
Distinct Plasma Immune Profile in ALS Implicates sTNFR-II in pAMPK/Leptin Homeostasis
 , , and
, , and
Abstract
:1. Introduction
2. Results
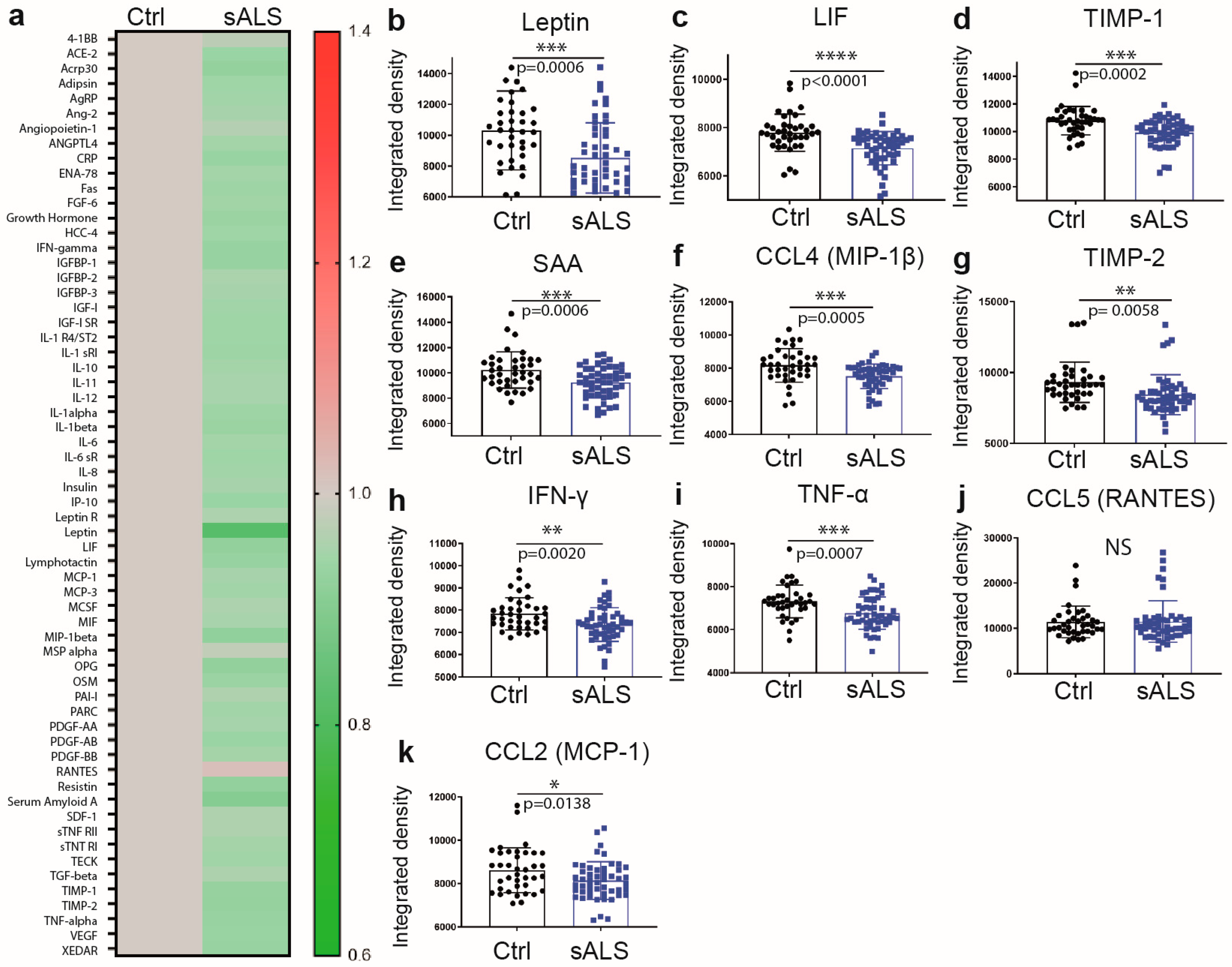
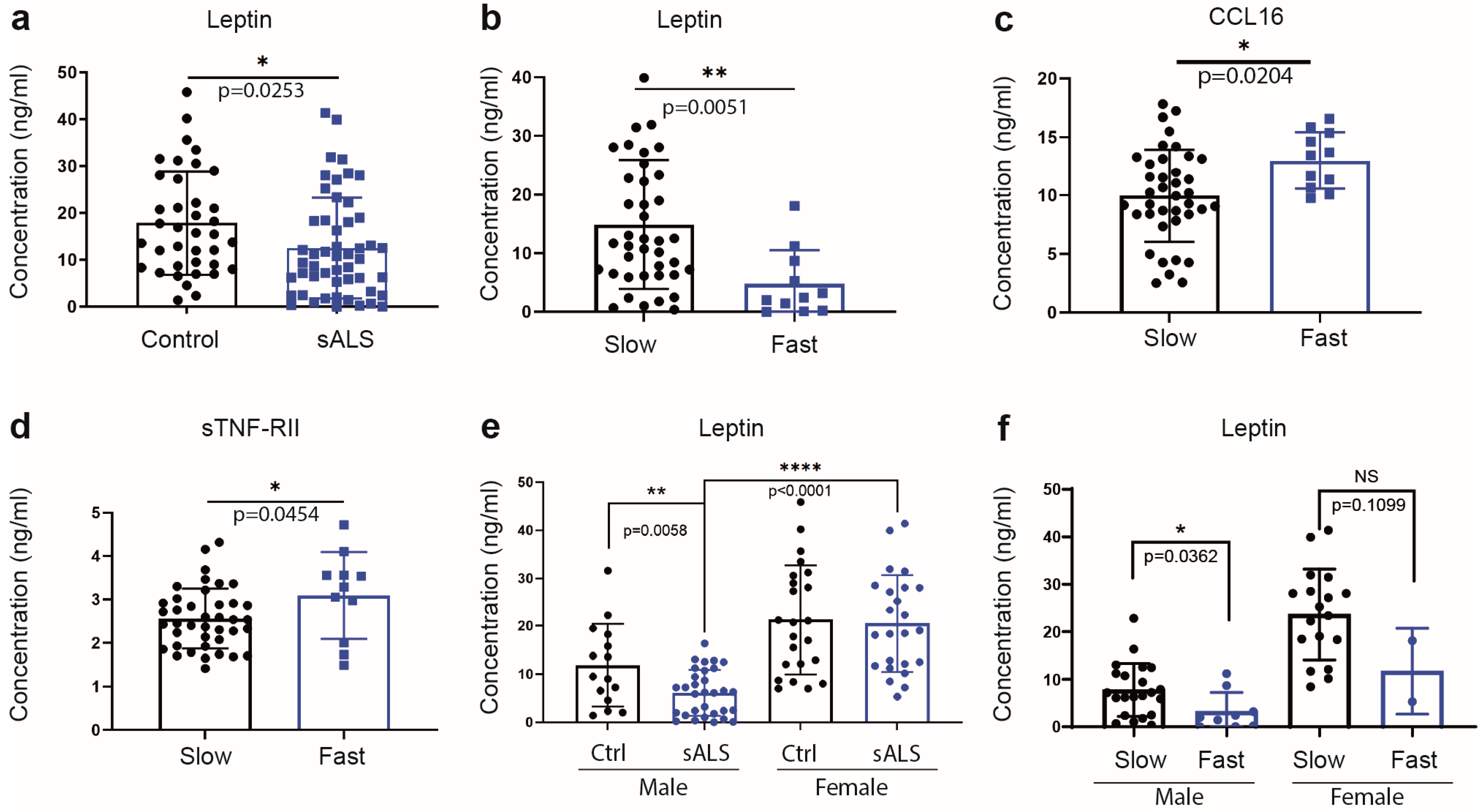
2.1. Reduced Levels of Immune Mediators and the Metabolic Sensor Leptin in Plasma of Sporadic ALS Patients
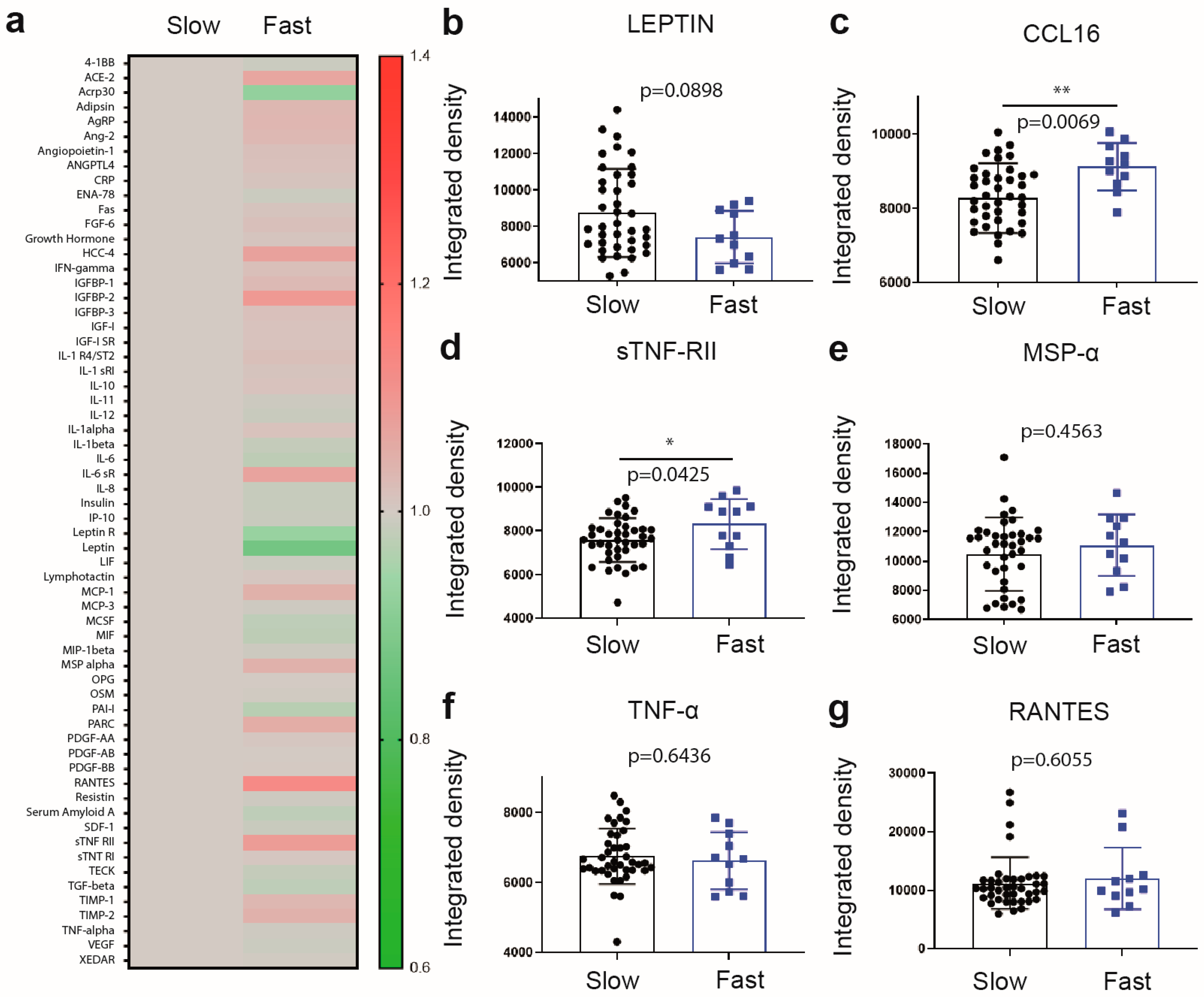
2.2. Differential Increase in Plasma Levels of sTNF-RII and CCL16 Is Associated with Fast Progressing Disease
