332, as well as an accurate deposition of C17 along the basement membrane zone (BMZ) of 3D-skin equivalents upon epidermal stratification. However, as the downstream analyses of
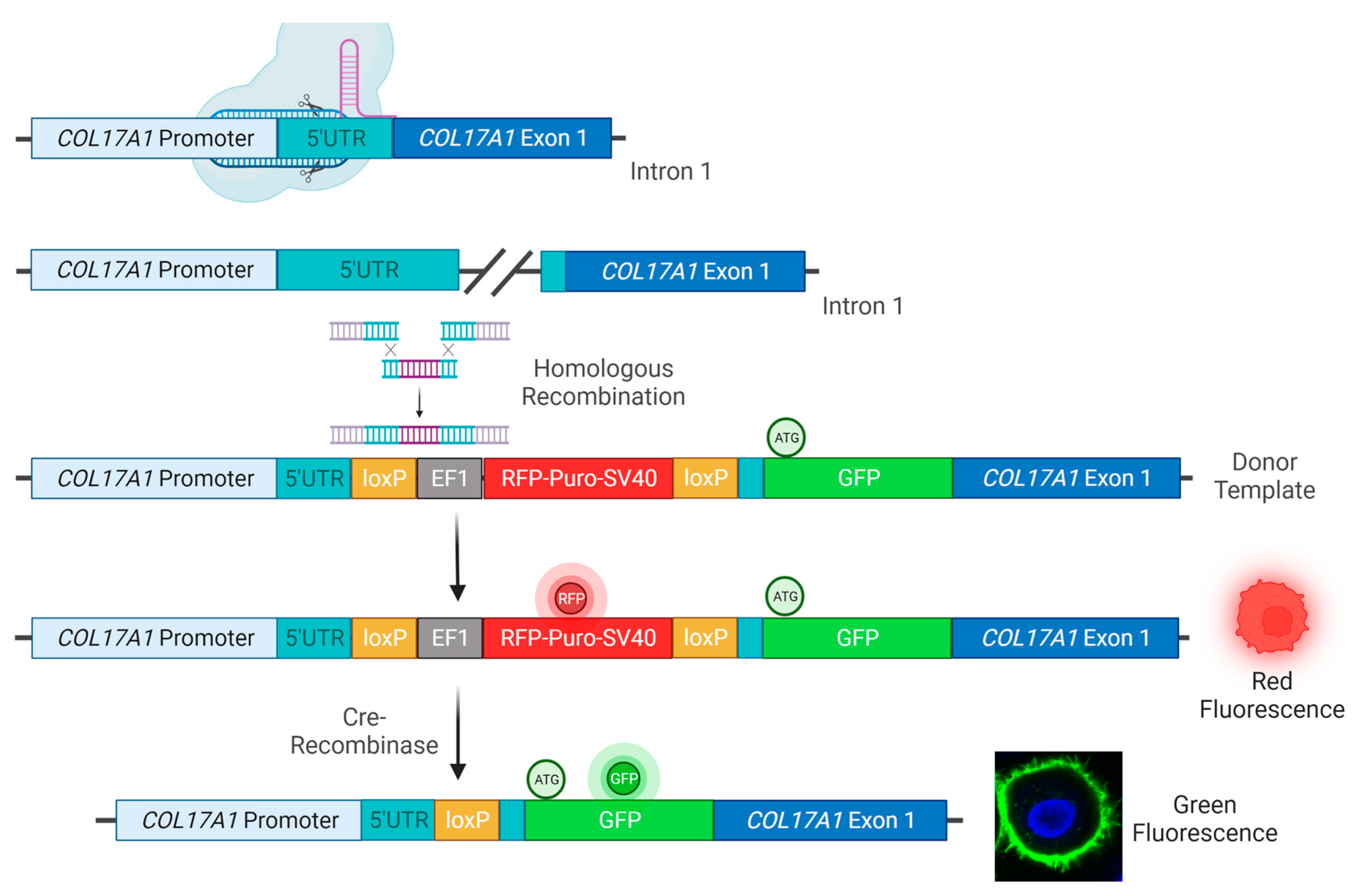
COL17A1 editing outcomes are time-consuming and costly, we developed a screening system to facilitate and accelerate the detection of C17 restoration, based on the incorporation of the GFP reporter cassette directly upstream of the coding sequence for C17 in both wild-type (WT) and JEB human keratinocytes, keeping the expression of the chimeric gene under the control of the endogenous
COL17A1 promotor.
