
In Silico Binding of 2-Aminocyclobutanones to SARS-CoV-2 Nsp13 Helicase and Demonstration of Antiviral Activity

 ,
,  and
and
Abstract
:1. Introduction
2. Results
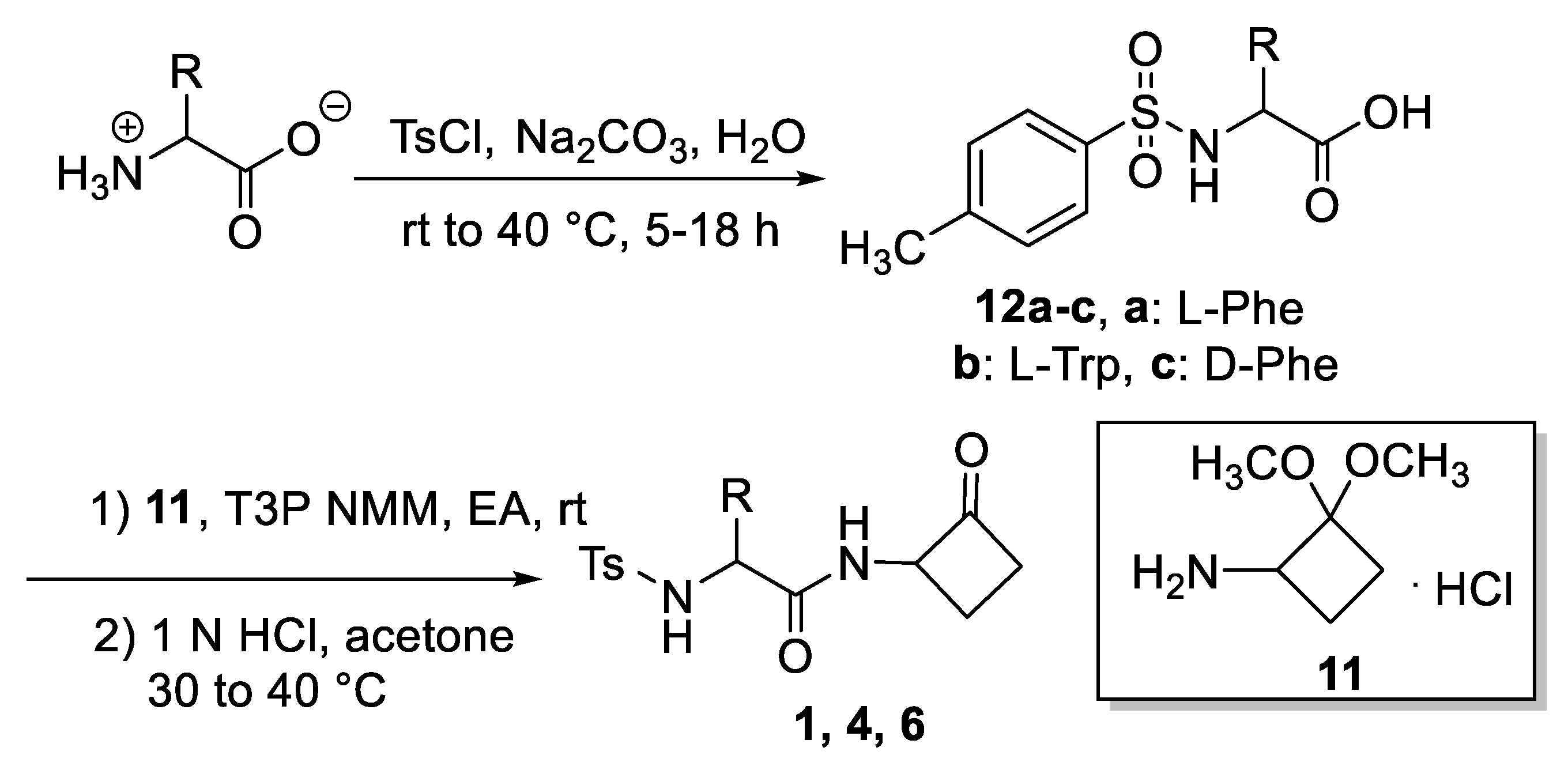
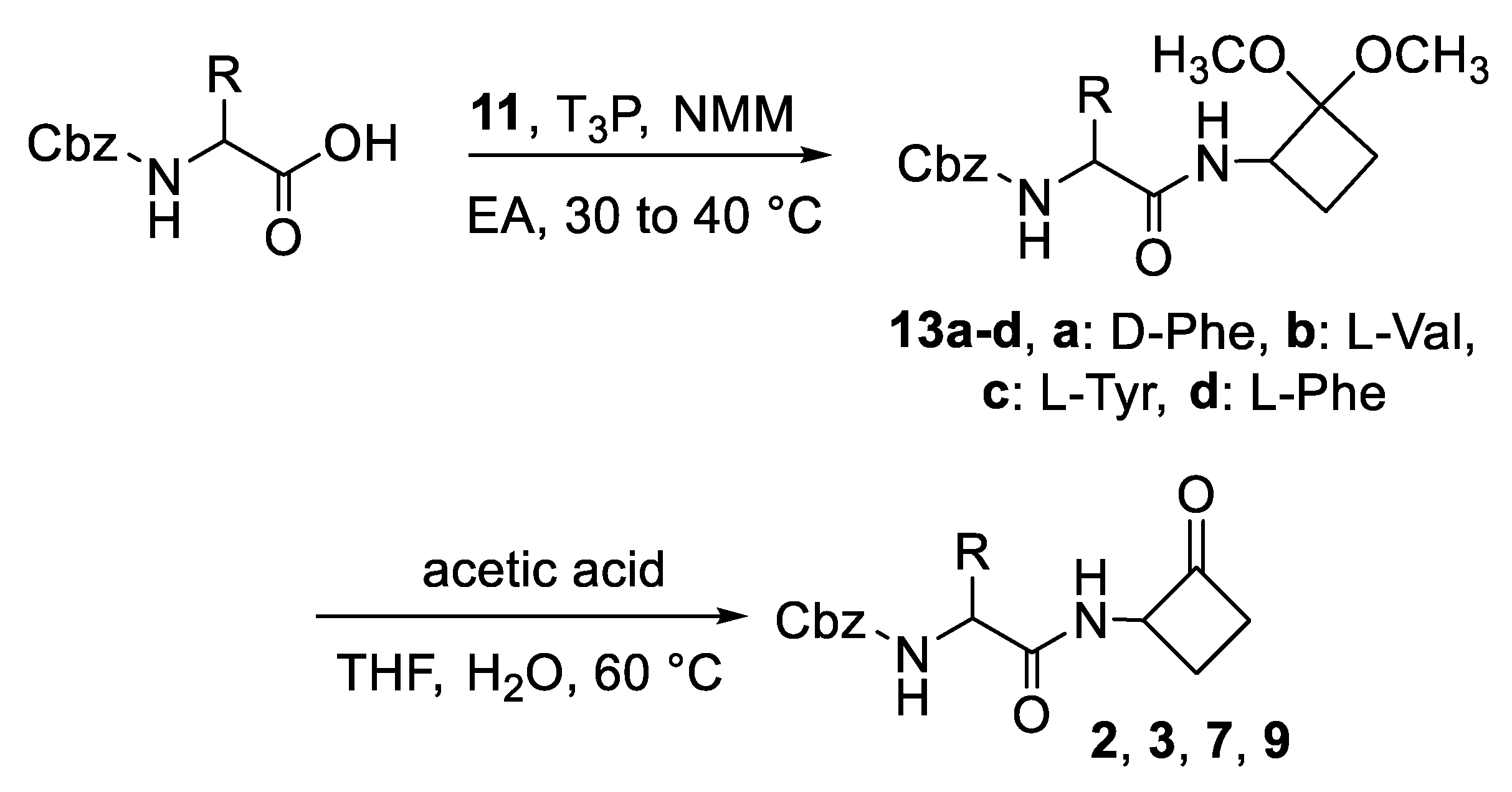
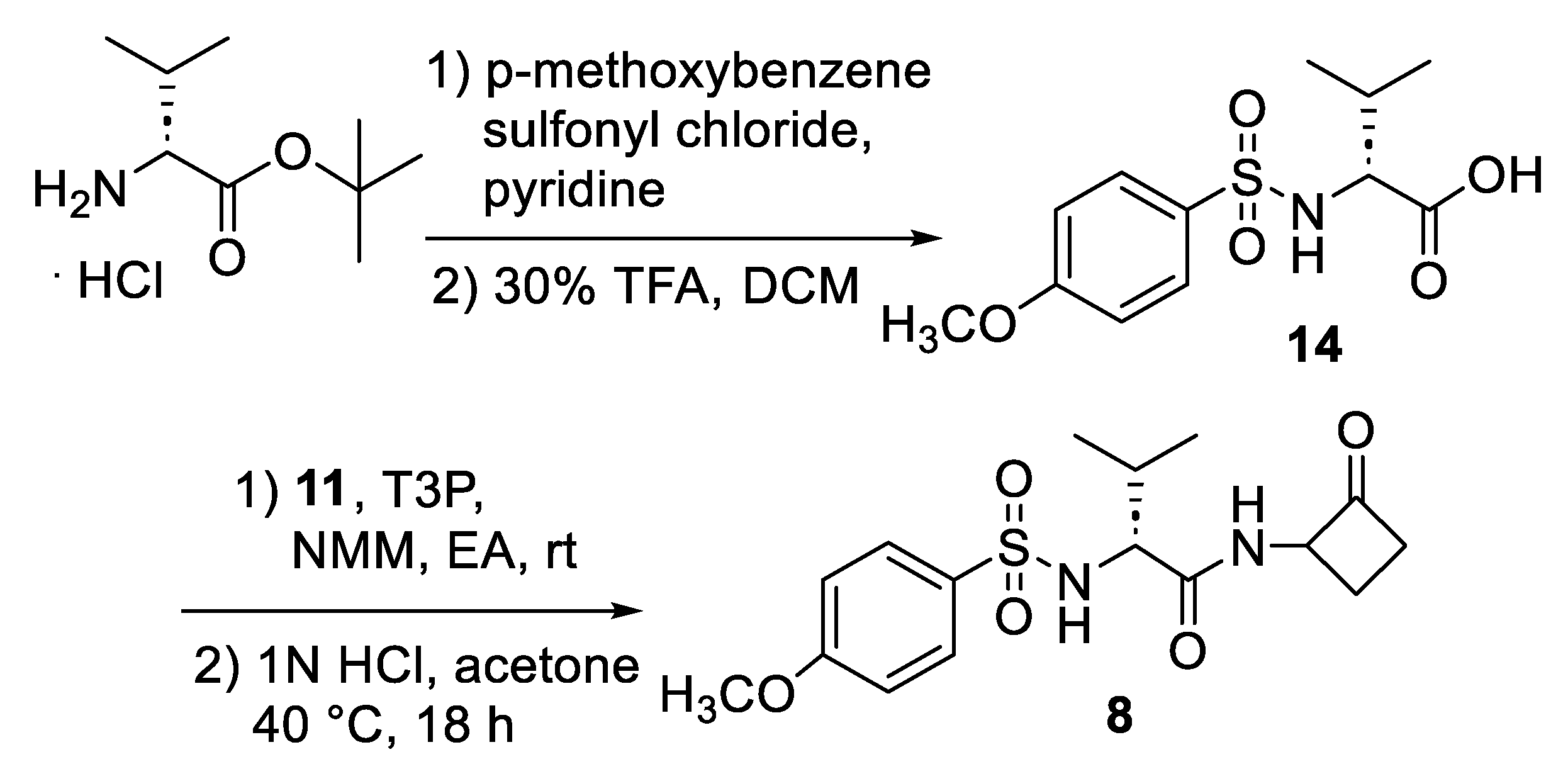
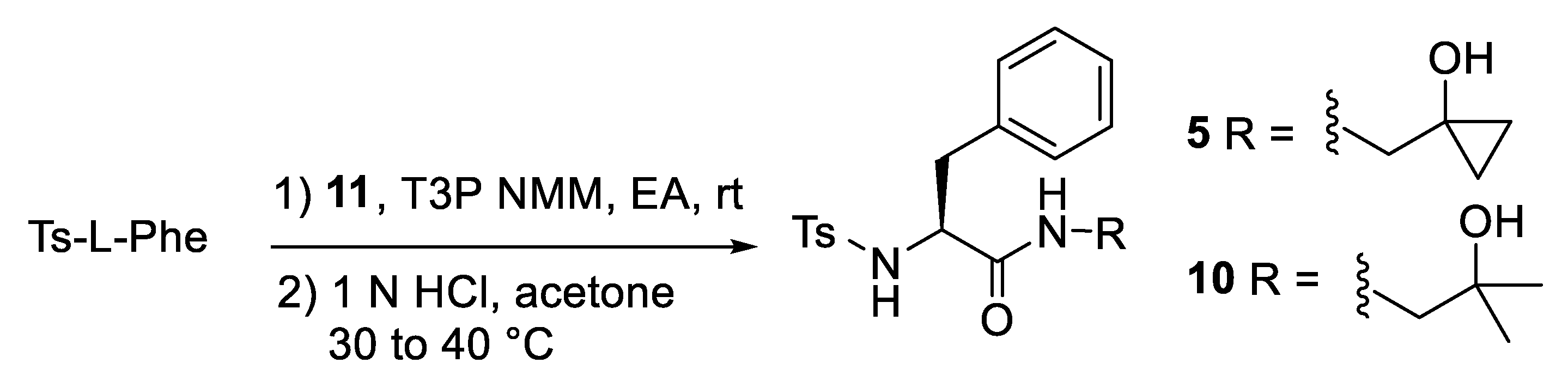
2.1. Synthesis
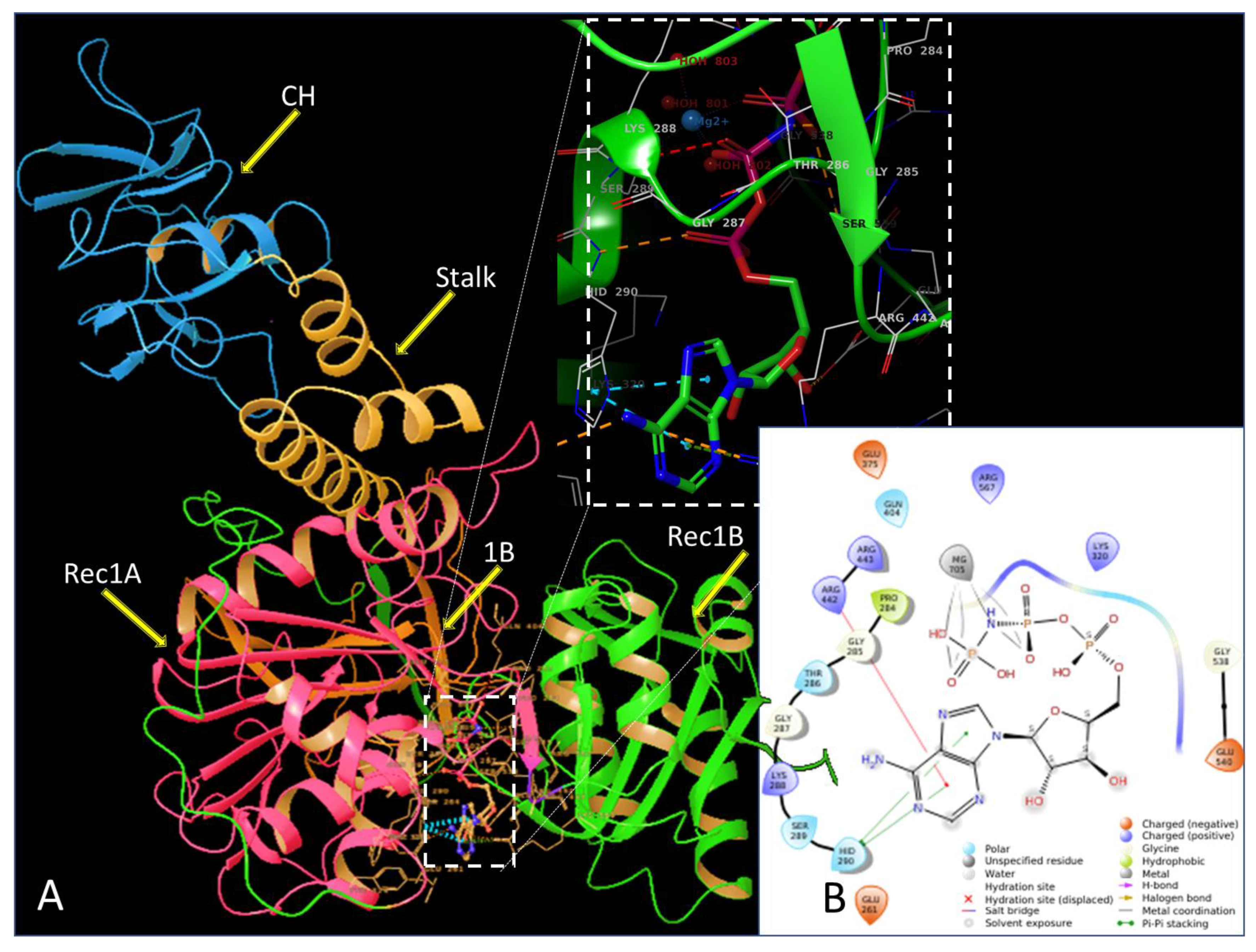
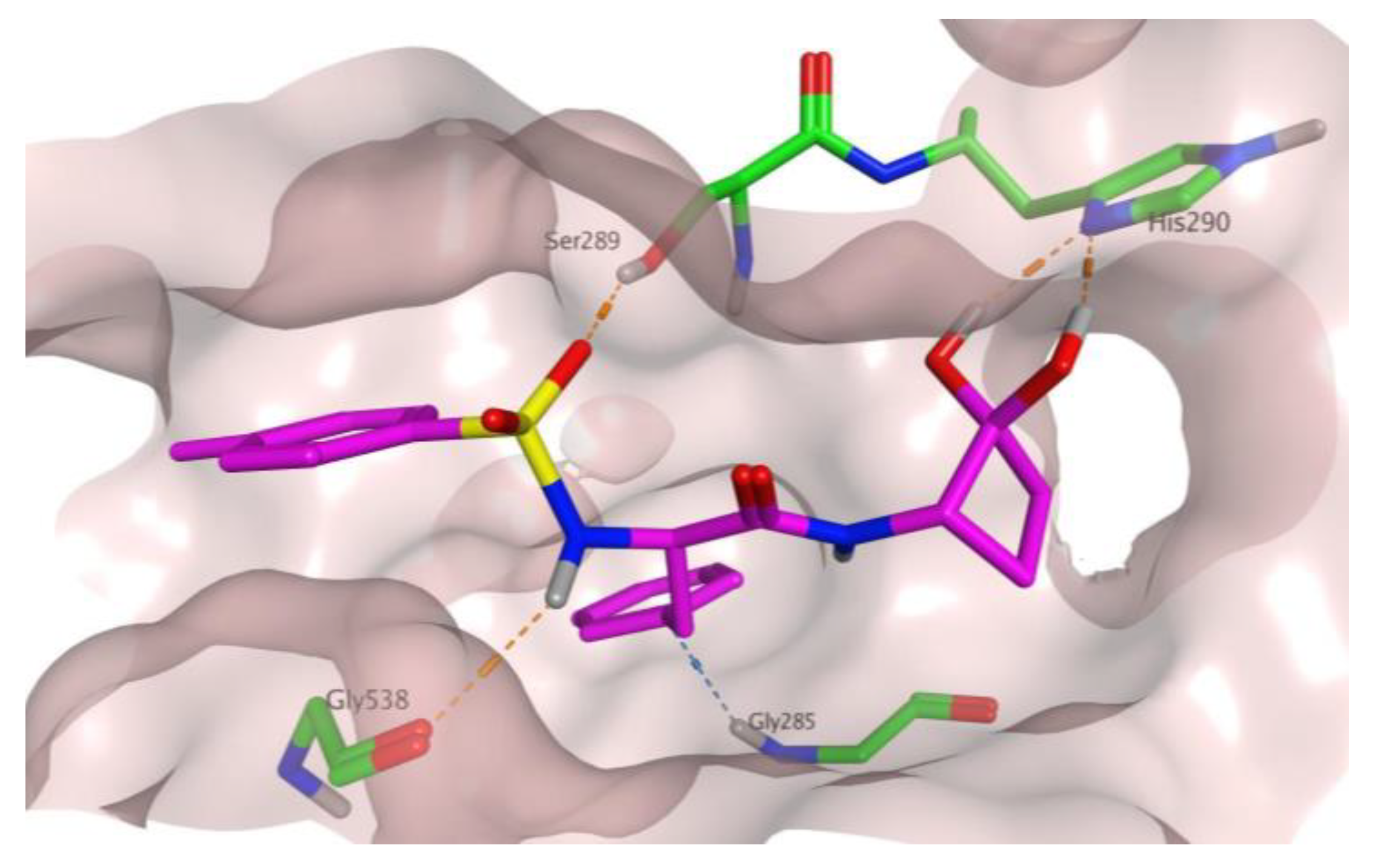
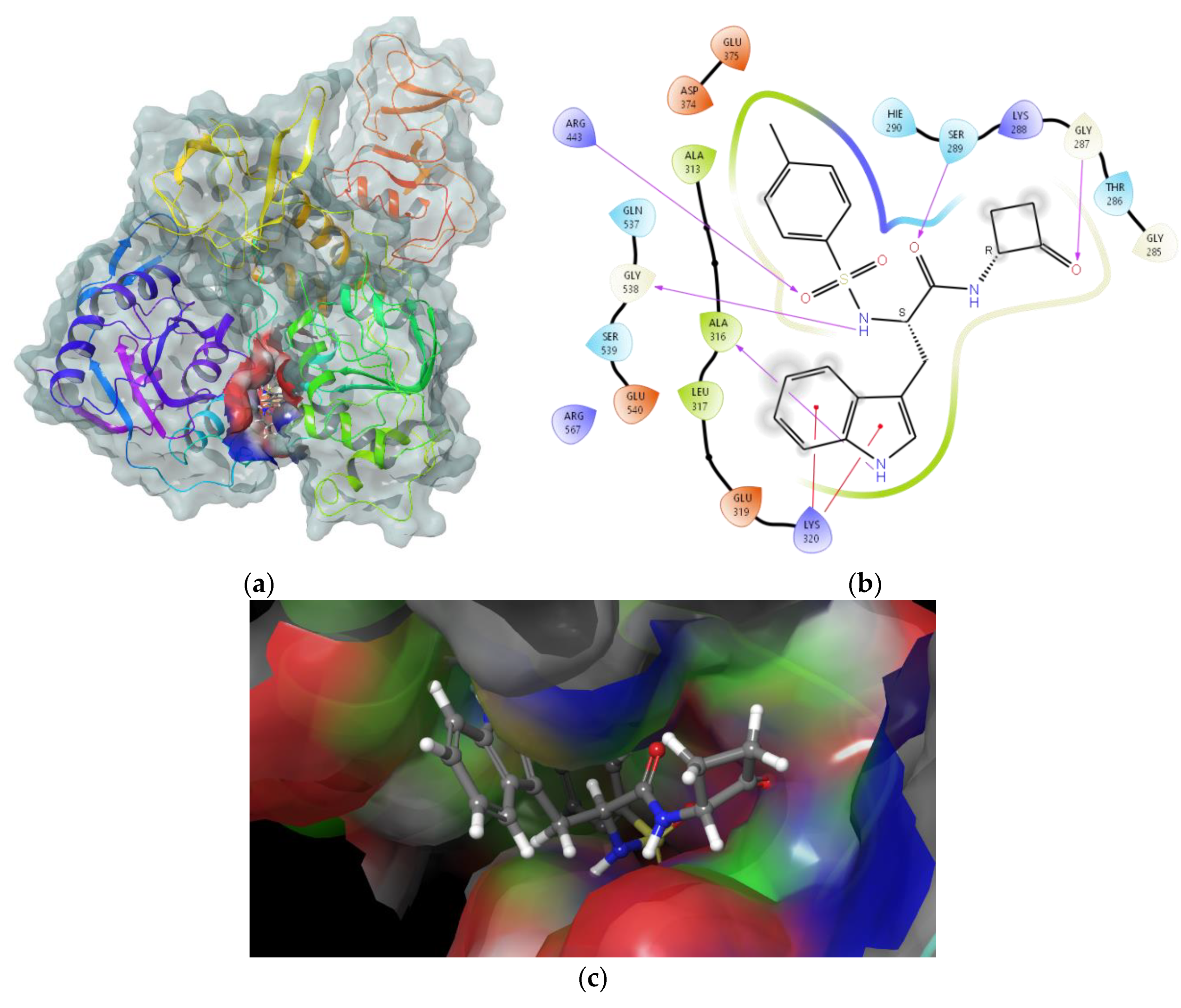
2.2. In Silico Evaluation of Cyclobutanone Derivatives
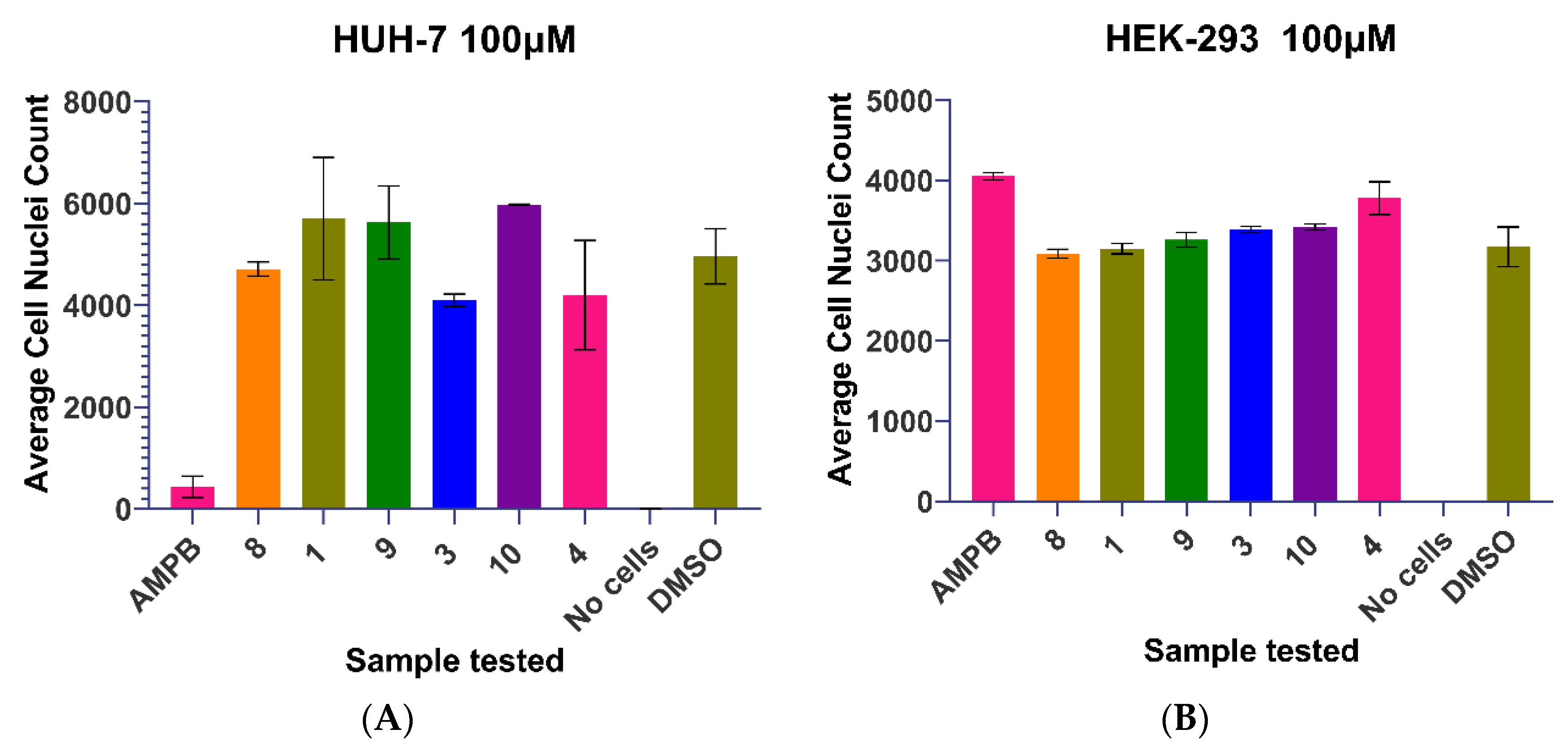
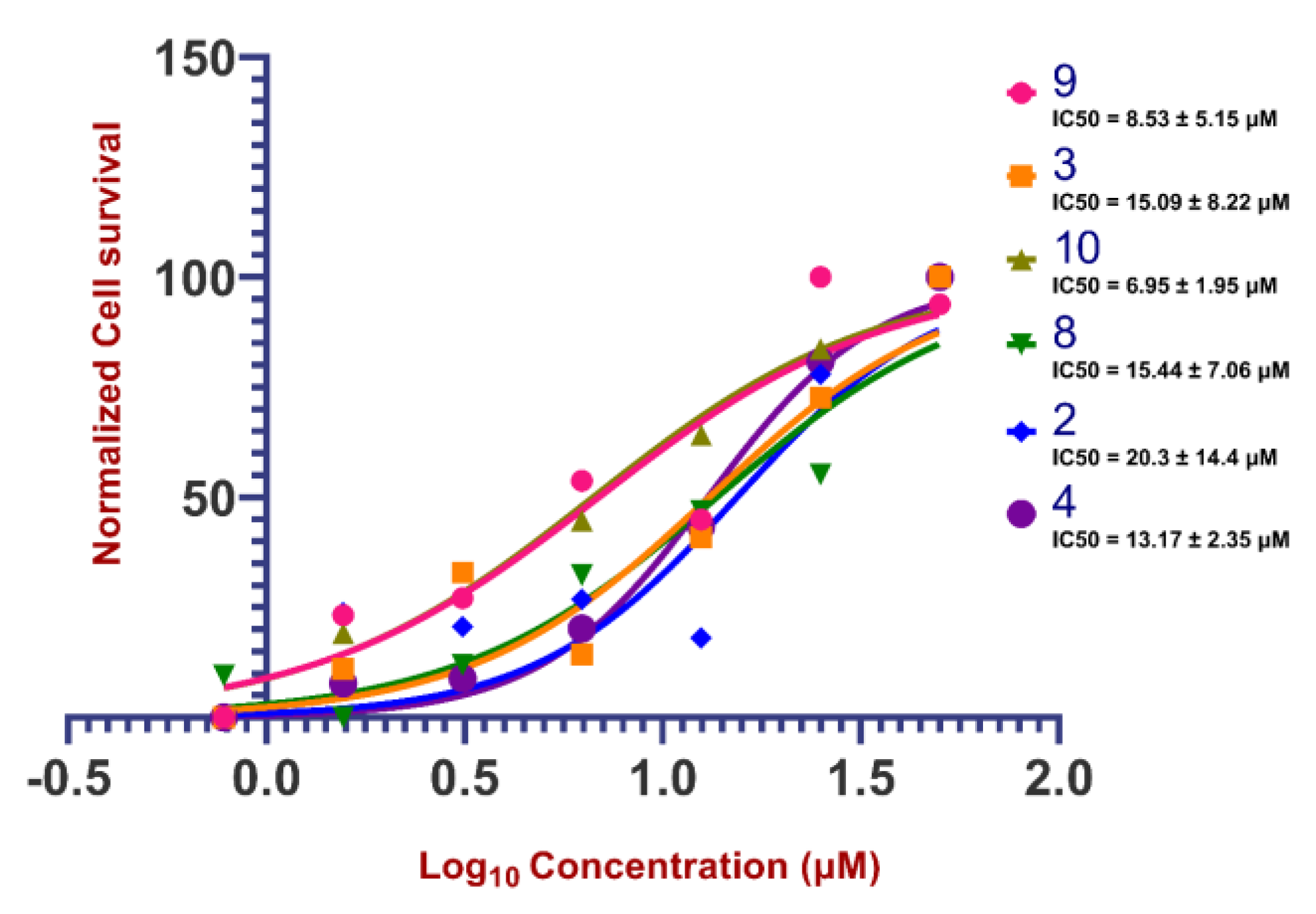
2.3. Biological Activities of Cyclobutanone Derivatives
3. Discussion
4. Methods and Materials
4.1. Organic Synthesis Methods
4.2. In Silico Methods
4.3. In Vitro Methods
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Huang, C.; Wang, Y.; Li, X.; Ren, L.; Zhao, J.; Hu, Y.; Zhang, L.; Fan, G.; Xu, J.; Gu, X. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet 2020, 395, 497–506. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, R.; Zhao, X.; Li, J.; Niu, P.; Yang, B.; Wu, H.; Wang, W.; Song, H.; Huang, B.; Zhu, N.; et al. Genomic characterisation and epidemiology of 2019 novel coronavirus: Implications for virus origins and receptor binding. Lancet 2020, 395, 565–574. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anonymous. WHO Coronavirus (COVID-19) Dashboard. Available online: https://covid19.who.int/ (accessed on 13 February 2023).
- Gupta, Y.; Maciorowski, D.; Medernach, B.; Becker, D.P.; Durvasula, R.; Libertin, C.R.; Kempaiah, P. Iron dysregulation in COVID-19 and reciprocal evolution of SARS-CoV-2: Natura nihil frustra facit. J. Cell. Biochem. 2022, 123, 601–619. [Google Scholar] [CrossRef] [PubMed]
- Whitcomb, J.M.; Parkin, N.T.; Chappey, C.; Hellmann, N.S.; Petropoulos, C.J. Broad nucleoside reverse-transcriptase inhibitor cross-resistance in human immunodeficiency virus type 1 clinical isolates. J. Infect. Dis. 2003, 188, 992–1000. [Google Scholar]
- Koizumi, Y.; Iwami, S. Mathematical modeling of multi-drugs therapy: A challenge for determining the optimal combinations of antiviral drugs. Theor. Biol. Med. Model. 2014, 11, 41. [Google Scholar] [CrossRef] [Green Version]
- Malone, B.; Urakova, N.; Snijder, E.J.; Campbell, E.A. Structures and functions of coronavirus replication–transcription complexes and their relevance for SARS-CoV-2 drug design. Nat. Rev. Mol. Cell Biol. 2022, 23, 21–39. [Google Scholar] [CrossRef]