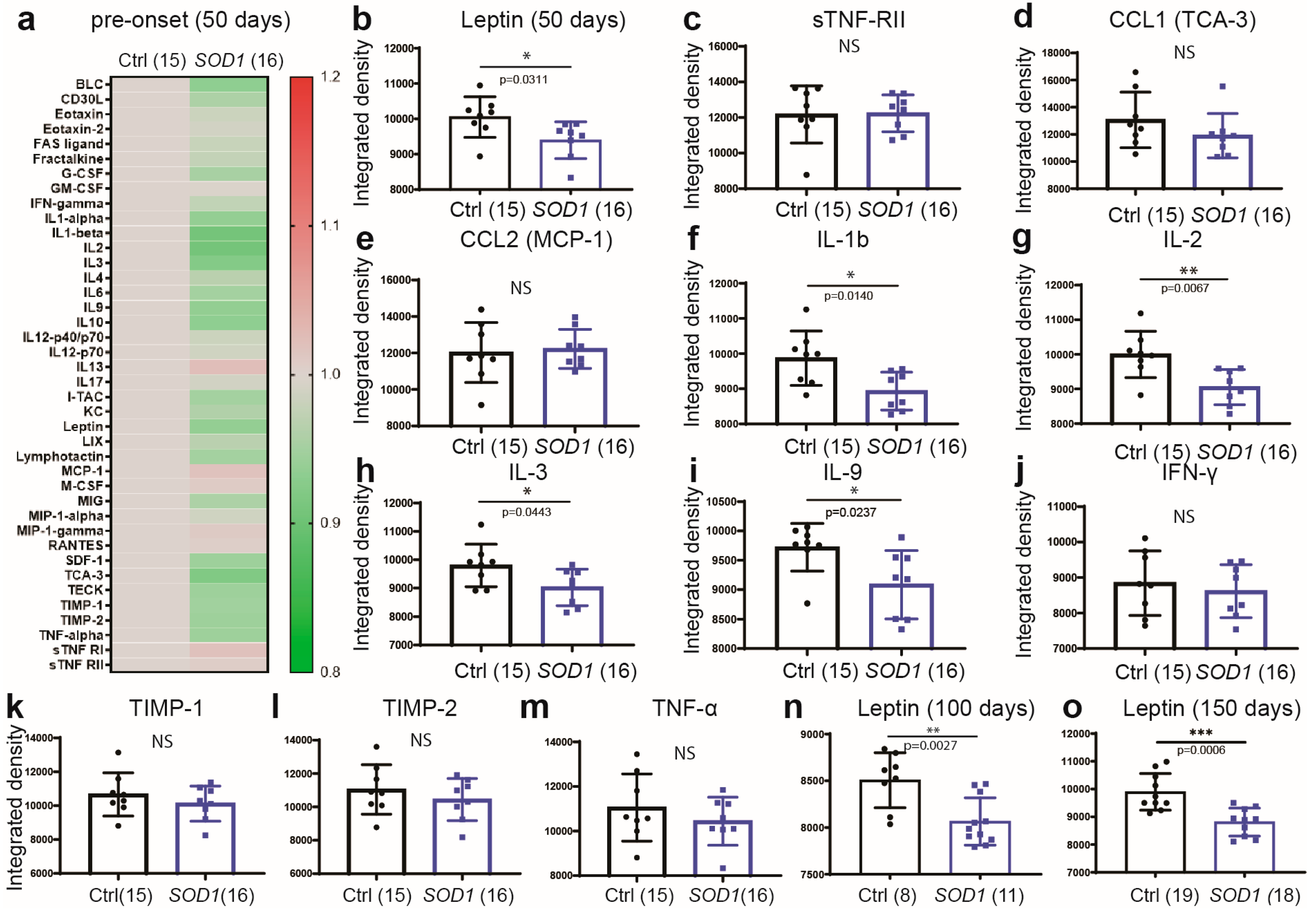
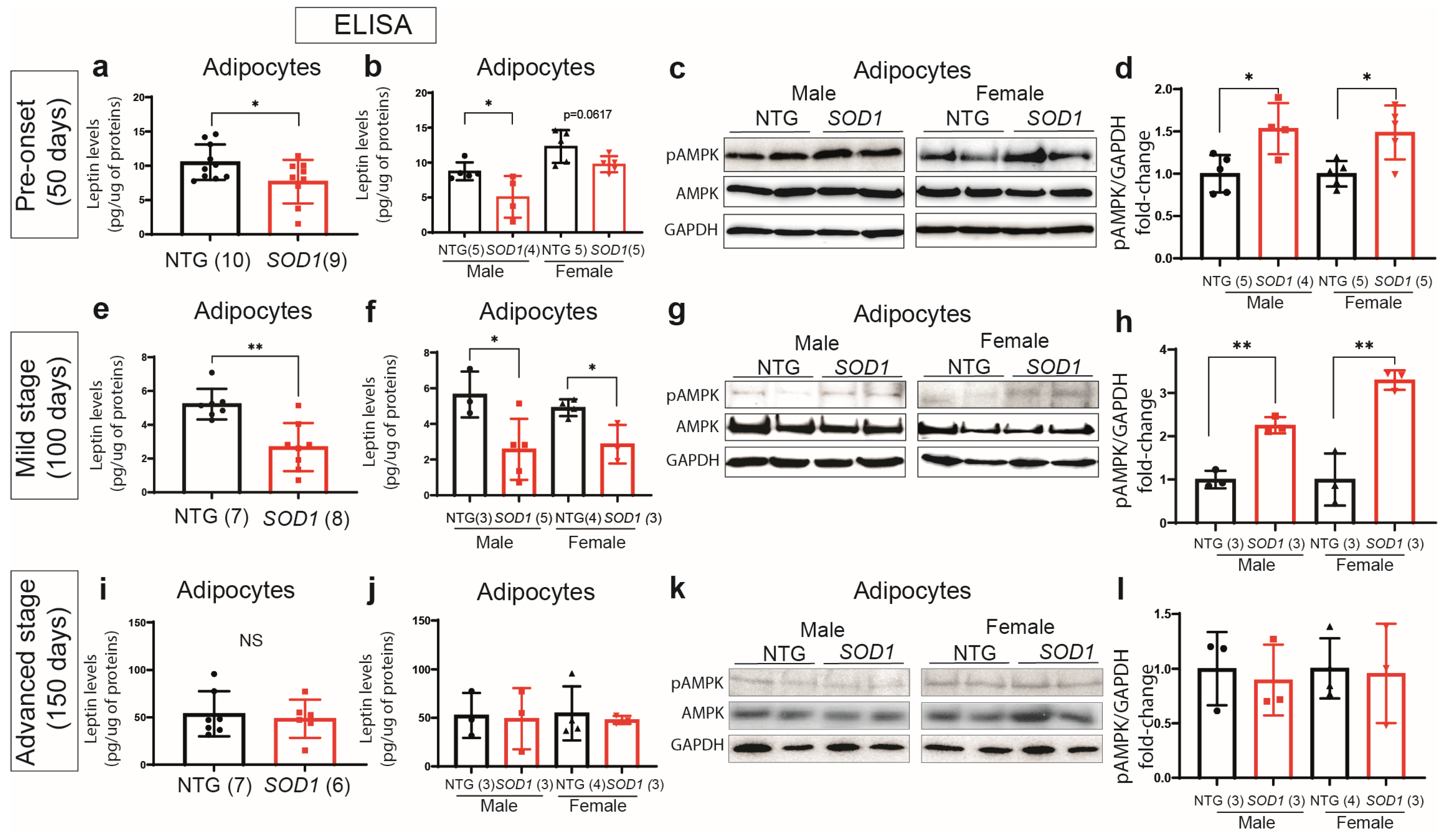
2.3. Early Deregulation of Leptin in Plasma of SOD1G93A Mice
2.4. Downregulation of Leptin Correlates with Hyperactivation of the AMPK Pathway in Adipose Tissue of the SOD1G93Amice
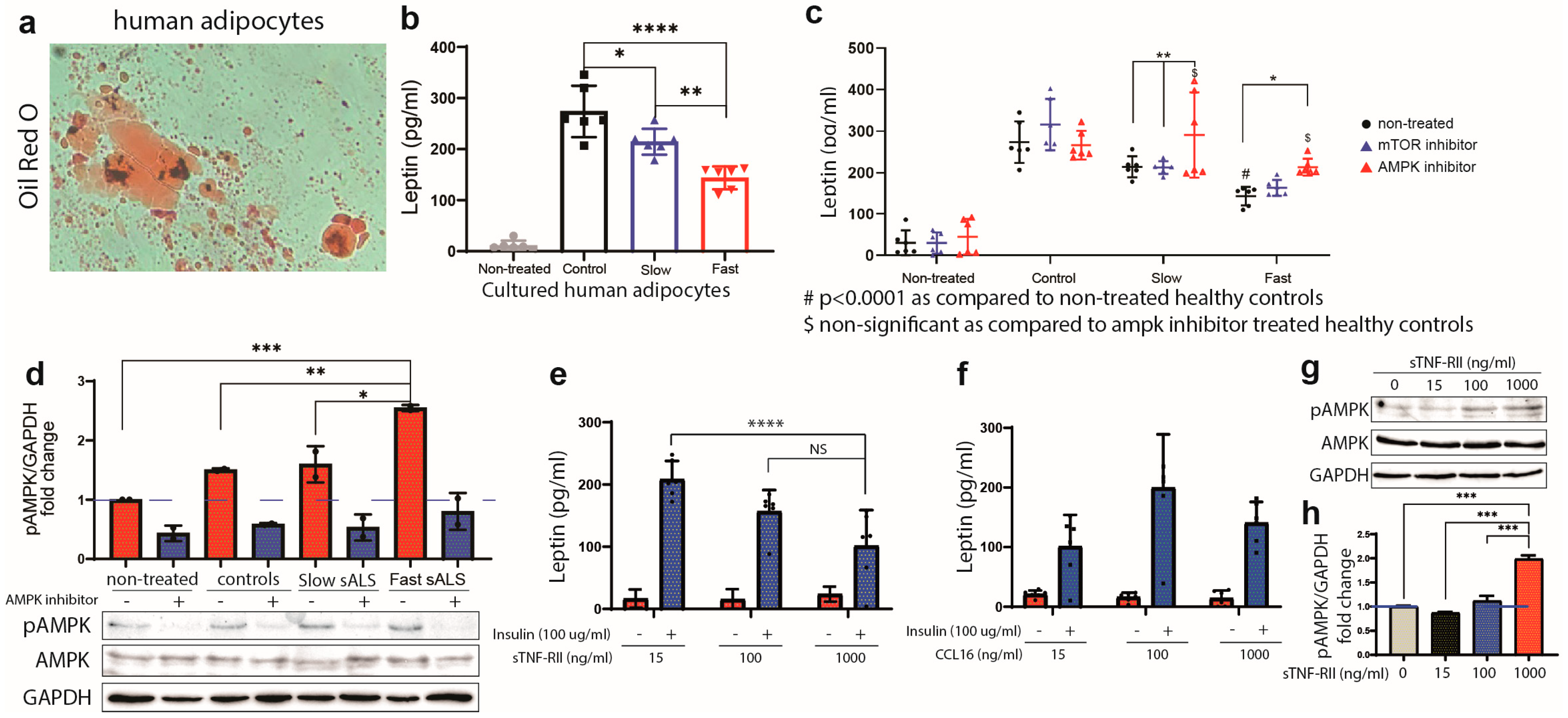
2.5. Production of Leptin in Human Adipocytes Is Regulated by AMPK
2.6. sTNF-RII Reduces Leptin Production in Human Adipocytes
3. Discussion
4. Materials and Methods