3. Discussion
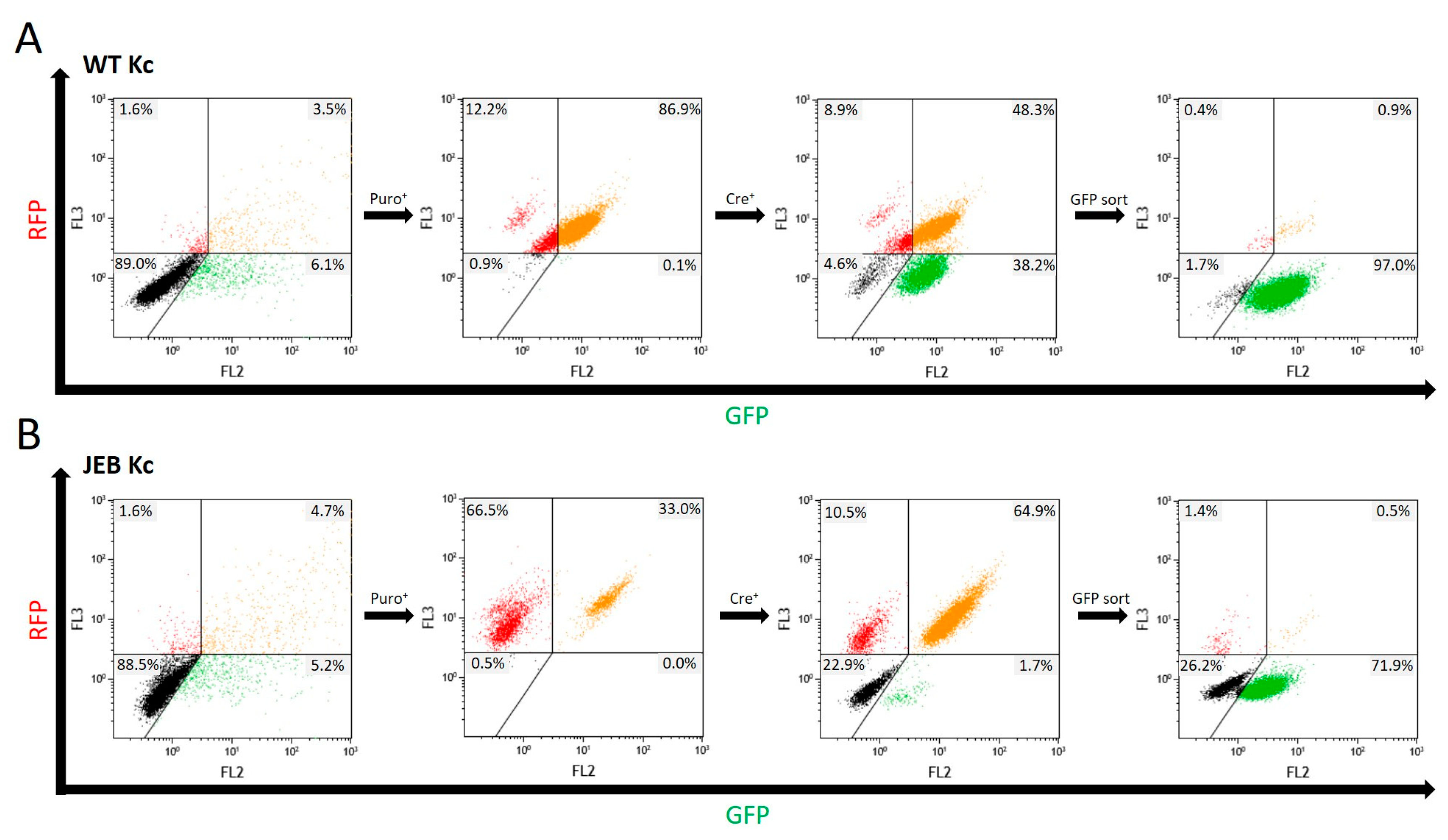
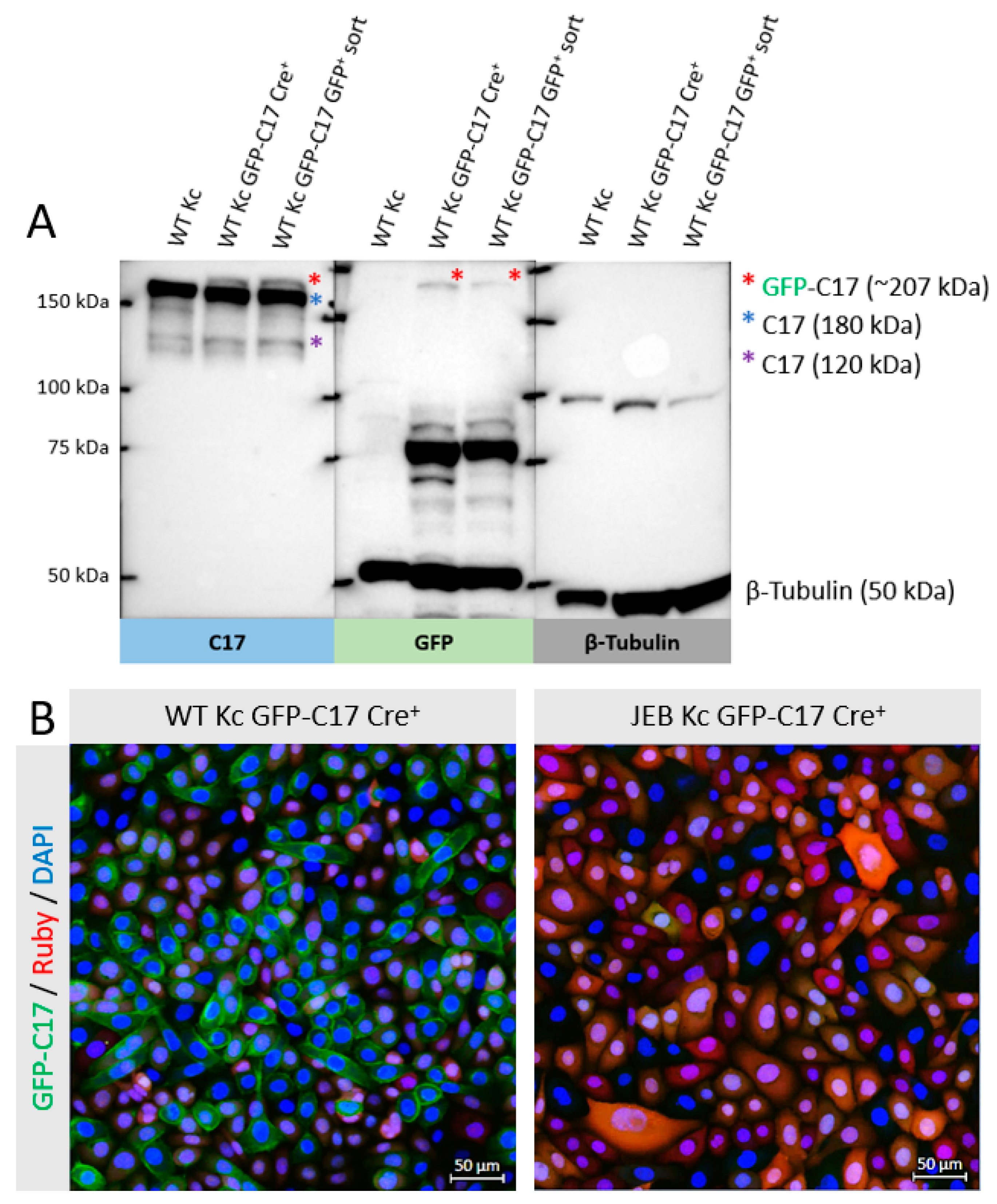
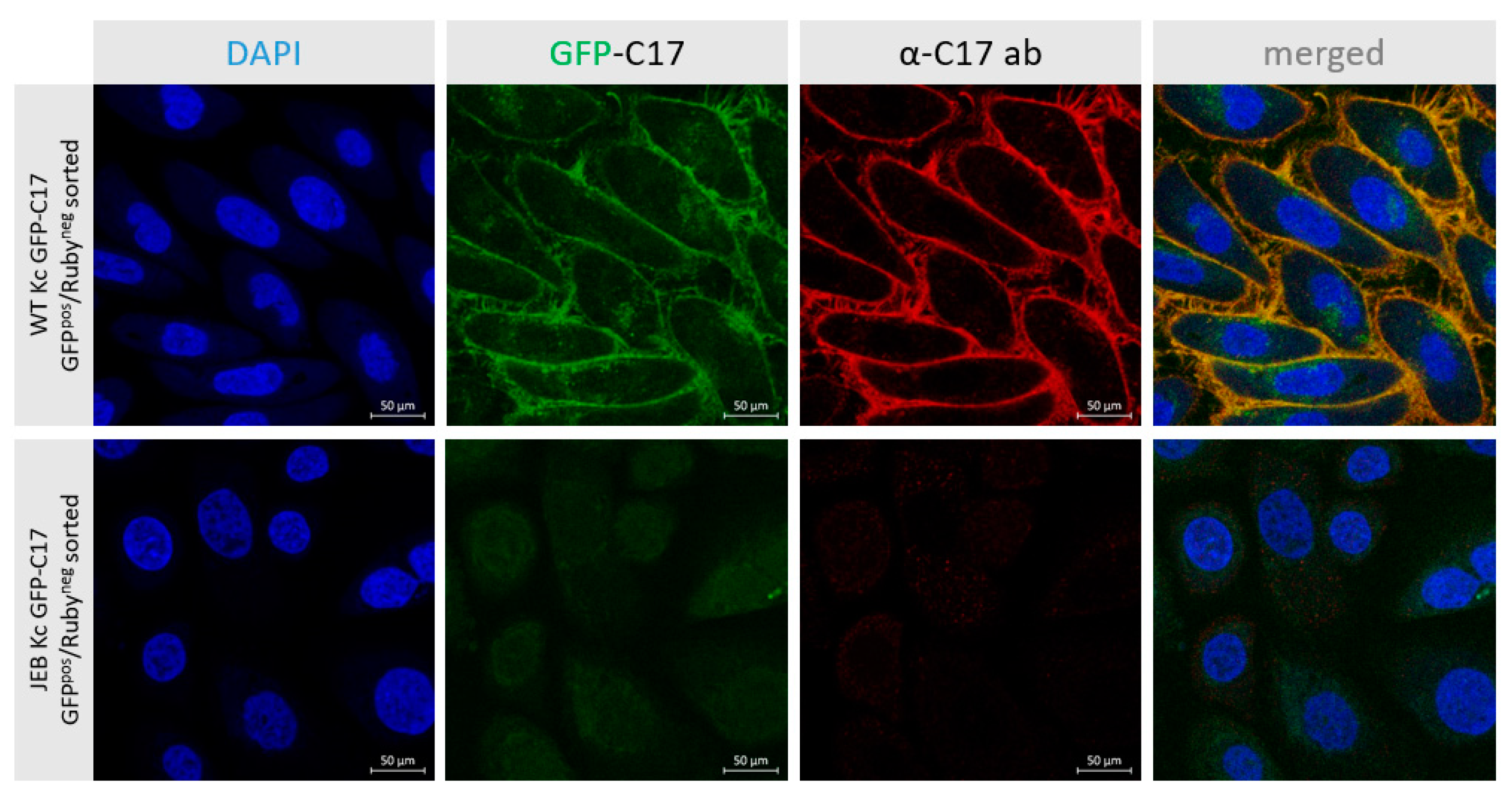
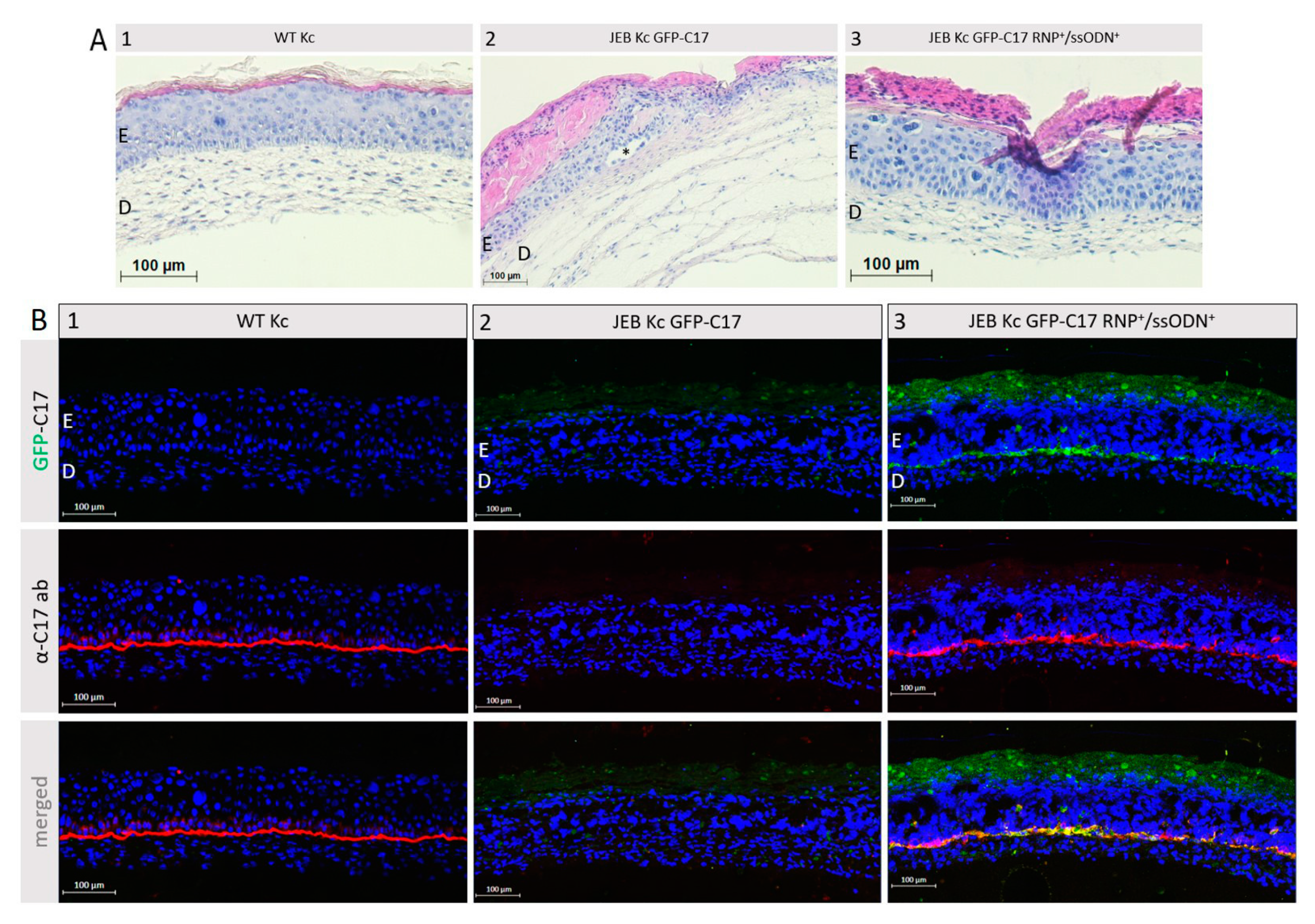
In this study, we generated a reporter cell line that would be suitable for conducting protein-protein interaction studies or, for our purpose, the selection and comparison of gene editing strategies/molecules for junctional EB. The direct labeling of mutant C17 with GFP enables a fast screening procedure for COL17A1-specific gene editing molecules, as the repair outcome can be subsequently analyzed live upon treatment of the cells according to the respective fluorescence signal generated. Currently, in our