
Anticancer Small-Molecule Agents Targeting Eukaryotic Elongation Factor 1A: State of the Art

Abstract
:1. Introduction
2. Recent Advances in Anticancer eEF1A-Targeting Agents
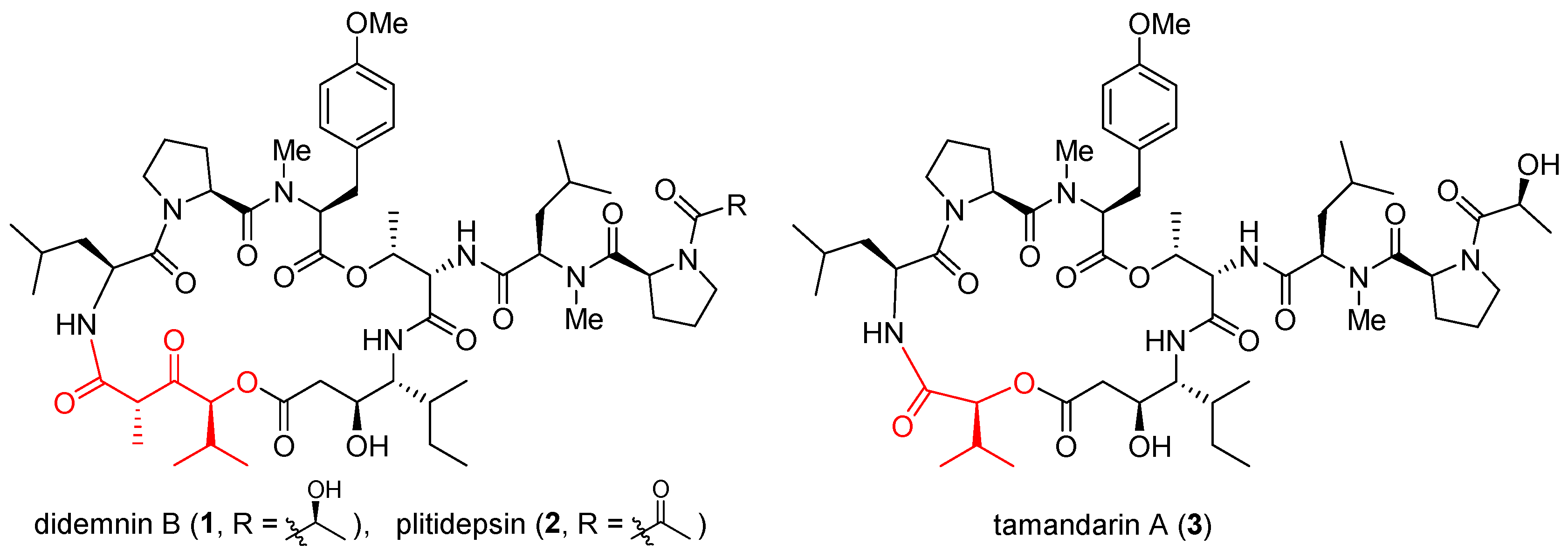
2.1. Didemnins and Tamandarins
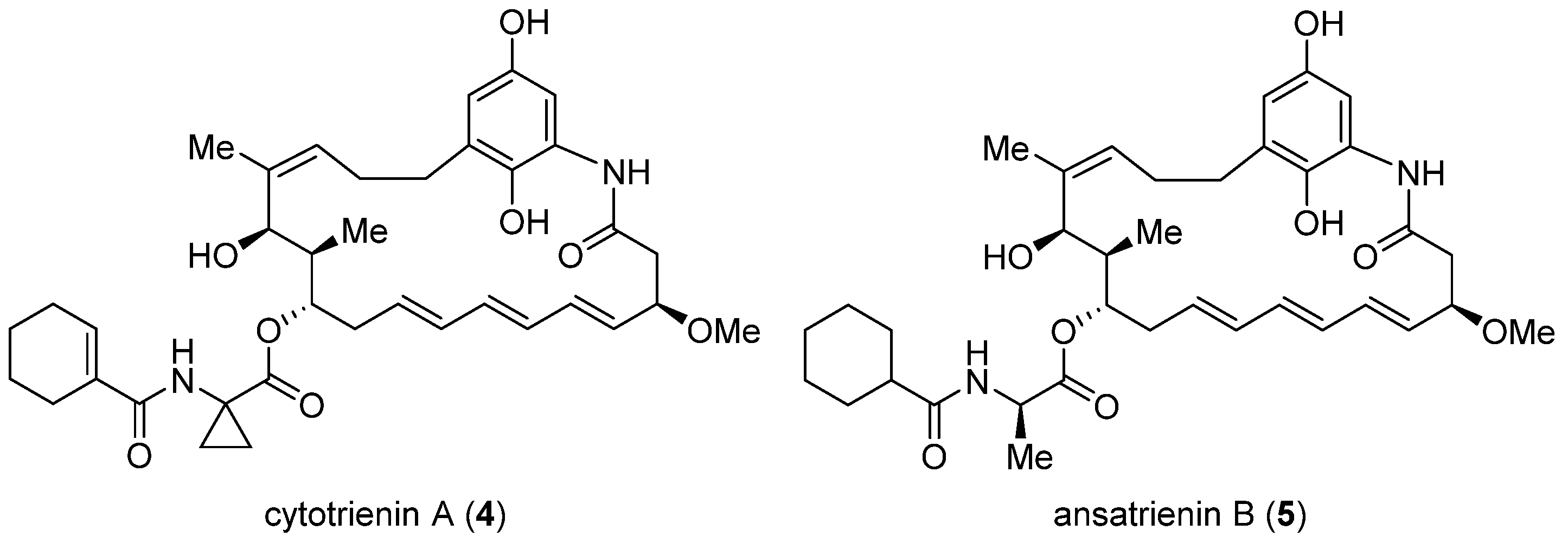
2.2. Cytotrienin A and Ansatrienin B
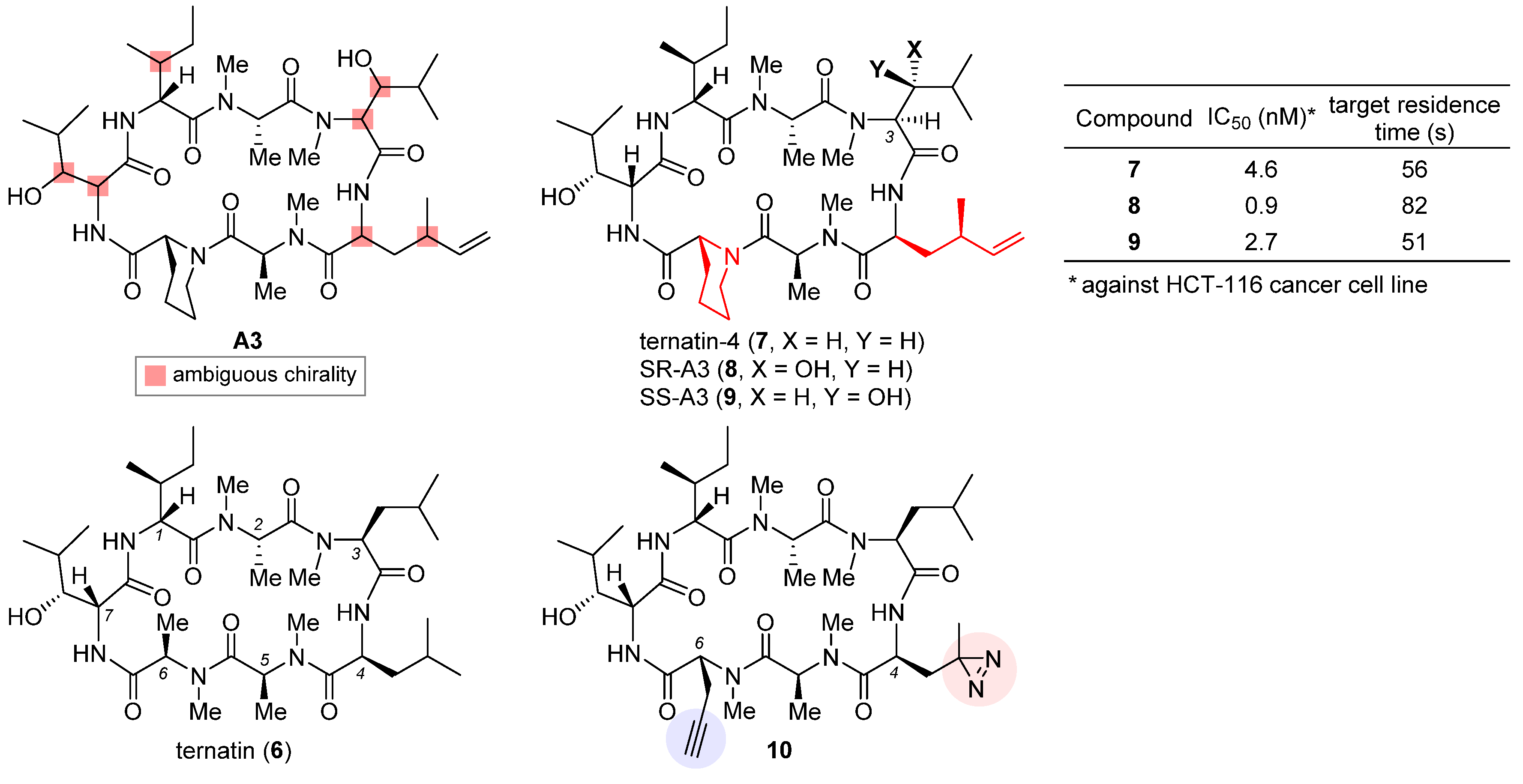
2.3. Ternatin-4
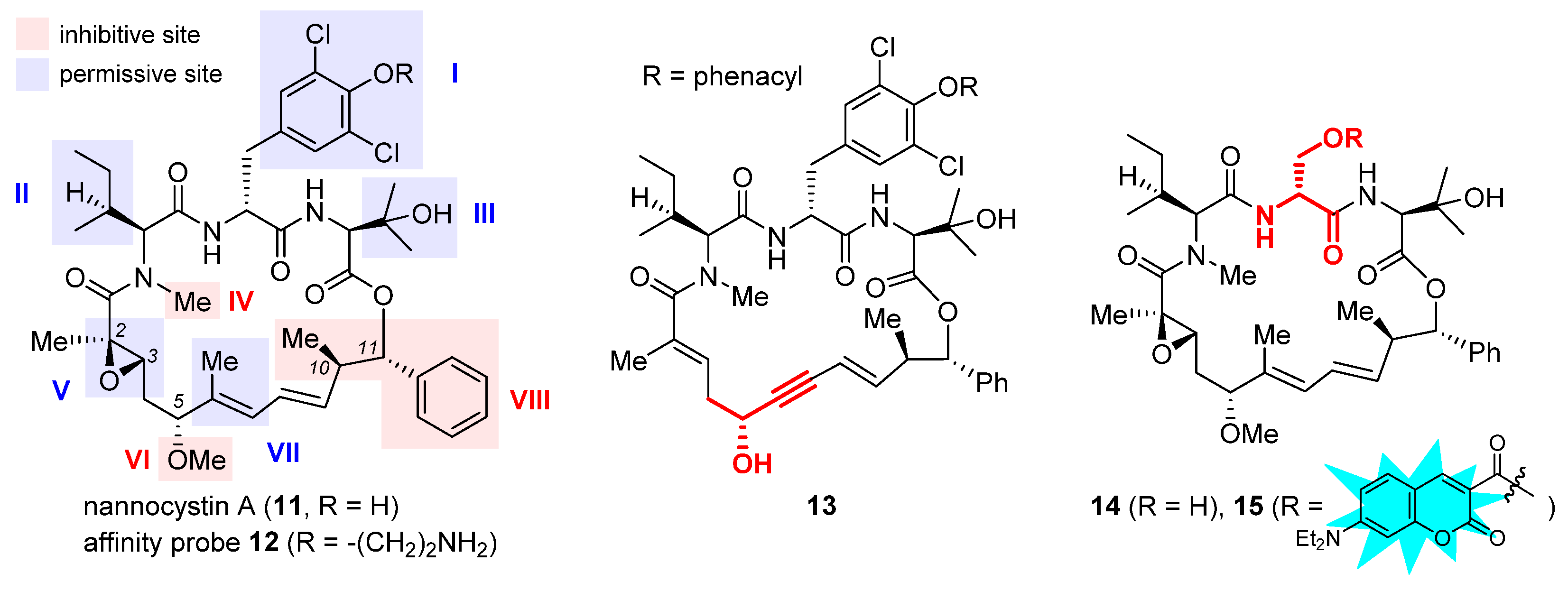
2.4. Nannocystin A
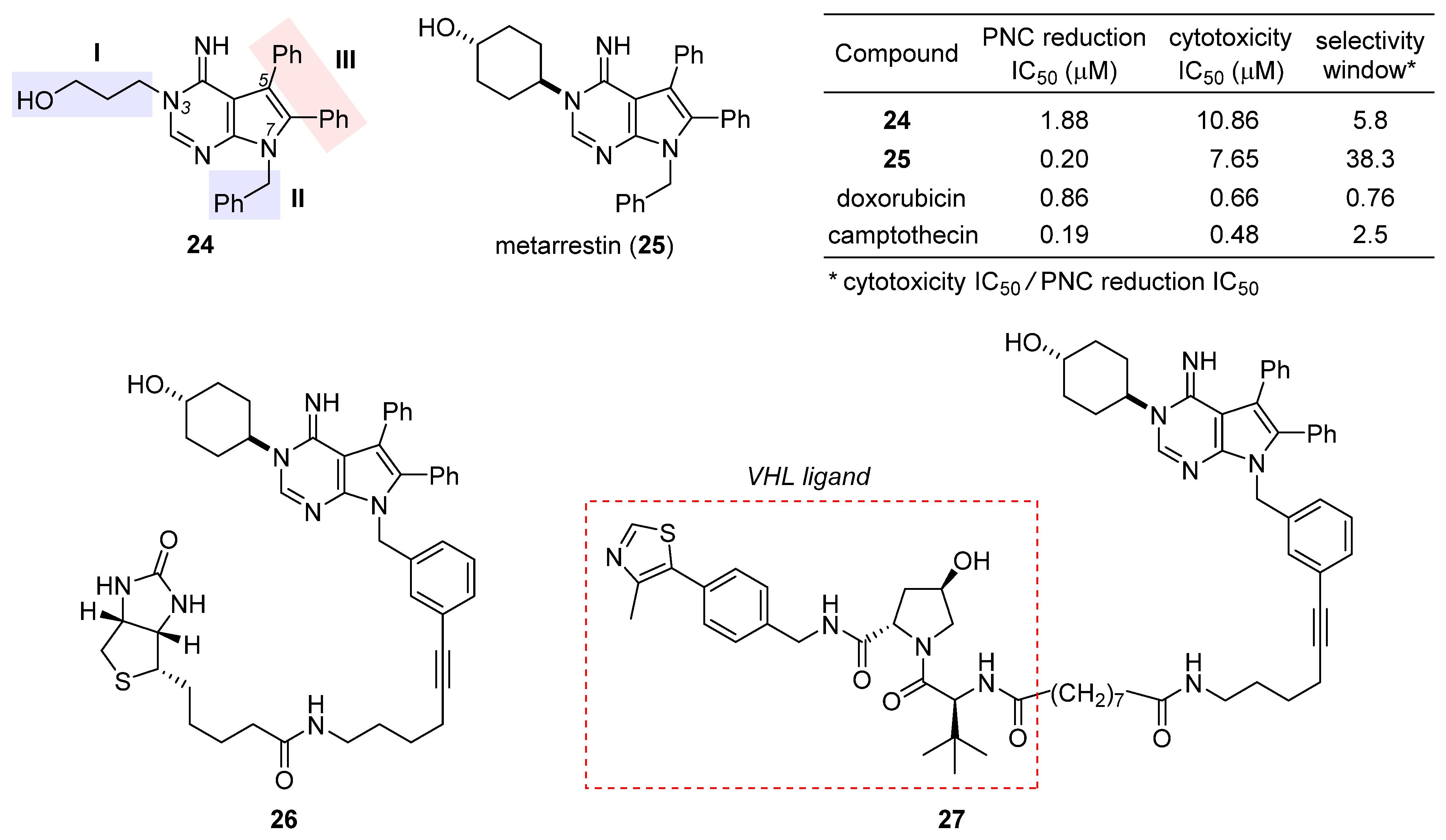
2.5. Metarrestin
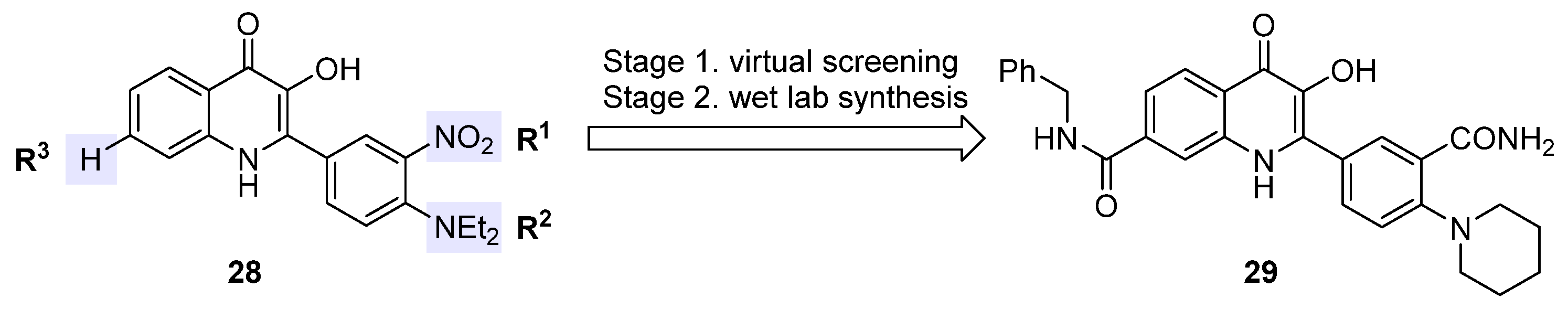
2.6. 2-Phenyl-3-Hydroxy-4(1H)-Quinolinones
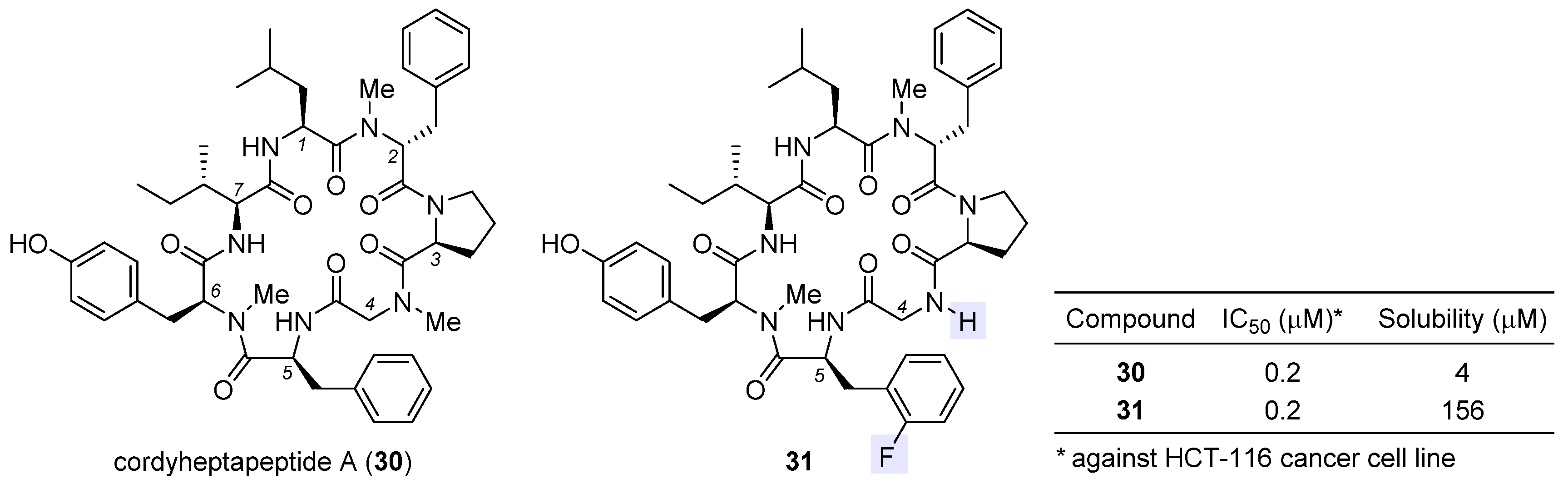
2.7. Cordyheptapeptide A
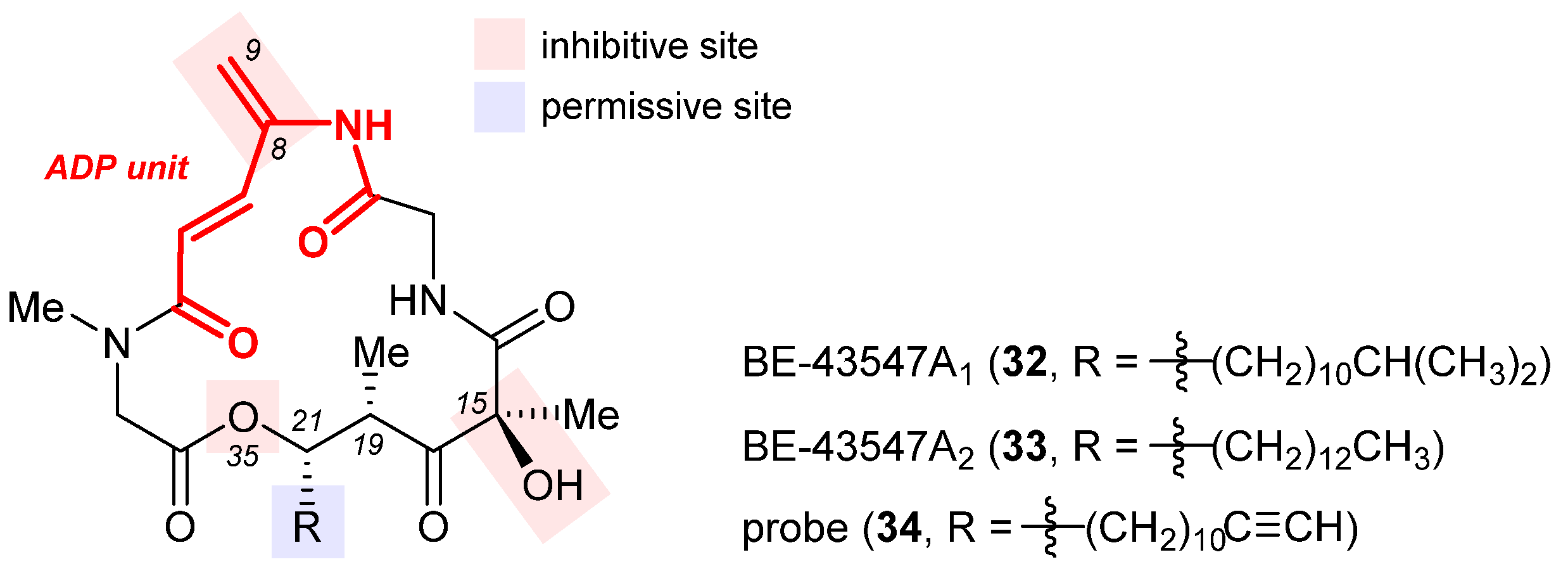
2.8. BE-43547A2
3. Conclusions and Future Perspectives

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Anticancer Efficacy and Selectivity | Reference |
|---|---|---|
| Didemnins |
| [36,41,50,51,52,53,54,55,56] |
| Tamandarins |
| [36,49] |
| Cytotrienin A (4) |
| [61,64] |
| Ansatrienin B (5) |
| [142] |
| Narciclasine |
| [39] |
| Synthetic flavonoids |
| [40] |
| Ternatin-4 (7) |
| [68] |
| Nannocystin A (11) |
| [73,91] |
| Metarrestin (25) |
| [42,100,101,102,103,104] |
| Synthetic quinolinones |
| [110] |
| Cordyheptapeptide A (30) |
| [114] |
| BE-43547A2 (33) |
| [127,130] |
Author Contributions
Funding
Conflicts of Interest
References
- Burrell, R.A.; McGranahan, N.; Bartek, J.; Swanton, C. The causes and consequences of genetic heterogeneity in cancer evolution. Nature 2013, 501, 338–345. [Google Scholar] [CrossRef] [PubMed]
- Kitano, H. Cancer as a robust system: Implications for anticancer therapy. Nat. Rev. Cancer 2004, 4, 227–235. [Google Scholar] [CrossRef] [PubMed]
- Barker, H.E.; Paget, J.T.; Khan, A.A.; Harrington, K.J. The tumour microenvironment after radiotherapy: Mechanisms of resistance and recurrence. Nat. Rev. Cancer 2015, 15, 409–425. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Avendaño, C.; Menendez, J.C. Medicinal Chemistry of Anticancer Drugs, 2nd ed.; Elsevier: Amsterdam, The Netherlands, 2015. [Google Scholar]
- Papież, M.A.; Krzyściak, W. Biological therapies in the treatment of cancer—Update and new directions. Int. J. Mol. Sci. 2021, 22, 11694. [Google Scholar] [CrossRef]
- Kaiser, M.; Semeraro, M.D.; Herrmann, M.; Absenger, G.; Gerger, A.; Renner, W. Immune aging and immunotherapy in cancer. Int. J. Mol. Sci. 2021, 22, 7016. [Google Scholar] [CrossRef]
- Diwan, D.; Cheng, L.; Usmani, Z.; Sharma, M.; Holden, N.; Willoughby, N.; Sangwan, N.; Baadhe, R.R.; Liu, C.; Gupta, V.K. Microbial cancer therapeutics: A promising approach. Semin. Cancer Biol. 2022, 86, 931–950. [Google Scholar] [CrossRef]
- Sawant, S.S.; Patil, S.M.; Gupta, V.; Kunda, N.K. Microbes as medicines: Harnessing the power of bacteria in advancing cancer treatment. Int. J. Mol. Sci. 2020, 21, 7575. [Google Scholar] [CrossRef]