- Bassett, M.; Salemi, M.; Rife Magalis, B. Lessons Learned and Yet-to-Be Learned on the Importance of RNA Structure in SARS-CoV-2 Replication. Microbiol. Mol. Biol. Rev. 2022, 86, e00057-21. [Google Scholar] [CrossRef]
- Gupta, Y.; Savytskyi, O.V.; Coban, M.; Venugopal, A.; Pleqi, V.; Weber, C.A.; Chitale, R.; Durvasula, R.; Hopkins, C.; Kempaiah, P. Protein structure-based in-silico approaches to drug discovery: Guide to COVID-19 therapeutics. Mol. Aspects Med. 2022, 91, 101151. [Google Scholar] [CrossRef]
- Kumar, S.; Gupta, Y.; Zak, S.E.; Upadhyay, C.; Sharma, N.; Herbert, A.S.; Durvasula, R.; Potemkin, V.; Dye, J.M.; Kempaiah, P. A novel compound active against SARS-CoV-2 targeting uridylate-specific endoribonuclease (NendoU/NSP15): In silico and in vitro investigations. RSC Med. Chem. 2021, 12, 1757–1764. [Google Scholar] [CrossRef]
- Gupta, Y.; Kumar, S.; Zak, S.E.; Jones, K.A.; Upadhyay, C.; Sharma, N.; Azizi, S.; Kathayat, R.S.; Herbert, A.S.; Durvasula, R. Antiviral evaluation of hydroxyethylamine analogs: Inhibitors of SARS-CoV-2 main protease (3CLpro), a virtual screening and simulation approach. Bioorg. Med. Chem. 2021, 47, 116393. [Google Scholar] [CrossRef]
- Bellus, D.; Ernst, B. New synthetic methods. Cyclobutanones and cyclobutenones in the synthesis of natural and synthetic products. Angew. Chem. 1988, 100, 820–850. [Google Scholar]
- Namyslo, J.C.; Kaufmann, D.E. The Application of Cyclobutane Derivatives in Organic Synthesis. Chem. Rev. 2003, 103, 1485–1537. [Google Scholar] [CrossRef]
- Leung, D.; Abbenante, G.; Fairlie, D.P. Protease inhibitors: Current status and future prospects. J. Med. Chem. 2000, 43, 305–341. [Google Scholar] [CrossRef]
- Pauls, H.W.; Cheng, B.; Reid, L.S. 2-(Peptidamido)cyclobutanones: A novel strategy for the inhibition of serine elastases. Bioorg. Chem. 1992, 20, 124–134. [Google Scholar] [CrossRef]
- Stewart, A.C.; Clifton, I.J.; Adlington, R.M.; Baldwin, J.E.; Rutledge, P.J. A cyclobutanone analogue mimics penicillin in binding to isopenicillin N synthase. ChemBioChem 2007, 8, 2003–2007. [Google Scholar] [CrossRef]
- Johnson, J.W.; Gretes, M.; Goodfellow, V.J.; Marrone, L.; Heynen, M.L.; Strynadka, N.C.J.; Dmitrienko, G.I. Cyclobutanone analogues of β-lactams revisited: Insights into conformational requirements for inhibition of serine- and metallo-β-lactamases. J. Am. Chem. Soc. 2010, 132, 2558–2560. [Google Scholar] [CrossRef]