JEB-screening cell line, we have a mixture of cells carrying GFP-COL17A1 on one or both alleles. Subsequent CRISPR/Cas9-mediated C17 repair can lead to functional correction in none, one, or both alleles. Depending on which type of correction takes place on which allele, different staining patterns can be observed, resulting in heterogenous outcomes in bulk-treated cells as seen in our immunofluorescence analyses of JEB keratinocyte monolayers, as well as of 3D skin equivalents. Only edits at the GFP-COL17A1 allele that restores the reading frame would result in enhanced reporter expression, whereas edits at the parental (non-GFP) allele that result in functional protein expression can only be detected by additional C17-antibody staining. As such, based on GFP expression alone, we would underestimate the functional correction efficiencies achieved by our gene editing strategies. In order to increase the robustness of our screening system, we are now planning to isolate and expand single-cell clones in which the GFP cassette is accurately fused to COL17A1 in a homozygous manner. This will decrease the heterogeneity of the outcomes observed and enable us to directly distinguish and estimate total protein restoration (increased GFP expression) from functional protein restoration (membrane GFP expression), thereby further enhancing the utility of the cell line.
Once optimized the fluorescent JEB cell line can be used for protein restoration analysis upon gene editing in vivo using appropriate mouse models as recently described [
13]. Currently, it is necessary to conduct time- and resource-intensive immunofluorescence stainings on fixed keratinocytes in order to evaluate the expression and localization of C17 within JEB Kc [
7,
8]. By using our novel GFP-C17 cell line we are now able to perform live cell imaging directly after treatment, thereby facilitating and accelerating the analysis of new gene editing platforms. This includes recently developed technologies such as prime editing, as well as future gene editing technologies [
14].