- Zhong, L.; Li, Y.; Xiong, L.; Wang, W.; Wu, M.; Yuan, T.; Yang, W.; Tian, C.; Miao, Z.; Wang, T. Small molecules in targeted cancer therapy: Advances, challenges, and future perspectives. Signal Transduct. Target Ther. 2021, 6, 201. [Google Scholar] [CrossRef]
- Ohishi, T.; Kaneko, M.K.; Yoshida, Y.; Takashima, A.; Kato, Y.; Kawada, M. Current Targeted Therapy for Metastatic Colorectal Cancer. Int. J. Mol. Sci. 2023, 24, 1702. [Google Scholar] [CrossRef]
- Lau, K.H.; Tan, A.M.; Shi, Y. New and emerging targeted therapies for advanced breast cancer. Int. J. Mol. Sci. 2022, 23, 2288. [Google Scholar] [CrossRef]
- Negrutskii, B.S.; El’skaya, A.V. Eukaryotic translation elongation factor 1α: Structure, expression, functions, and possible role in aminoacyl-tRNA channeling. Prog. Nucleic Acid Res. Mol. Biol. 1998, 60, 47–78. [Google Scholar]
- Sanders, J.; Brandsma, M.; Janssen, G.M.C.; Dijk, J.; Moeller, W. Immunofluorescence studies of human fibroblasts demonstrate the presence of the complex of elongation factor-1βγδ in the endoplasmic reticulum. J. Cell Sci. 1996, 109, 1113–1117. [Google Scholar] [CrossRef] [PubMed]
- Kjær, S.; Wind, T.; Ravn, P.; Østergaard, M.; Clark, B.F.C.; Nissim, A. Generation and epitope mapping of high-affinity scFv to eukaryotic elongation factor 1A by dual application of phage display. Eur. J. Biochem. 2001, 268, 3407–3415. [Google Scholar] [CrossRef]
- Migliaccio, N.; Ruggiero, I.; Martucci, N.M.; Sanges, C.; Arbucci, S.; Tatè, R.; Rippa, E.; Arcari, P.; Lamberti, A. New insights on the interaction between the isoforms 1 and 2 of human translation elongation factor 1A. Biochimie 2015, 118, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Dever, T.E.; Dinman, J.D.; Green, R. Translation elongation and recoding in eukaryotes. Cold Spring Harb. Perspect. Biol. 2018, 10, a032649. [Google Scholar] [CrossRef] [PubMed]
- Mateyak, M.K.; Kinzy, T.G. eEF1A: Thinking outside the ribosome. J. Biol. Chem. 2010, 285, 21209–21213. [Google Scholar] [CrossRef] [Green Version]
- Sasikumar, A.N.; Perez, W.B.; Kinzy, T.G. The many roles of the eukaryotic elongation factor 1 complex. Wiley Interdiscip. Rev. RNA 2012, 3, 543–555. [Google Scholar] [CrossRef] [Green Version]
- Li, D.; Wei, T.; Abbott, C.M.; Harrich, D. The unexpected roles of eukaryotic translation elongation factors in RNA virus replication and pathogenesis. Microbiol. Mol. Biol. Rev. 2013, 77, 253–266. [Google Scholar] [CrossRef] [Green Version]
- Carriles, A.A.; Mills, A.; Muñoz-Alonso, M.-J.; Gutiérrez, D.; Domínguez, J.M.; Hermoso, J.A.; Gago, F. Structural Cues for Understanding eEF1A2 Moonlighting. ChemBioChem 2021, 22, 374–391. [Google Scholar] [CrossRef]
- Thornton, S.; Anand, N.; Purcell, D.; Lee, J. Not just for housekeeping: Protein initiation and elongation factors in cell growth and tumorigenesis. J. Mol. Med. 2003, 81, 536–548. [Google Scholar] [CrossRef]