- Abboud, M.I.; Kosmopoulou, M.; Krismanich, A.; Johnson, J.W.; Hinchliffe, P.; Brem, J.; Claridge, T.D.; Spencer, J.; Schofield, C.; Dmitrienko, G.I. Cyclobutanone Mimics of Intermediates in Metallo-β-Lactamase Catalysis. Chem.-A Eur. J. 2017, 24, 5734–5737. [Google Scholar] [CrossRef] [Green Version]
- Filippova, E.V.; Weston, L.A.; Kuhn, M.L.; Geissler, B.; Gehring, A.M.; Armoush, N.; Adkins, C.T.; Minasov, G.; Dubrovska, I.; Shuvalova, L.; et al. Large Scale Structural Rearrangement of a Serine Hydrolase from Francisella tularensis Facilitates Catalysis. J. Biol. Chem. 2013, 288, 10522–10535. [Google Scholar] [CrossRef] [Green Version]
- Shu, T.; Huang, M.; Wu, D.; Ren, Y.; Zhang, X.; Han, Y.; Mu, J.; Wang, R.; Qiu, Y.; Zhang, D.; et al. SARS-Coronavirus-2 Nsp13 Possesses NTPase and RNA Helicase Activities That Can Be Inhibited by Bismuth Salts. Virol. Sin. 2020, 35, 321–329. [Google Scholar] [CrossRef]
- Lan, T.C.; Allan, M.F.; Malsick, L.E.; Woo, J.Z.; Zhu, C.; Zhang, F.; Khandwala, S.; Nyeo, S.S.; Sun, Y.; Guo, J.U. Secondary structural ensembles of the SARS-CoV-2 RNA genome in infected cells. Nat. Commun. 2022, 13, 1128. [Google Scholar] [CrossRef]
- Ariumi, Y. Host cellular RNA helicases regulate SARS-CoV-2 infection. J. Virol. 2022, 96, e00002-22. [Google Scholar] [CrossRef] [PubMed]
- Vesuna, F.; Akhrymuk, I.; Smith, A.; Winnard, P.T.; Lin, S.; Scharpf, R.; Kehn-Hall, K.; Raman, V. RK-33, a small molecule inhibitor of host RNA helicase DDX3, suppresses multiple variants of SARS-CoV-2. bioRxiv 2022. [Google Scholar] [CrossRef] [PubMed]
- White, M.A.; Lin, W.; Cheng, X. Discovery of COVID-19 Inhibitors Targeting the SARS-CoV-2 Nsp13 Helicase. J. Phys. Chem. Lett. 2020, 11, 9144–9151. [Google Scholar] [CrossRef] [PubMed]
- Spratt, A.N.; Gallazzi, F.; Quinn, T.P.; Lorson, C.L.; Sönnerborg, A.; Singh, K. Coronavirus helicases: Attractive and unique targets of antiviral drug-development and therapeutic patents. Expert Opin. Ther. Pat. 2021, 31, 339–350. [Google Scholar] [CrossRef] [PubMed]
- Gupta, Y.; Maciorowski, D.; Zak, S.E.; Jones, K.A.; Kathayat, R.S.; Azizi, S.; Mathur, R.; Pearce, C.M.; Ilc, D.J.; Husein, H. Bisindolylmaleimide IX: A novel anti-SARS-CoV2 agent targeting viral main protease 3CLpro demonstrated by virtual screening pipeline and in-vitro validation assays. Methods 2021, 195, 57–71. [Google Scholar] [CrossRef]