4. Materials and Methods
4.1. Cell Culture & Cell Lines
JEB keratinocytes (JEB-282-Kc) were isolated from a patient biopsy and carry a homozygous frameshift mutation (c.3899-3900delCT/c.3899-3900delCT) within exon 52 of
COL17A1 [
7,
8]. Wild-type keratinocytes (hKc-1090 and WT-200-Kc) and wild-type fibroblasts (HC-941-Fibs) were isolated from healthy donors upon informed consent. All cells were subsequently immortalized through transduction of the human papillomavirus (HPV) proteins E6 and E7 and grown at 37 °C and 5% CO
2 in a humidified incubator [
15]. WT Kc as well as JEB Kc were cultured in CnT-Prime Epithelial Proliferation Medium (CELLnTEC, Bern, Switzerland) with primocin (InvivoGen, Toulouse, France). Wild-type fibroblasts were cultivated in CnT Fibroblast Medium (CELLnTEC, Bern, Switzerland) with primocin (InvivoGen, Toulouse, France).
4.2. Transfection and Electroporation of Keratinocytes and Fibroblasts
The sgRNA was amplified from the genomic DNA of WT Kc using the following primer combination: fw primer: 5′-ATCCGGTTATCAGCTTCAACAGTG-3′; rv primer: 5′-GGTTATCAGCTTCAACAGTGGTTT-3′. The PCR product was cloned as double-stranded DNA oligonucleotide into the CMV-hspCas9-T2A-GFP-H1-gRNA linearized SmartNucleaseTM and SmartNickaseTM vector (System Biosciences, Palo Alto, CA, USA) according to the manufacturer’s protocol.
The homology arms (HA) for homologous recombination were amplified from genomic DNA isolated from WT Kc using a forward primer (5′- GATCGAATTCGAATGTTTCATACTTTGAGAAGAGTAAAGTTCCATAC-3′) and a reverse primer (5′-GATCAGATCTTGTTGAAGCTGATAACCAAAAACATGATTTCAAGAATT-3′) specific for the 5′HA and a forward primer (5′-GATCGGATCCGGCGCAGGAGCCGGCGCCACCGATGTAACCAAGAAAAACAAACGAGATGGAAC-3′) and a reverse primer (5′-GATCGCATGCTGACTTAGAGTTGGTAGGAGGAGAGTTTAAATG-3′) specific for the 3′HA, respectively. The PCR products were cloned into HR Targeting Vector HR110PA-1 (MCS1-EF1α-RFP-T2A-Puro-pA-MCS2) (System Biosciences, Palo Alto, CA, USA), carrying an RFP-Puromycin selection cassette flanked by loxP sites. The GFP sequence was amplified from pMXs-IRES-GFP Retroviral Vector (Cell Biolabs, San Diego, CA, USA) using a GFP-specific forward (5′-GATCGGATCCGTGGGGACCTATAATTACTAAAAACAATGATAAGTATGACTATCATGGTTTCTGATTTTTCCTGCAGGTGGCTATGGTATGGTGAGCAAGGGCGAGGAGC-3′) and reverse (5′-GATCGGATCCGGCGCCTGCTCCCTTGTACAGCTCGTCCATGCC-3′) primer. The nucleotide sequence 5′-ggagcaggcgccGGATCCggcgcaggagccggcgccacc-3′ encoding the Gly-Ser-Linker for GFP-C17 fusion was included in the 3′HA forward and GFP reverse primer, respectively.
Keratinocytes were grown to a confluency of 80–90% in 6-well plates and transfected with 1.5 µg of each Cas9/sgRNA-expressing plasmids and HR donor plasmid and 0.3 µL Xfect™ Transfection Reagent per µg plasmid DNA according to the manufacturer’s protocol (Takara Bio, Kusatu, Japan).