- Lamberti, A.; Caraglia, M.; Longo, O.; Marra, M.; Abbruzzese, A.; Arcari, P. The translation elongation factor 1A in tumorigenesis, signal transduction and apoptosis: Review article. Amino Acids 2004, 26, 443–448. [Google Scholar] [CrossRef]
- Abbas, W.; Kumar, A.; Herbein, G. The eEF1A Proteins: At the Crossroads of Oncogenesis, Apoptosis, and Viral Infections. Front. Oncol. 2015, 5, 75. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hussey, G.S.; Chaudhury, A.; Dawson, A.E.; Lindner, D.J.; Knudsen, C.R.; Wilce, M.C.; Merrick, W.C.; Howe, P.H. Identification of an mRNP Complex Regulating Tumorigenesis at the Translational Elongation Step. Mol. Cell 2011, 41, 419–431. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, S.; Hausmann, S.; Carlson, S.M.; Fuentes, M.E.; Francis, J.W.; Pillai, R.; Lofgren, S.M.; Hulea, L.; Tandoc, K.; Lu, J.; et al. METTL13 Methylation of eEF1A Increases Translational Output to Promote Tumorigenesis. Cell 2019, 176, 491–504. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kobayashi, D.; Tokuda, T.; Sato, K.; Okanishi, H.; Nagayama, M.; Hirayama-Kurogi, M.; Ohtsuki, S.; Araki, N. Identification of a specific translational machinery via TCTP-EF1A2 interaction regulating NF1-associated tumor growth by affinity purification and data-independent mass spectrometry acquisition (AP-DIA). Mol. Cell. Proteom. 2019, 18, 245–262. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amiri, A.; Noei, F.; Jeganathan, S.; Kulkarni, G.; Pinke, D.E.; Lee, J.M. eEF1A2 activates Akt and stimulates Akt-dependent actin remodeling, invasion and migration. Oncogene 2007, 26, 3027–3040. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Z.; Qi, C.F.; Shin, D.M.; Zingone, A.; Newbery, H.J.; Kovalchuk, A.L.; Abbott, C.M.; Morse, H.C.I. Eef1a2 promotes cell growth, inhibits apoptosis and activates JAK/STAT and AKT signaling in mouse plasmacytomas. PLoS ONE 2010, 5, e10755. [Google Scholar] [CrossRef] [Green Version]
- Pellegrino, R.; Calvisi, D.F.; Neumann, O.; Kolluru, V.; Wesely, J.; Chen, X.; Wang, C.; Wuestefeld, T.; Ladu, S.; Elgohary, N.; et al. EEF1A2 inactivates p53 by way of PI3K/AKT/mTOR-dependent stabilization of MDM4 in hepatocellular carcinoma. Hepatology 2014, 59, 1886–1899. [Google Scholar] [CrossRef] [Green Version]
- Losada, A.; Munoz-Alonso, M.J.; Martinez-Diez, M.; Gago, F.; Dominguez, J.M.; Martinez-Leal, J.F.; Galmarini, C.M. Binding of eEF1A2 to the RNA-dependent protein kinase PKR modulates its activity and promotes tumor cell survival. Br. J. Cancer 2018, 119, 1410–1420. [Google Scholar] [CrossRef] [Green Version]
- Itagaki, K.; Sasada, M.; Iyoda, T.; Imaizumi, T.; Haga, M.; Kuga, A.; Inomata, H.; Fukai, F.; Miyazaki, S.; Kondo, Y.; et al. Exposure of the cryptic de-adhesive site FNIII14 in fibronectin molecule and its binding to membrane-type eEF1A induce migration and invasion of cancer cells via β1-integrin inactivation. Am. J. Cancer Res. 2020, 10, 3990–4004. [Google Scholar]