- Newman, J.A.; Douangamath, A.; Yadzani, S.; Yosaatmadja, Y.; Aimon, A.; Brandão-Neto, J.; Dunnett, L.; Gorrie-Stone, T.; Skyner, R.; Fearon, D. Structure, mechanism and crystallographic fragment screening of the SARS-CoV-2 NSP13 helicase. Nat. Commun. 2021, 12, 4848. [Google Scholar] [CrossRef]
- Habeeb Mohammad, T.S.; Reidl, C.T.; Zeller, M.; Becker, D.P. Synthesis of a protected 2-aminocyclobutanone as a modular transition state synthon for medicinal chemistry. Tetrahedron Lett. 2020, 61, 151632. [Google Scholar] [CrossRef]
- Dror, O.; Schneidman-Duhovny, D.; Inbar, Y.; Nussinov, R.; Wolfson, H.J. Novel Approach for Efficient Pharmacophore-Based Virtual Screening: Method and Applications. J. Chem. Inf. Model. 2009, 49, 2333–2343. [Google Scholar] [CrossRef] [Green Version]
- Molecular Operating Environment (MOE); Chemical Computing Group ULC: Montreal, QC, Canada, 2022; Available online: https://www.chemcomp.com/Research-Citing_MOE.htm (accessed on 13 February 2023).
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 2001, 46, 3–26. [Google Scholar] [CrossRef]
- Veber, D.F.; Johnson, S.R.; Cheng, H.Y.; Smith, B.R.; Ward, K.W.; Kopple, K.D. Molecular properties that influence the oral bioavailability of drug candidates. J. Med. Chem. 2002, 45, 2615–2623. [Google Scholar] [CrossRef]
- Ugwu, D.I.; Okoro, U.C.; Mishra, N.K. Synthesis, characterization and in vitro antitrypanosomal activities of new carboxamides bearing quinoline moiety. PLoS ONE 2018, 13, e0191234. [Google Scholar] [CrossRef] [Green Version]
- Lopez Bernal, J.; Andrews, N.; Gower, C.; Gallagher, E.; Simmons, R.; Thelwall, S.; Stowe, J.; Tessier, E.; Groves, N.; Dabrera, G. Effectiveness of COVID-19 vaccines against the B. 1.617. 2 (Delta) variant. N. Engl. J. Med. 2021, 385, 585–594. [Google Scholar] [CrossRef]
- Chi, W.; Li, Y.; Huang, H.; Chan, T.E.H.; Chow, S.; Su, J.; Ferrall, L.; Hung, C.; Wu, T. COVID-19 vaccine update: Vaccine effectiveness, SARS-CoV-2 variants, boosters, adverse effects, and immune correlates of protection. J. Biomed. Sci. 2022, 29, 82. [Google Scholar] [CrossRef]
- Wen, W.; Chen, C.; Tang, J.; Wang, C.; Zhou, M.; Cheng, Y.; Zhou, X.; Wu, Q.; Zhang, X.; Feng, Z. Efficacy and safety of three new oral antiviral treatment (molnupiravir, fluvoxamine and Paxlovid) for COVID-19: A meta-analysis. Ann. Med. 2022, 54, 516–523. [Google Scholar] [CrossRef]