For restoration of C17 in the JEB GFP-C17
mut screening cell line, the cells were trypsinized and washed with PBS prior to nucleofection using the Neon™ Transfection System 10 μL Kit (Invitrogen, Waltham, MA, USA). For RNP complexing, 0.3 µL of spCas9 (IDT, Coralville, IA, USA) and 0.486 µL of sgRNA (IDT, Coralville, IA, USA) were mixed and incubated for 10 min at RT. Then, 250 ng of the HR donor template was added to the RNPs. 3 × 10
5 keratinocytes were resuspended in buffer “R” provided in the Neon Transfection Kit, mixed with pre-complexed RNPs, and electroporated using the Neon transfection system according to Petkovic et al. [
8]. Treated cells were then seeded into antibiotic-free CnT-Prime Medium (CELLnTEC, Bern, Switzerland) in 6-well plates. After 1 day of incubation, primocin (InvivoGen, Toulouse, France) was added.
4.3. Flow Cytometric Analysis and FACS
Transfection efficiency was evaluated via flow cytometric analysis 48 h post-transfection using a Beckman Coulter FC500 FACS analyzer (Beckman Coulter, Brea, CA, USA). Data analysis was performed using the Kaluza software (Beckman Coulter, Brea, CA, USA). Statistical analysis was performed using GraphPad Prism 9 (GraphPad Software, La Jolla, CA, USA) for paired Student’s t-tests.
For Flow Cytometry mediated sorting of GFPpos cells, cells were trypsinized and washed with PBS. They were maintained in PBS and sorted using the FACS Aria III cell sorter (BD Biosciences, Franklin Lakes, NJ, USA).
4.4. Immunofluorescence Staining of C17 in Keratinocytes
5 × 104 cells were seeded into chamber slides and fixed with 4% Para-formaldehyde for 10 min at RT upon reaching confluency. After three washing steps with 1× PBS, cells were incubated with anti-collagen type XVII rabbit monoclonal antibody (#184996; Abcam, Cambridge, UK), diluted 1:1000 in 1× blocking reagent (Roche Diagnostics, Vienna, Austria) for 1 h at RT. Cells were incubated for 1 h with Alexa-Fluor-488 goat anti-rabbit or Alexa-Fluor-594 goat-anti-rabbit (Thermo Fisher Scientific, Waltham, MA, USA), diluted 1:400 in PBS, in combination with 4′, 6-Diamidino-2-phenylindol (DAPI) (1:2000) (VWR, Vienna, Austria). Cells were stored in PBS and analyzed using the confocal laser scanning microscope Axio Observer Z1 attached to LSM700 (Carl Zeiss, Jena, Germany).
4.5. DNA Isolation and Integration PCR
Genomic DNA of keratinocytes was isolated using the ReliaPrep™ Blood gDNA Miniprep System kit (Promega, Madison, WI, USA), according to the manufacturer’s protocol. Integration of the donor template was detected by PCR analysis using a specific forward primer (5′-GGGCGACACCCTGGTGAACC-3′) binding within the GFP sequence and reverse primer (5′- CCTGGGCAACAGAGCAAGACTCCGTCTC-‘3) binding Intron 2 of COL17A1. As well as with specific forward primer (5′-GGGACGGGACCCTTGCTGTGCATTCC-3′) binding Intron 1 of COL17A1 and reverse primer (5′-GGTTCACCAGGGTGTCGCCC-3′) binding GFP.
4.6. Protein Isolation and Western Blot Analysis
Protein lysates were collected from cell pellets using radioimmunoprecipitation assay (RIPA) buffer (Santa Cruz Biotechnology, Heidelberg, Germany). For loading onto an 8% BisTris-Gel (Invitrogen, Waltham, MA, USA), cell lysates were mixed with a 4× loading buffer (0.25 M Tris, 8% SGS, 30% glycerol, 0.02% bromophenol blue [pH 6.8]) and denatured at 95 °C for 5 min. Western blot analysis was performed as previously described [
7,
16]. The nitrocellulose membrane was blocked with 10× blocking reagent from Roche Diagnostics (Roche Diagnostics GmbH, Mannheim, Germany) diluted 1:10 in Tris-buffered saline with 0.2% Tween (TBS-T) for 1 h at room temperature. C17 was detected via a monoclonal anti-C17 antibody (#184996) (Abcam, Cambridge, UK) at a dilution of 1:2000 in TBS-T. The membrane was incubated overnight at 4 °C with the primary antibody. GFP detection was performed using a GFP-specific antibody (MBL International Corporation, Woburn, MA, USA) diluted 1:300 in blocking reagent (Roche Diagnostics GmbH, Mannheim, Germany) (diluted 1:10 in TBS-T). Antibody staining against β-tubulin (ab6064; Abcam, Cambridge, UK) was used as loading control in a dilution of 1:2000. A goat anti-rabbit HRP-labelled antibody (Dako, Santa Clara, CA, USA) was used as a secondary antibody. The membrane was incubated with the secondary antibody for 1 h at a dilution of 1:300 in TBS-T. Protein band visualization was performed using the Immobilon Western Chemiluminescent HRP Substrate (Merck, Darmstadt, Germany) and the ChemiDoc XRS Imager (BioRad, Hercules, CA, USA).