- Jia, L.; Ge, X.; Du, C.; Chen, L.; Zhou, Y.; Xiong, W.; Xiang, J.; Li, G.; Xiao, G.; Fang, L.; et al. EEF1A2 interacts with HSP90AB1 to promote lung adenocarcinoma metastasis via enhancing TGF-β/SMAD signalling. Brit. J. Cancer 2021, 124, 1301–1311. [Google Scholar] [CrossRef]
- Mills, A.; Gago, F. On the Need to Tell Apart Fraternal Twins eEF1A1 and eEF1A2, and Their Respective Outfits. Int. J. Mol. Sci. 2021, 22, 6973, Human eFE1A has two predominant isoforms eEF1A1 and eEF1A2. According to the literature, some agents were reported to target eEF1A without knowing whether it is eEF1A1, eEF1A2, or both, whereas others target only one isoform. For a recent discussion about these two isoforms. [Google Scholar] [CrossRef]
- Crews, C.M.; Collins, J.L.; Lane, W.S.; Snapper, M.L.; Schreiber, S.L. GTP-dependent binding of the antiproliferative agent didemnin to elongation factor 1α. J. Biol. Chem. 1994, 269, 15411–15414. [Google Scholar] [CrossRef]
- Losada, A.; Munoz-Alonso, M.J.; Garcia, C.; Sanchez-Murcia, P.A.; Martinez-Leal, J.F.; Dominguez, J.M.; Lillo, M.P.; Gago, F.; Galmarini, C.M. Translation Elongation Factor eEF1A2 is a Novel Anticancer Target for the Marine Natural Product Plitidepsin. Sci. Rep. 2016, 6, 35100. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Currano, J.N.; Carroll, P.J.; Joullie, M.M. Didemnins, tamandarins and related natural products. Nat. Prod. Rep. 2012, 29, 404–424. [Google Scholar] [CrossRef] [PubMed]
- Lindqvist, L.; Robert, F.; Merrick, W.; Kakeya, H.; Fraser, C.; Osada, H.; Pelletier, J. Inhibition of translation by cytotrienin A--a member of the ansamycin family. RNA 2010, 16, 2404–2413. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carelli, J.D.; Sethofer, S.G.; Smith, G.A.; Miller, H.R.; Simard, J.L.; Merrick, W.C.; Jain, R.K.; Ross, N.T.; Taunton, J. Ternatin and improved synthetic variants kill cancer cells by targeting the elongation factor-1A ternary complex. eLife 2015, 4, e10222. [Google Scholar] [CrossRef] [PubMed]
- Van Goietsenoven, G.; Hutton, J.; Becker, J.P.; Lallemand, B.; Robert, F.; Lefranc, F.; Pirker, C.; Vandenbussche, G.; Van Antwerpen, P.; Evidente, A.; et al. Targeting of eEF1A with Amaryllidaceae isocarbostyrils as a strategy to combat melanomas. FASEB J. 2010, 24, 4575–4584. [Google Scholar] [CrossRef] [Green Version]
- Yao, N.; Chen, C.Y.; Wu, C.Y.; Motonishi, K.; Kung, H.J.; Lam, K.S. Novel flavonoids with antiproliferative activities against breast cancer cells. J. Med. Chem. 2011, 54, 4339–4349. [Google Scholar] [CrossRef] [Green Version]
- Jimenez, P.C.; Wilke, D.V.; Branco, P.C.; Bauermeister, A.; Rezende-Teixeira, P.; Gaudencio, S.P.; Costa-Lotufo, L.V. Enriching cancer pharmacology with drugs of marine origin. Brit. J. Pharmacol. 2020, 177, 3–27. [Google Scholar] [CrossRef] [Green Version]
- Metarrestin (ML-246) in Subjects with Metastatic Solid Tumors. 2020. Available online: https://ClinicalTrials.gov/show/NCT04222413 (accessed on 2 March 2023).