- Miller, J.; Hachmann, N.P.; Collier, A.Y.; Lasrado, N.; Mazurek, C.R.; Patio, R.C.; Powers, O.; Surve, N.; Theiler, J.; Korber, B. Substantial Neutralization Escape by SARS-CoV-2 Omicron Variants BQ. 1.1 and XBB. N. Engl. J. Med. 2023, 388, 662–664. [Google Scholar] [CrossRef]
- Parums, D.V. The XBB. 1.5 (‘Kraken’) Subvariant of Omicron SARS-CoV-2 and its Rapid Global Spread. Med. Sci. Monit. 2023, 29, e939580. [Google Scholar] [CrossRef]
- Gonçalves, R.; Couto, J.; Ferreirinha, P.; Costa, J.M.; Silvério, D.; Silva, M.L.; Fernandes, A.I.; Madureira, P.; Alves, N.L.; Lamas, S. SARS-CoV-2 variants induce distinct disease and impact in the bone marrow and thymus of mice. iScience 2023, 26, 105972. [Google Scholar] [CrossRef]
- Xie, Y.; Choi, T.; Al-Aly, Z. Nirmatrelvir and the risk of post-acute sequelae of COVID-19. medRxiv 2022. [Google Scholar] [CrossRef]
- Gupta, Y.; Maciorowski, D.; Mathur, R.; Pearce, C.M.; Ilc, D.J.; Husein, H.; Bharti, A.; Becker, D.; Brijesh, R.; Bradfute, S.B.; et al. Revealing SARS-CoV-2 Functional Druggability Through Multi-Target Cadd Screening of Repurposable Drugs. Preprints 2020, 2020050199. [Google Scholar] [CrossRef]
- Liu, Y.; Rocklöv, J. The effective reproductive number of the Omicron variant of SARS-CoV-2 is several times relative to Delta. J. Travel Med. 2022, 29, taac037. [Google Scholar] [CrossRef]
- Halfmann, P.J.; Iida, S.; Iwatsuki-Horimoto, K.; Maemura, T.; Kiso, M.; Scheaffer, S.M.; Darling, T.L.; Joshi, A.; Loeber, S.; Singh, G. SARS-CoV-2 Omicron virus causes attenuated disease in mice and hamsters. Nature 2022, 603, 687–692. [Google Scholar] [CrossRef] [PubMed]
- Bojkova, D.; Widera, M.; Ciesek, S.; Wass, M.N.; Michaelis, M.; Cinatl, J. Reduced interferon antagonism but similar drug sensitivity in Omicron variant compared to Delta variant of SARS-CoV-2 isolates. Cell Res. 2022, 32, 319–321. [Google Scholar] [CrossRef] [PubMed]
- Munoz, L. Non-kinase targets of protein kinase inhibitors. Nat. Rev. Drug Discov. 2017, 16, 424–440. [Google Scholar] [CrossRef] [PubMed]
- Traut, T.W. Physiological concentrations of purines and pyrimidines. Mol. Cell. Biochem. 1994, 140, 1–22. [Google Scholar] [CrossRef]
- Attwood, M.M.; Fabbro, D.; Sokolov, A.V.; Knapp, S.; Schiöth, H.B. Trends in kinase drug discovery: Targets, indications and inhibitor design. Nat. Rev. Drug Discov. 2021, 20, 839–861. [Google Scholar] [CrossRef]