4.7. Generation of Skin Equivalents
A human fibrin scaffold was used for the generation of skin equivalents. 1 × 105 fibroblasts (HC-941-Fibs) were immersed in a fibrinogen scaffold consisting of DMEM with 20% FCS, fibrinogen (F4883; Sigma–Aldrich, St. Louis, MO, USA) in 0.9% NaCl (final concentration = 25 mg/mL), thrombin (T8885; Sigma–Aldrich, St. Louis, MO, USA) dissolved in 25 mM CaCl2 and aprotinin (A6279; Sigma–Aldrich, St. Louis, MO, USA). The scaffold was directly prepared in Falcon® permeable support inserts with a 0.4 µm transparent PET membrane (Corning, New York, NY, USA) and placed in BioCoat™ Deep-Well Plates (6-well (Corning, New York, NY, USA) for 1 h at 37 °C and 5% CO2. 1 × 106 keratinocytes (HC-941-Kc, JEB-282-Kc GFP-C17mut or JEB-282-Kc GFP-C17mut RNP/ssODN treated) per well were seeded on top of the matrix and grown to confluence in DMEM:Ham’s F-12 Green’s keratinocyte medium. Skin equivalents were then raised to an air-liquid interface and cultured for 28 days to allow stratification, before harvesting. SEs were either fixed in OCT (Scigen, Tissue-Plus, Paramount, CA, USA) for cryosectioning or placed into 4% PFA for subsequent paraffin embedding.
4.8. Immunofluorescence Staining of Skin Equivalents
Cryosections of 8 µm were fixed with acetone:methanol (1:1) at −20 °C for 15 min and washed twice with PBS for 5 min. The slides were then incubated for 1.5 h with a human-specific anti-C17 antibody (#184996; Abcam, Cambridge, United Kingdom) diluted 1:500 in 1× blocking reagent (Roche Diagnostics GmbH, Mannheim, Germany) in TBS-T. After three washing steps with PBS, secondary antibody Alexa Fluor 549 goat anti-rabbit IgG (H + L) (Thermo Fisher Scientific, Waltham, MA, USA) diluted 1:500 in PBS and DAPI (4′,6-Diamidino-2-phenylindol) (Thermo Fisher Scientific, Waltham, MA, USA) diluted 1:1000 in PBS for 1 h at room temperature. After three washing steps with PBS for 5 min, cryosections were covered with a DAKO fluorescent mounting medium (Agilent, Santa Clara, CA, USA). For laminin-332 staining, slides were incubated for 1.5 h with a human-specific anti-laminin alpha 3/laminin-5 antibody (#MAB2144; R&D systems, Minneapolis, MN, USA) diluted 1:2000 in 1× blocking reagent (Roche Diagnostics GmbH, Mannheim, Germany) in TBS-TX. Staining with secondary antibody and DAPI was performed in the same way as for C17. Cryosections were analyzed using the confocal laser scanning microscope Axio Observer Z1 attached to LSM700 (Carl Zeiss).
4.9. H&E Staining of Skin Equivalents
Paraffin sections of 8 µm were used for hematoxylin and eosin stainings, where hematoxylin stains the cell nuclei and eosin stains the extracellular matrix and cytoplasm. After 30 washing steps with H2O, slides were re-incubated for 6 min in Mayer’s hemalum solution (Merck, Kenilworth, IL, USA) followed by another 30 washing steps in H2O. The cryosections are dipped 10 times in a 0.3% HCl/EtOH solution, 30 times in H2O and further incubated for 2 min in a 0.5% Eosin G solution (Merck, Kenilworth, IL, USA). Slides were then washed 30 times in H2O, 30 times in isopropanol, and 30 times in HistoChoice® Clearing Agent (Merck, Kenilworth, IL, USA). Finally, paraffin sections were covered with ROTI®Histokitt (Carl Roth, Karlsruhe, Germany) and stored at 4 °C.

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}