- Dmitriev, S.E.; Vladimirov, D.O.; Lashkevich, K.A. A Quick Guide to Small-Molecule Inhibitors of Eukaryotic Protein Synthesis. Biochemistry (Moscow) 2020, 85, 1389–1421. [Google Scholar] [CrossRef] [PubMed]
- Brönstrup, M.; Sasse, F. Natural products targeting the elongation phase of eukaryotic protein biosynthesis. Nat. Prod. Rep. 2020, 37, 752–762. [Google Scholar] [CrossRef]
- Burgers, L.D.; Fuerst, R. Natural products as drugs and tools for influencing core processes of eukaryotic mRNA translation. Pharmacol. Res. 2021, 170, 105535. [Google Scholar] [CrossRef]
- Fan, A.; Sharp, P.P. Inhibitors of Eukaryotic Translational Machinery as Therapeutic Agents. J. Med. Chem. 2021, 64, 2436–2465. [Google Scholar] [CrossRef]
- Zhang, J.-N.; Xia, Y.-X.; Zhang, H.-J. Natural cyclopeptides as anticancer agents in the last 20 years. Int. J. Mol. Sci. 2021, 22, 3973. [Google Scholar] [CrossRef] [PubMed]
- Rinehart, K.L., Jr.; Gloer, J.B.; Hughes, R.G., Jr.; Renis, H.E.; McGovren, J.P.; Swynenberg, E.B.; Stringfellow, D.A.; Kuentzel, S.L.; Li, L.H. Didemnins: Antiviral and antitumor depsipeptides from a caribbean tunicate. Science 1981, 212, 933–935. [Google Scholar] [CrossRef] [PubMed]
- Vervoort, H.; Fenical, W.; de Epifanio, R. Tamandarins A and B: New cytotoxic depsipeptides from a Brazilian ascidian of the family Didemnidae. J. Org. Chem. 2000, 65, 782–792. [Google Scholar] [CrossRef]
- Leisch, M.; Egle, A.; Greil, R. Plitidepsin: A potential new treatment for relapsed/refractory multiple myeloma. Future Oncol. 2019, 15, 109–120. [Google Scholar] [CrossRef]
- Chun, H.G.; Davies, B.; Hoth, D.; Suffness, M.; Plowman, J.; Flora, K.; Grieshaber, C.; Leyland-Jones, B. Didemnin B. The first marine compound entering clinical trials as an antineoplastic agent. Investig. New Drugs 1986, 4, 279–284. [Google Scholar]
- Vera, M.D.; Joullie, M.M. Natural products as probes of cell biology: 20 years of didemnin research. Med. Res. Rev. 2002, 22, 102–145. [Google Scholar] [CrossRef]
- Le Tourneau, C.; Raymond, E.; Faivre, S. Aplidine: A paradigm of how to handle the activity and toxicity of a novel marine anticancer poison. Curr. Pharm. Des. 2007, 13, 3427–3439. [Google Scholar] [CrossRef]
- Muñoz-Alonso, M.J.; González-Santiago, L.; Martínez, T.; Losada, A.; Galmarini, C.M.; Muñoz, A. The mechanism of action of plitidepsin. Curr. Opin. Investig. Drugs 2009, 10, 536–542. [Google Scholar] [PubMed]
- Danu, A.; Willekens, C.; Ribrag, V. Plitidepsin: An orphan drug. Expert Opin. Orphan Drugs 2013, 1, 569–580. [Google Scholar] [CrossRef]
- Alonso-Alvarez, S.; Pardal, E.; Sanchez-Nieto, D.; Navarro, M.; Caballero, M.D.; Mateos, M.V.; Martin, A. Plitidepsin: Design, development, and potential place in therapy. Drug Des. Dev. Ther. 2017, 11, 253–264. [Google Scholar] [CrossRef] [Green Version]
- Potts, M.B.; Kim, H.S.; Fisher, K.W.; Hu, Y.; Carrasco, Y.P.; Bulut, G.B.; Ou, Y.-H.; Herrera-Herrera, M.L.; Cubillos, F.; Mendiratta, S.; et al. Using functional signature ontology (FUSION) to identify mechanisms of action for natural products. Sci. Signal. 2013, 6, ra90. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Potts, M.B.; McMillan, E.A.; Rosales, T.I.; Kim, H.S.; Ou, Y.-H.; Toombs, J.E.; Brekken, R.A.; Minden, M.D.; MacMillan, J.B.; White, M.A. Mode of action and pharmacogenomic biomarkers for exceptional responders to didemnin B. Nat. Chem. Biol. 2015, 11, 401–408. [Google Scholar] [CrossRef] [Green Version]
- Losada, A.; Berlanga, J.J.; Molina-Guijarro, J.M.; Jimenez-Ruiz, A.; Gago, F.; Aviles, P.; de Haro, C.; Martinez-Leal, J.F. Generation of endoplasmic reticulum stress and inhibition of autophagy by plitidepsin induces proteotoxic apoptosis in cancer cells. Biochem. Pharmacol. 2020, 172, 113744. [Google Scholar] [CrossRef]
- Kakeya, H.; Zhang, H.P.; Kobinata, K.; Onose, R.; Onozawa, C.; Kudo, T.; Osada, H. Cytotrienin A, a novel apoptosis inducer in human leukemia HL-60 cells. J. Antibiot. 1997, 50, 370–372. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.P.; Kakeya, H.; Osada, H. Novel triene-ansamycins, cytotrienins A and B, inducing apoptosis on human leukemia HL-60 cells. Tetrahedron Lett. 1997, 38, 1789–1792. [Google Scholar] [CrossRef]
- Kakeya, H.; Onose, R.; Osada, H. Caspase-mediated activation of a 36-kDa myelin basic protein kinase during anticancer drug-induced apoptosis. Cancer Res. 1998, 58, 4888–4894. [Google Scholar] [PubMed]
- Watabe, M.; Kakeya, H.; Onose, R.; Osada, H. Activation of MST/Krs and c-Jun N-terminal kinases by different signaling pathways during cytotrienin A-induced apoptosis. J. Biol. Chem. 2000, 275, 8766–8771. [Google Scholar] [CrossRef] [Green Version]
- Yamada, Y.; Taketani, S.; Osada, H.; Kataoka, T. Cytotrienin A, a translation inhibitor that induces ectodomain shedding of TNF receptor 1 via activation of ERK and p38 MAP kinase. Eur. J. Pharmacol. 2011, 667, 113–119. [Google Scholar] [CrossRef]
- Blunt, J.; Cole, T.; Munro, M.; Sun, L.; Weber, J.-F.R.; Ramasamy, K.; Abu Bakar, H.; Abdul Majeed, A.B.B. Bioactive Compounds Derived from Endophytic Aspergillus Fungus Strain Isolated from Garcinia Scortechinii. WO2010062159A1, 3 June 2010. [Google Scholar]
- Briggs, L.H.; Locker, R.H. 459. Flavonols from the bark of Melicope ternata. Part I. The isolation of four new flavonols, meliternatin, meliternin, ternatin, and wharangin. J. Chem. Soc. 1949, 2157–2162, The term ternatin also refers to a flavone-type natural product, 5-dihydroxy-3,3ʹ,7,8-tetramethoxyflavone (CAS registry number: 571-71-1) first reported in 1949. [Google Scholar] [CrossRef]
- Shimokawa, K.; Mashima, I.; Asai, A.; Yamada, K.; Kita, M.; Uemura, D. (−)-Ternatin, a highly N-methylated cyclic heptapeptide that inhibits fat accumulation: Structure and synthesis. Tetrahedron Lett. 2006, 47, 4445–4448. [Google Scholar] [CrossRef]
- Wang, H.-Y.; Yang, H.; Holm, M.; Tom, H.; Oltion, K.; Al-Khdhairawi, A.A.Q.; Weber, J.-F.F.; Blanchard, S.C.; Ruggero, D.; Taunton, J. Synthesis and single-molecule imaging reveal stereospecific enhancement of binding kinetics by the antitumour eEF1A antagonist SR-A3. Nat. Chem. 2022, 14, 1443–1450. [Google Scholar] [CrossRef]
- Copeland, R.A. The drug–target residence time model: A 10-year retrospective. Nat. Rev. Drug Discov. 2016, 15, 87–95. [Google Scholar] [CrossRef]
- Juette, M.F.; Carelli, J.D.; Rundlet, E.J.; Brown, A.; Shao, S.; Ferguson, A.; Wasserman, M.R.; Holm, M.; Taunton, J.; Blanchard, S.C. Didemnin B and ternatin-4 differentially inhibit conformational changes in eEF1A required for aminoacyl-tRNA accommodation into mammalian ribosomes. eLife 2022, 11, e81608. [Google Scholar] [CrossRef] [PubMed]