- Marques, S.M.; Nuti, E.; Rossello, A.; Supuran, C.T.; Tuccinardi, T.; Martinelli, A.; Santos, M.A. Dual Inhibitors of Matrix Metalloproteinases and Carbonic Anhydrases: Iminodiacetyl-Based Hydroxamate−Benzenesulfonamide Conjugates. J. Med. Chem. 2008, 51, 7968–7979. [Google Scholar] [CrossRef]
- Rose, P.W.; Prlić, A.; Altunkaya, A.; Bi, C.; Bradley, A.R.; Christie, C.H.; Costanzo, L.D.; Duarte, J.M.; Dutta, S.; Feng, Z. The RCSB protein data bank: Integrative view of protein, gene and 3D structural information. Nucleic Acids Res. 2017, 45, D271–D281. [Google Scholar]
- Velankar, S.; Alhroub, Y.; Alili, A.; Best, C.; Boutselakis, H.C.; Caboche, S.; Conroy, M.J.; Dana, J.M.; Van Ginkel, G.; Golovin, A. PDBe: Protein data bank in Europe. Nucleic Acids Res. 2010, 39, D402–D410. [Google Scholar] [CrossRef] [Green Version]
- Schrödinger, LLC. Schrödinger Suite; Schrödinger, LLC: New York, NY, USA, 2017. [Google Scholar]
- Gupta, Y.; Maciorowski, D.; Zak, S.E.; Kulkarni, C.V.; Herbert, A.S.; Durvasula, R.; Fareed, J.; Dye, J.M.; Kempaiah, P. Heparin: A simplistic repurposing to prevent SARS-CoV-2 transmission in light of its in-vitro nanomolar efficacy. Int. J. Biol. Macromol. 2021, 183, 203–212. [Google Scholar] [CrossRef]
- Yazdi, A.K.; Pakarian, P.; Perveen, S.; Hajian, T.; Santhakumar, V.; Bolotokova, A.; Li, F.; Vedadi, M. Kinetic Characterization of SARS-CoV-2 nsp13 ATPase Activity and Discovery of Small-Molecule Inhibitors. ACS Infect. Dis. 2022, 8, 1533–1542. [Google Scholar] [CrossRef]
- Tanner, J.A.; Zheng, B.; Zhou, J.; Watt, R.M.; Jiang, J.; Wong, K.; Lin, Y.; Lu, L.; He, M.; Kung, H. The adamantane-derived bananins are potent inhibitors of the helicase activities and replication of SARS coronavirus. Chem. Biol. 2005, 12, 303–311. [Google Scholar] [CrossRef] [Green Version]
- Kim, M.K.; Yu, M.; Park, H.R.; Kim, K.B.; Lee, C.; Cho, S.Y.; Kang, J.; Yoon, H.; Kim, D.; Choo, H. 2,6-Bis-arylmethyloxy-5-hydroxychromones with antiviral activity against both hepatitis C virus (HCV) and SARS-associated coronavirus (SCV). Eur. J. Med. Chem. 2011, 46, 5698–5704. [Google Scholar] [CrossRef]
- Canal, B.; McClure, A.W.; Curran, J.F.; Wu, M.; Ulferts, R.; Weissmann, F.; Zeng, J.; Bertolin, A.P.; Milligan, J.C.; Basu, S. Identifying SARS-CoV-2 antiviral compounds by screening for small molecule inhibitors of nsp14/nsp10 exoribonuclease. Biochem. J. 2021, 478, 2445–2464. [Google Scholar] [CrossRef]
- Lu, L.; Peng, Y.; Yao, H.; Wang, Y.; Li, J.; Yang, Y.; Lin, Z. Punicalagin as an allosteric NSP13 helicase inhibitor potently suppresses SARS-CoV-2 replication in vitro. Antivir. Res. 2022, 206, 105389. [Google Scholar] [CrossRef]
- Muturi, E.; Hong, W.; Li, J.; Yang, W.; He, J.; Wei, H.; Yang, H. Effects of simeprevir on the replication of SARS-CoV-2 in vitro and in transgenic hACE2 mice. Int. J. Antimicrob. Agents 2022, 59, 106499. [Google Scholar] [CrossRef]
- Mehyar, N.; Mashhour, A.; Islam, I.; Alhadrami, H.; Tolah, A.; Alghanem, B.; Alkhaldi, S.; Somaie, B.; Al Ghobain, M.; Alobaida, Y. Discovery of Zafirlukast as a novel SARS-CoV-2 helicase inhibitor using in silico modelling and a FRET-based assay. SAR QSAR Environ. Res. 2021, 32, 963–983. [Google Scholar] [CrossRef]
- Corona, A.; Wycisk, K.; Talarico, C.; Manelfi, C.; Milia, J.; Cannalire, R.; Esposito, F.; Gribbon, P.; Zaliani, A.; Iaconis, D. Natural compounds inhibit SARS-CoV-2 nsp13 unwinding and ATPase enzyme activities. ACS Pharmacol. Transl. Sci. 2022, 5, 226–239. [Google Scholar] [CrossRef]
- Zeng, J.; Weissmann, F.; Bertolin, A.P.; Posse, V.; Canal, B.; Ulferts, R.; Wu, M.; Harvey, R.; Hussain, S.; Milligan, J.C. Identifying SARS-CoV-2 antiviral compounds by screening for small molecule inhibitors of nsp13 helicase. Biochem. J. 2021, 478, 2405–2423. [Google Scholar] [CrossRef]
- Chen, T.; Fei, C.; Chen, Y.; Sargsyan, K.; Chang, C.; Yuan, H.S.; Lim, C. Synergistic inhibition of SARS-CoV-2 replication using disulfiram/ebselen and remdesivir. ACS Pharmacol. Transl. Sci. 2021, 4, 898–907. [Google Scholar] [CrossRef]
- Yuan, S.; Wang, R.; Chan, J.F.; Zhang, A.J.; Cheng, T.; Chik, K.K.; Ye, Z.; Wang, S.; Lee, A.C.; Jin, L. Metallodrug ranitidine bismuth citrate suppresses SARS-CoV-2 replication and relieves virus-associated pneumonia in Syrian hamsters. Nat. Microbiol. 2020, 5, 1439–1448. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| # | Structure a | Glide Score | dG Bind Energy | cLogP b | mp (°C) | Cell c 5 μM | Cell d 50 μM |
|---|---|---|---|---|---|---|---|
| 1 |  | 2.6 | 177–180 | 39.3 | 49.8 | ||
| 1a | −4.012242 | −29.14638 | |||||
| 1b | −6.751353 | −28.84719 | |||||
| 1c | −3.765231 | −27.9652 | |||||
| 1d | −4.447791 | −20.97472 | |||||
| 2 |  | 2.9 | 98–100 | 24.2 | 34.7 | ||
| 2a | −6.161241 | −34.03249 | |||||
| 2b | −4.512567 | −42.28711 | |||||