- Oltion, K.; Carelli, J.D.; Yang, T.; See, S.K.; Wang, H.-Y.; Kampmann, M.; Taunton, J. An E3 ligase network engages GCN1 to promote the degradation of translation factors on stalled ribosomes. Cell 2023, 186, 346–362. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, H.; Kogler, H.; Heyse, W.; Matter, H.; Caspers, M.; Schummer, D.; Klemke-Jahn, C.; Bauer, A.; Penarier, G.; Debussche, L.; et al. Discovery, Structure Elucidation, and Biological Characterization of Nannocystin A, a Macrocyclic Myxobacterial Metabolite with Potent Antiproliferative Properties. Angew. Chem. Int. Ed. 2015, 54, 10145–10148. [Google Scholar] [CrossRef]
- Krastel, P.; Roggo, S.; Schirle, M.; Ross, N.T.; Perruccio, F.; Aspesi, P., Jr.; Aust, T.; Buntin, K.; Estoppey, D.; Liechty, B.; et al. Nannocystin A: An Elongation Factor 1 Inhibitor from Myxobacteria with Differential Anti-Cancer Properties. Angew. Chem. Int. Ed. 2015, 54, 10149–10154. [Google Scholar] [CrossRef]
- Liao, L.; Zhou, J.; Xu, Z.; Ye, T. Concise Total Synthesis of Nannocystin A. Angew. Chem. Int. Ed. 2016, 55, 13263–13266. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Wang, Z. Total Syntheses of Nannocystins A and A0, Two Elongation Factor 1 Inhibitors. Org. Lett. 2016, 18, 4702–4705. [Google Scholar] [CrossRef]
- Yang, Z.; Xu, X.; Yang, C.H.; Tian, Y.; Chen, X.; Lian, L.; Pan, W.; Su, X.; Zhang, W.; Chen, Y. Total Synthesis of Nannocystin A. Org. Lett. 2016, 18, 5768–5770. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.H.; Liu, R.; Liu, B. Total synthesis of nannocystin Ax. Chem. Commun. 2017, 53, 5549–5552. [Google Scholar] [CrossRef] [PubMed]
- Poock, C.; Kalesse, M. Total Synthesis of Nannocystin Ax. Org. Lett. 2017, 19, 4536–4539. [Google Scholar] [CrossRef]
- Liu, Q.; Hu, P.; He, Y. Asymmetric Total Synthesis of Nannocystin A. J. Org. Chem. 2017, 82, 9217–9222. [Google Scholar] [CrossRef]
- Meng, Z.; Souillart, L.; Monks, B.; Huwyler, N.; Herrmann, J.; Mueller, R.; Furstner, A. A “Motif-Oriented” Total Synthesis of Nannocystin Ax. Preparation and Biological Assessment of Analogues. J. Org. Chem. 2018, 83, 6977–6994. [Google Scholar] [CrossRef]
- Wang, Z. The Chemical Syntheses of Nannocystins. Synthesis 2019, 51, 2252–2260. [Google Scholar] [CrossRef]
- Zhang, W. From Target-Oriented to Motif-Oriented: A Case Study on Nannocystin Total Synthesis. Molecules 2020, 25, 5327. [Google Scholar] [CrossRef]
- Fürstner, A. Lessons from Natural Product Total Synthesis: Macrocyclization and Postcyclization Strategies. Acc. Chem. Res. 2021, 54, 861–874. [Google Scholar] [CrossRef]
- Paul, D.; Das, S.; Saha, S.; Sharma, H.; Goswami, R.K. Intramolecular Heck Reaction in Total Synthesis of Natural Products: An Update. Eur. J. Org. Chem. 2021, 2021, 2057–2076. [Google Scholar] [CrossRef]
- Zhang, W. Heck macrocyclization in natural product total synthesis. Nat. Prod. Rep. 2021, 38, 1109–1135. [Google Scholar] [CrossRef]
- Tian, Y.; Xu, X.; Ding, Y.; Hao, X.; Bai, Y.; Tang, Y.; Zhang, X.; Li, Q.; Yang, Z.; Zhang, W.; et al. Synthesis and biological evaluation of nannocystin analogues toward understanding the binding role of the (2R,3S)-Epoxide in nannocystin A. Eur. J. Med. Chem. 2018, 150, 626–632. [Google Scholar] [CrossRef] [PubMed]
- Tian, Y.; Ding, Y.; Xu, X.; Bai, Y.; Tang, Y.; Hao, X.; Zhang, W.; Chen, Y. Total synthesis and biological evaluation of nannocystin analogues modified at the polyketide phenyl moiety. Tetrahedron Lett. 2018, 59, 3206–3209. [Google Scholar] [CrossRef]
- Tian, Y.; Wang, J.; Liu, W.; Yuan, X.; Tang, Y.; Li, J.; Chen, Y.; Zhang, W. Stereodivergent total synthesis of Br-nannocystins underpinning the polyketide (10R,11S) configuration as a key determinant of potency. J. Mol. Struct. 2019, 1181, 568–578. [Google Scholar] [CrossRef]
- Liu, Q.; Yang, X.; Ji, J.; Zhang, S.-L.; He, Y. Novel nannocystin A analogues as anticancer therapeutics: Synthesis, biological evaluations and structure-activity relationship studies. Eur. J. Med. Chem. 2019, 170, 99–111. [Google Scholar] [CrossRef]
- Sun, C.; Liu, R.; Xia, M.; Hou, Y.; Wang, X.; Lu, J.-J.; Liu, B.; Chen, X. Nannocystin Ax, a natural elongation factor 1α inhibitor from Nannocystis sp., suppresses epithelial-mesenchymal transition, adhesion and migration in lung cancer cells. Toxicol. Appl. Pharmacol. 2021, 420, 115535. [Google Scholar] [CrossRef]
- Hou, Y.; Liu, R.; Xia, M.; Sun, C.; Zhong, B.; Yu, J.; Ai, N.; Lu, J.-J.; Ge, W.; Liu, B.; et al. Nannocystin ax, an eEF1A inhibitor, induces G1 cell cycle arrest and caspase-independent apoptosis through cyclin D1 downregulation in colon cancer in vivo. Pharmacol. Res. 2021, 173, 105870. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Tian, Y.; Yuan, X.; Xie, F.; Yu, S.; Cai, J.; Sun, B.; Shan, C.; Zhang, W. Site-directed Late-Stage Diversification of Macrocyclic Nannocystins Facilitating Anticancer SAR and Mode of Action Studies. RSC Med. Chem. 2023, 14, 299–312. [Google Scholar] [CrossRef]
- Gemmer, M.; Chaillet, M.L.; van Loenhout, J.; Cuevas Arenas, R.; Vismpas, D.; Gröllers-Mulderij, M.; Koh, F.A.; Albanese, P.; Scheltema, R.A.; Howes, S.C.; et al. Visualization of translation and protein biogenesis at the ER membrane. Nature 2023, 614, 160–167. [Google Scholar] [CrossRef]
- Pollock, C.; Huang, S. The perinucleolar compartment. Cold Spring Harbor Perspect. Biol. 2010, 2, a000679. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kamath, R.V.; Thor, A.D.; Wang, C.; Edgerton, S.M.; Slusarczyk, A.; Leary, D.J.; Wang, J.; Wiley, E.L.; Jovanovic, B.; Wu, Q.; et al. Perinucleolar compartment prevalence has an independent prognostic value for breast cancer. Cancer Res. 2005, 65, 246–253. [Google Scholar] [CrossRef] [PubMed]
- Norton, J.T.; Pollock, C.B.; Wang, C.; Schink, J.C.; Kim, J.J.; Huang, S. Perinucleolar compartment prevalence is a phenotypic pancancer marker of malignancy. Cancer 2008, 113, 861–869. [Google Scholar] [CrossRef] [Green Version]
- Norton, J.T.; Wang, C.; Gjidoda, A.; Henry, R.W.; Huang, S. The Perinucleolar Compartment Is Directly Associated with DNA. J. Biol. Chem. 2009, 284, 4090–4101. [Google Scholar] [CrossRef] [Green Version]