| 2c | −3.59575 | −25.1313 | |||||
| 2d | −4.938537 | −38.28715 | |||||
| 3 |  | 2.1 | 140–144 | 37.9 | 54.9 | ||
| 3a | −4.096235 | −47.9141 | |||||
| 3b | −5.27515 | −55.17853 | |||||
| 3c | −3.818151 | −37.84926 | |||||
| 3d | −6.073736 | −56.36429 | |||||
| 4 |  | 2.7 | 200–201 | 54.3 | 70.9 | ||
| 4d | −5.859944 | −37.83419 | |||||
| 5 |  | −5.79524 | −28.19968 | 2.2 | 131–132 | 30.0 | 30.7 |
| 6 |  | 2.6 | 190–192 | 26.6 | 35.9 | ||
| 6a | −3.5261 | −21.16836 | |||||
| 6b | −5.719611 | −24.16271 | |||||
| 6c | −3.891619 | −24.29386 | |||||
| 6d | −4.905462 | −23.3425 | |||||
| 7 |  | 2.5 | 154–155 | −2 | 28.1 | ||
| 7a | −4.050345 | −39.41216 | |||||
| 7b | −5.690338 | −34.44169 | |||||
| 7c | −4.872337 | −36.31399 | |||||
| 7d | −5.418732 | −30.65196 | |||||
| 8 |  | 1.3 | 179–181 | 32.0 | 49.1 | ||
| 8b | −5.27903 | −18.91634 | |||||
| 9 |  | 2.9 | 100–101 | 41.6 | 57.5 | ||
| 9a | −4.949141 | −31.68122 | |||||
| 9c | −5.038913 | −38.05829 | |||||
| 10 |  | −3.923168 | −29.98338 | 2.6 | 131–133 | 36.7 | 59.2 |
| Compound | IC50 (μM) Values Forinhibition of SARS-CoV-2 Mediated Cell Death | ||
|---|---|---|---|
| Wild Type | Delta | Omicron | |
| 1 | 15.39 | --- | --- |
| 2 | 6.89 | --- | --- |
| 3 | 13.10 | --- | --- |
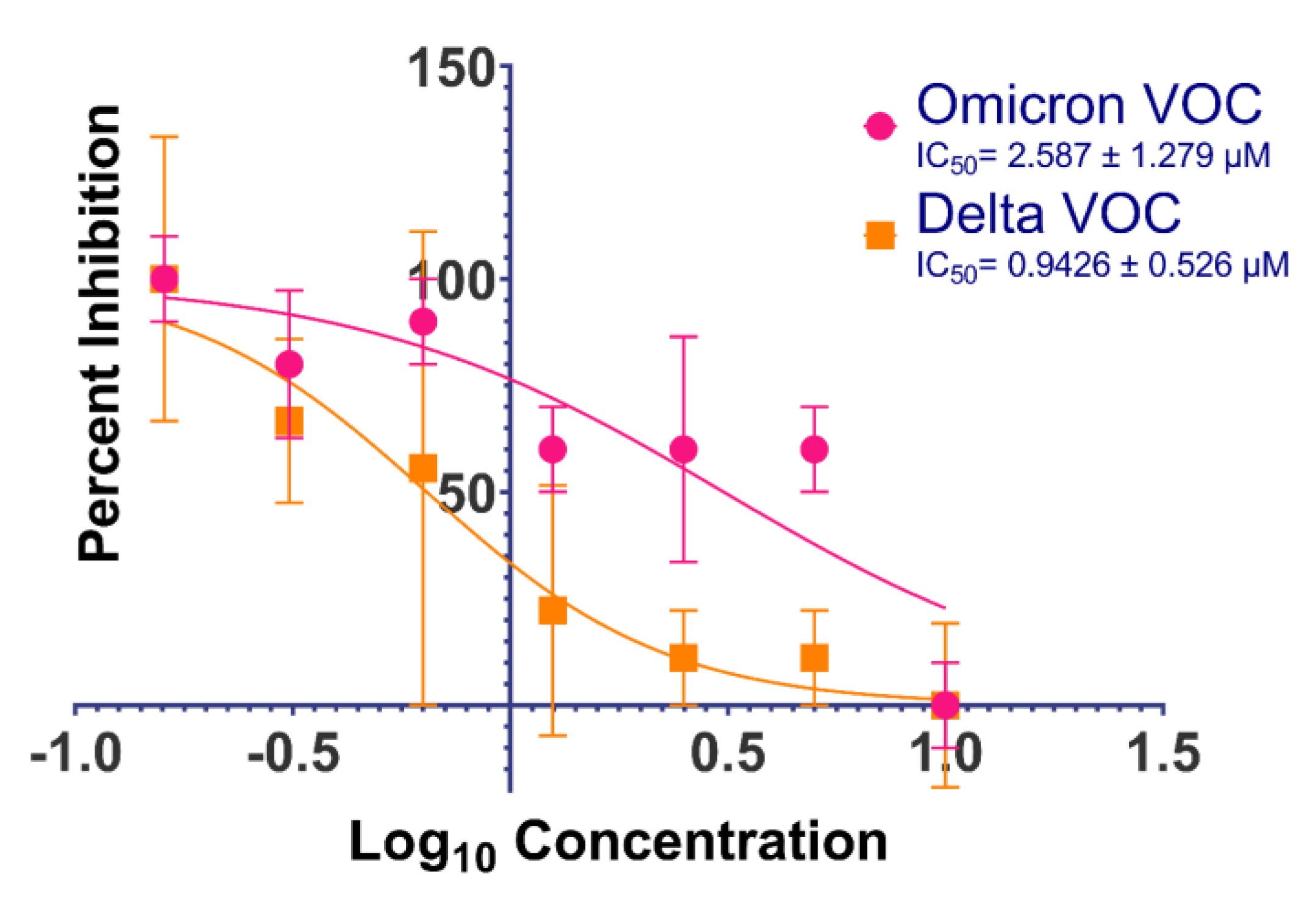
| 4 | 13.01 | 0.22 | 1.58 |
| 8 | 13.69 | --- | --- |
| 10 | 6.68 | --- | --- |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mohammad, T.S.H.; Gupta, Y.; Reidl, C.T.; Nicolaescu, V.; Gula, H.; Durvasula, R.; Kempaiah, P.; Becker, D.P. In Silico Binding of 2-Aminocyclobutanones to SARS-CoV-2 Nsp13 Helicase and Demonstration of Antiviral Activity. Int. J. Mol. Sci. 2023, 24, 5120. https://doi.org/10.3390/ijms24065120
Mohammad TSH, Gupta Y, Reidl CT, Nicolaescu V, Gula H, Durvasula R, Kempaiah P, Becker DP. In Silico Binding of 2-Aminocyclobutanones to SARS-CoV-2 Nsp13 Helicase and Demonstration of Antiviral Activity. International Journal of Molecular Sciences. 2023; 24(6):5120. https://doi.org/10.3390/ijms24065120
Chicago/Turabian StyleMohammad, Thahani S. Habeeb, Yash Gupta, Cory T. Reidl, Vlad Nicolaescu, Haley Gula, Ravi Durvasula, Prakasha Kempaiah, and Daniel P. Becker. 2023. "In Silico Binding of 2-Aminocyclobutanones to SARS-CoV-2 Nsp13 Helicase and Demonstration of Antiviral Activity" International Journal of Molecular Sciences 24, no. 6: 5120. https://doi.org/10.3390/ijms24065120