- Huang, S.; Deerinck, T.J.; Ellisman, M.H.; Spector, D.L. The Dynamic Organization of the Perinucleolar Compartment in the Cell Nucleus. J. Cell Biol. 1997, 137, 965–974. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Norton, J.T.; Titus, S.A.; Dexter, D.; Austin, C.P.; Zheng, W.; Huang, S. Automated High-Content Screening for Compounds That Disassemble the Perinucleolar Compartment. J. Biomol. Screen. 2009, 14, 1045–1053. [Google Scholar] [CrossRef] [Green Version]
- Frankowski, K.J.; Wang, C.; Patnaik, S.; Schoenen, F.J.; Southall, N.; Li, D.; Teper, Y.; Sun, W.; Kandela, I.; Hu, D.; et al. Metarrestin, a perinucleolar compartment inhibitor, effectively suppresses metastasis. Sci. Transl. Med. 2018, 10, eaap8307. [Google Scholar] [CrossRef] [Green Version]
- Frankowski, K.J.; Patnaik, S.; Wang, C.; Southall, N.; Dutta, D.; De, S.; Li, D.; Dextras, C.; Lin, Y.-H.; Bryant-Connah, M.; et al. Discovery and Optimization of Pyrrolopyrimidine Derivatives as Selective Disruptors of the Perinucleolar Compartment, a Marker of Tumor Progression toward Metastasis. J. Med. Chem. 2022, 65, 8303–8331. [Google Scholar] [CrossRef]
- Vilimas, T.; Wang, A.Q.; Patnaik, S.; Hughes, E.A.; Singleton, M.D.; Knotts, Z.; Li, D.; Frankowski, K.; Schlomer, J.J.; Guerin, T.M.; et al. Pharmacokinetic evaluation of the PNC disassembler metarrestin in wild-type and Pdx1-Cre; LSL-KrasG12D/+; Tp53R172H/+ (KPC) mice, a genetically engineered model of pancreatic cancer. Cancer Chemoth. Pharm. 2018, 82, 1067–1080. [Google Scholar] [CrossRef] [Green Version]
- Padilha, E.C.; Shah, P.; Wang, A.Q.; Singleton, M.D.; Hughes, E.A.; Li, D.; Rice, K.A.; Konrath, K.M.; Patnaik, S.; Marugan, J. Metabolism and pharmacokinetics characterization of metarrestin in multiple species. Cancer Chemother. Pharm. 2020, 85, 805–816. [Google Scholar] [CrossRef]
- Bourdi, M.; Rudloff, U.; Patnaik, S.; Marugan, J.; Terse, P.S. Safety assessment of metarrestin in dogs: A clinical candidate targeting a subnuclear structure unique to metastatic cancer cells. Regul. Toxicol. Pharm. 2020, 116, 104716. [Google Scholar] [CrossRef]
- Békés, M.; Langley, D.R.; Crews, C.M. PROTAC targeted protein degraders: The past is prologue. Nat. Rev. Drug Discov. 2022, 21, 181–200. [Google Scholar] [CrossRef]
- Pedrucci, F.; Pappalardo, C.; Marzaro, G.; Ferri, N.; Ferlin, A.; De Toni, L. Proteolysis Targeting Chimeric Molecules: Tuning Molecular Strategies for a Clinically Sound Listening. Int. J. Mol. Sci. 2022, 23, 6630. [Google Scholar] [CrossRef] [PubMed]
- Yao, T.; Xiao, H.; Wang, H.; Xu, X. Recent Advances in PROTACs for Drug Targeted Protein Research. Int. J. Mol. Sci. 2022, 23, 10328. [Google Scholar] [CrossRef]
- Jin, J.; Kabir, M.; Sun, N.; Kaniskan, H.U. Preparation of Heterobifunctional Compounds as Degraders of eEF1A2. WO2022159650A1, 28 July 2022. [Google Scholar]
- Tash, J.S.; Chakrasali, R.; Jakkaraj, S.R.; Hughes, J.; Smith, S.K.; Hornbaker, K.; Heckert, L.L.; Ozturk, S.B.; Hadden, M.K.; Kinzy, T.G.; et al. Gamendazole, an orally active indazole carboxylic acid male contraceptive agent, targets HSP90AB1 (HSP90BETA) and EEF1A1 (eEF1A), and stimulates Il1a transcription in rat sertoli cells. Biol. Reprod. 2008, 78, 1139–1152. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burglová, K.; Rylová, G.; Markos, A.; Prichystalova, H.; Soural, M.; Petracek, M.; Medvedikova, M.; Tejral, G.; Sopko, B.; Hradil, P.; et al. Identification of Eukaryotic Translation Elongation Factor 1-α 1 Gamendazole-Binding Site for Binding of 3-Hydroxy-4(1H)-quinolinones as Novel Ligands with Anticancer Activity. J. Med. Chem. 2018, 61, 3027–3036. [Google Scholar] [CrossRef] [PubMed]
- Isaka, M.; Srisanoh, U.; Lartpornmatulee, N.; Boonruangprapa, T. ES-242 derivatives and cycloheptapeptides from Cordyceps sp. strains BCC 16173 and BCC 16176. J. Nat. Prod. 2007, 70, 1601–1604. [Google Scholar] [CrossRef]
- Rukachaisirikul, V.; Chantaruk, S.; Tansakul, C.; Saithong, S.; Chaicharernwimonkoon, L.; Pakawatchai, C.; Isaka, M.; Intereya, K. A cyclopeptide from the insect pathogenic fungus Cordyceps sp. BCC 1788. J. Nat. Prod. 2006, 69, 305–307. [Google Scholar] [CrossRef]
- Kumar, S.; Dahiya, R.; Khokra, S.L.; Mourya, R.; Chennupati, S.V.; Maharaj, S. Total synthesis and pharmacological investigation of cordyheptapeptide A. Molecules 2017, 22, 682. [Google Scholar] [CrossRef] [Green Version]
- Klein, V.G.; Bray, W.M.; Wang, H.-Y.; Edmondson, Q.; Schwochert, J.; Ono, S.; Naylor, M.R.; Turmon, A.C.; Faris, J.H.; Okada, O.; et al. Identifying the Cellular Target of Cordyheptapeptide A and Synthetic Derivatives. ACS Chem. Biol. 2021, 16, 1354–1364. [Google Scholar] [CrossRef] [PubMed]
- Naylor, M.R.; Ly, A.M.; Handford, M.J.; Ramos, D.P.; Pye, C.R.; Furukawa, A.; Klein, V.G.; Noland, R.P.; Edmondson, Q.; Turmon, A.C.; et al. Lipophilic Permeability Efficiency Reconciles the Opposing Roles of Lipophilicity in Membrane Permeability and Aqueous Solubility. J. Med. Chem. 2018, 61, 11169–11182. [Google Scholar] [CrossRef]
- Chan, J.; Khan, S.N.; Harvey, I.; Merrick, W.; Pelletier, J. Eukaryotic protein synthesis inhibitors identified by comparison of cytotoxicity profiles. RNA 2004, 10, 528–543. [Google Scholar] [CrossRef] [Green Version]
- Schulze, C.J.; Bray, W.M.; Woerhmann, M.H.; Stuart, J.; Lokey, R.S.; Linington, R.G. “Function-first” lead discovery: Mode of action profiling of natural product libraries using image-based screening. Chem. Biol. 2013, 20, 285–295. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dieterich, D.C.; Link, A.J.; Graumann, J.; Tirrell, D.A.; Schuman, E.M. Selective identification of newly synthesized proteins in mammalian cells using bioorthogonal noncanonical amino acid tagging (BONCAT). Proc. Natl. Acad. Sci. USA 2006, 103, 9482–9487. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sánchez-Murcia, P.A.; Cortés-Cabrera, Á.; Gago, F. Structural rationale for the cross-resistance of tumor cells bearing the A399V variant of elongation factor eEF1A1 to the structurally unrelated didemnin B, ternatin, nannocystin A and ansatrienin B. J. Comput. Aided Mol. Des. 2017, 31, 915–928. [Google Scholar] [CrossRef]
- Nishioka, H.; Nakajima, S.; Nagashima, M.; Kojiri, K.; Suda, H. BE-43547 Series Substances, Their Manufacture with Streptomyces Species, and Their Use as Antitumor Agents. JP10147594A, 2 June 1998. [Google Scholar]
- Poulsen, T.B. Total Synthesis of Natural Products Containing Enamine or Enol Ether Derivatives. Acc. Chem. Res. 2021, 54, 1830–1842. [Google Scholar] [CrossRef]
- Villadsen, N.L.; Jacobsen, K.M.; Keiding, U.B.; Weibel, E.T.; Christiansen, B.; Vosegaard, T.; Bjerring, M.; Jensen, F.; Johannsen, M.; Tørring, T.; et al. Synthesis of ent-BE-43547A1 reveals a potent hypoxia-selective anticancer agent and uncovers the biosynthetic origin of the APD-CLD natural products. Nat. Chem. 2017, 9, 264–272. [Google Scholar] [CrossRef] [Green Version]
- Poulsen, T.B. A concise route to the macrocyclic core of the rakicidins. Chem. Commun. 2011, 47, 12837–12839. [Google Scholar] [CrossRef] [PubMed]
- Clement, L.L.; Tsakos, M.; Schaffert, E.S.; Scavenius, C.; Enghild, J.J.; Poulsen, T.B. The amido-pentadienoate-functionality of the rakicidins is a thiol reactive electrophile–development of a general synthetic strategy. Chem. Commun. 2015, 51, 12427–12430. [Google Scholar] [CrossRef] [PubMed]
- Tsakos, M.; Clement, L.L.; Schaffert, E.S.; Olsen, F.N.; Rupiani, S.; Djurhuus, R.; Yu, W.; Jacobsen, K.M.; Villadsen, N.L.; Poulsen, T.B. Total synthesis and biological evaluation of rakicidin A and discovery of a simplified bioactive analogue. Angew. Chem. Int. Ed. 2016, 55, 1030–1035. [Google Scholar] [CrossRef]
- Tsakos, M.; Jacobsen, K.M.; Yu, W.; Villadsen, N.L.; Poulsen, T.B. The rakicidin family of anticancer natural products–synthetic strategies towards a new class of hypoxia-selective cytotoxins. Synlett 2016, 27, 1898–1906. [Google Scholar] [CrossRef]
- Sun, Y.; Ding, Y.; Li, D.; Zhou, R.; Su, X.; Yang, J.; Guo, X.; Chong, C.; Wang, J.; Zhang, W.; et al. Cyclic Depsipeptide BE-43547A2: Synthesis and Activity against Pancreatic Cancer Stem Cells. Angew. Chem. Int. Ed. 2017, 56, 14627–14631. [Google Scholar] [CrossRef]
- Sun, Y.; Su, X.; Zhou, R.; Wang, D.; Zhao, Y.; Jiang, Y.; Wang, L.; Chen, Y. Total synthesis of BE-43547A2. Tetrahedron 2018, 74, 5955–5964. [Google Scholar] [CrossRef]
- Sun, Y.; Zhou, R.; Xu, H.; Wang, D.; Su, X.; Wang, C.; Ding, Y.; Wang, L.; Chen, Y. Syntheses and biological evaluation of BE-43547A2 analogs modified at O35 ester and C15-OH sites. Tetrahedron 2019, 75, 1808–1818. [Google Scholar] [CrossRef]
- Liu, C.; Wang, L.; Sun, Y.; Zhao, X.; Chen, T.; Su, X.; Guo, H.; Wang, Q.; Xi, X.; Ding, Y.; et al. Probe Synthesis Reveals Eukaryotic Translation Elongation Factor 1 Alpha 1 as the Anti-Pancreatic Cancer Target of BE-43547A2. Angew. Chem. Int. Ed. 2022, 61, e202206953. [Google Scholar] [CrossRef] [PubMed]
- Jacobsen, K.M.; Villadsen, N.L.; Toerring, T.; Nielsen, C.B.; Salomon, T.; Nielsen, M.M.; Tsakos, M.; Sibbersen, C.; Scavenius, C.; Nielsen, R.; et al. APD-Containing Cyclolipodepsipeptides Target Mitochondrial Function in Hypoxic Cancer Cells. Cell Chem. Biol. 2018, 25, 1337–1349. [Google Scholar] [CrossRef] [Green Version]
- Silvera, D.; Formenti, S.C.; Schneider, R.J. Translational control in cancer. Nat. Rev. Cancer 2010, 10, 254–266. [Google Scholar] [CrossRef]
- Bhat, M.; Robichaud, N.; Hulea, L.; Sonenberg, N.; Pelletier, J.; Topisirovic, I. Targeting the translation machinery in cancer. Nat. Rev. Drug Discov. 2015, 14, 261–278. [Google Scholar] [CrossRef] [PubMed]
- Chu, J.; Pelletier, J. Therapeutic opportunities in eukaryotic translation. Cold Spring Harbor Perspect. Biol. 2018, 10, a032995. [Google Scholar] [CrossRef] [PubMed]
- Gandhi, V.; Plunkett, W.; Cortes, J.E. Omacetaxine: A Protein Translation Inhibitor for Treatment of Chronic Myelogenous Leukemia. Clin. Cancer Res. 2014, 20, 1735–1740. [Google Scholar] [CrossRef] [Green Version]
- Varona, J.F.; Landete, P.; Lopez-Martin, J.A.; Estrada, V.; Paredes, R.; Guisado-Vasco, P.; Fernandez de Orueta, L.; Torralba, M.; Fortun, J.; Vates, R.; et al. Preclinical and randomized phase I studies of plitidepsin in adults hospitalized with COVID-19. Life Sci. Alliance 2022, 5, e202101200. [Google Scholar] [CrossRef] [PubMed]
- White, K.M.; Rosales, R.; Yildiz, S.; Kehrer, T.; Miorin, L.; Moreno, E.; Jangra, S.; Uccellini, M.B.; Rathnasinghe, R.; Coughlan, L.; et al. Plitidepsin has potent preclinical efficacy against SARS-CoV-2 by targeting the host protein eEF1A. Science 2021, 371, 926–931. [Google Scholar] [CrossRef] [PubMed]
- Sachse, M.; Tenorio, R.; de Castro, I.F.; Muñoz-Basagoiti, J.; Perez-Zsolt, D.; Raïch-Regué, D.; Rodon, J.; Losada, A.; Avilés, P.; Cuevas, C. Unraveling the antiviral activity of plitidepsin against SARS-CoV-2 by subcellular and morphological analysis. Antivir. Res. 2022, 200, 105270. [Google Scholar] [CrossRef]
- Bosutti, A.; Dapas, B.; Grassi, G.; Bussani, R.; Zanconati, F.; Giudici, F.; Bottin, C.; Pavan, N.; Trombetta, C.; Scaggiante, B. High eEF1A1 Protein Levels Mark Aggressive Prostate Cancers and the In Vitro Targeting of eEF1A1 Reveals the eEF1A1–actin Complex as a New Potential Target for Therapy. Int. J. Mol. Sci. 2022, 23, 4143. [Google Scholar] [CrossRef] [PubMed]
- Wu, A.; Tang, J.; Guo, Z.; Dai, Y.; Nie, J.; Hu, W.; Liu, N.; Ye, C.; Li, S.; Pei, H. Long non-coding RNA CRYBG3 promotes lung cancer metastasis via activating the eEF1A1/MDM2/MTBP axis. Int. J. Mol. Sci. 2021, 22, 3211. [Google Scholar] [CrossRef] [PubMed]
- Dugger, S.A.; Platt, A.; Goldstein, D.B. Drug development in the era of precision medicine. Nat. Rev. Drug Discov. 2018, 17, 183–196. [Google Scholar] [CrossRef]
- Li, H.; Chen, S.; Wang, J.; Zhang, M.; Wu, W.; Liu, W.; Sun, P. Ansafurantrienins, Unprecedented Ansatrienin Derivatives Formed via Photocatalytic Intramolecular [3+ 2] Oxidative Cycloaddition. Org. Lett. 2022, 24, 592–596. [Google Scholar] [CrossRef]










| Compound | X | R | IC50 (nM) |
|---|---|---|---|
| Nannocystin Ax (16) | Me | Me | 0.8 |
| 17 | H | 198 | |
| 18 | F | Me | 1.5 |
| 19 | H | 1345 | |
| 20 | H | Me | 4.3 |
| 21 | H | 1549 | |
| 22 | --- | Me | 22.2 |
| 23 | H | 1761 |
| Pancreatic Cancer Cells | In Vitro Cytotoxicity IC50 (μM) | In Vivo Tumor Inhibition Rate (%) |
|---|---|---|
| WT | 1.33 | 98.8 |
| KD | 11.62 | 20.7 |
| RE-KD | 0.80 | 93.2 |
| RE-C234S | 11.61 | 18.3 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, H.; Cai, J.; Yu, S.; Sun, B.; Zhang, W. Anticancer Small-Molecule Agents Targeting Eukaryotic Elongation Factor 1A: State of the Art. Int. J. Mol. Sci. 2023, 24, 5184. https://doi.org/10.3390/ijms24065184
Zhang H, Cai J, Yu S, Sun B, Zhang W. Anticancer Small-Molecule Agents Targeting Eukaryotic Elongation Factor 1A: State of the Art. International Journal of Molecular Sciences. 2023; 24(6):5184. https://doi.org/10.3390/ijms24065184
Chicago/Turabian StyleZhang, Han, Jiayou Cai, Siqi Yu, Bin Sun, and Weicheng Zhang. 2023. "Anticancer Small-Molecule Agents Targeting Eukaryotic Elongation Factor 1A: State of the Art" International Journal of Molecular Sciences 24, no. 6: 5184. https://doi.org/10.3390/ijms24065184