4.1. Recruitment and Samples Preparation
4.2. Human and Mouse Cytokines Array
4.3. Experimental Animals
4.4. Blood and Tissue Collection, Protein Extraction, and Immunoblotting
4.5. Pre-Adipocyte Culture and Treatment
Oil-Red O Coloration
4.6. ELISA
4.7. Statistics
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| ALS | Amyotrophic lateral sclerosis |
| Sals | Sporadic ALS |
| fALS | Familial ALS |
| ALSFRS-R | Revised ALS Functional Rating Scale |
| TDP-43 | Transactive response (TAR) DNA binding protein 43 |
| SOD1 | Superoxide Dismutase one |
| UBQLN2 | Ubiquiline-2 |
| sTNF-RII | Soluble tumor necrosis factor – receptor II |
| AMPK | AMP-activated protein kinase |
| BMI | Body mass index |
| LIF | Leukemia inhibitory factor |
| TIMP-1 | Tissue inhibitor of metalloproteinase 1 |
| TIMP-2 | Tissue inhibitor of metalloproteinase 2 |
| SAA | Serum amyloid A |
| CCL1 | C-C motif chemokine ligand 1 |
| CCL2 | C-C motif chemokine ligand 2 |
| CCL4 | C-C motif chemokine ligand 4 |
| CCL16 | C-C motif chemokine ligand 16 |
| IFN-γ | Interferon-gamma |
| TNF-α | Tumor necrosis factor-alpha |
| mTOR | Mammalian target of rapamycin |
| PI3K/AKT | phosphoinositide-3-kinase–protein kinase B |
References
- Al-Chalabi, A.; Hardiman, O.; Kiernan, M.C.; Chiò, A.; Rix-Brooks, B.; van den Berg, L.H. Amyotrophic lateral sclerosis: Moving towards a new classification system. Lancet Neurol. 2016, 15, 1182–1194. [Google Scholar] [CrossRef] [PubMed]
- Mejzini, R.; Flynn, L.L.; Pitout, I.L.; Fletcher, S.; Wilton, S.D.; Akkari, P.A. ALS Genetics, Mechanisms, and Therapeutics: Where Are We Now? Front Neurosci. 2019, 13, 1310. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chia, R.; Chiò, A.; Traynor, B.J. Novel genes associated with amyotrophic lateral sclerosis: Diagnostic and clinical implications. Lancet Neurol. 2018, 17, 94–102. [Google Scholar] [CrossRef] [PubMed]
- Fournier, C.; Glass, J.D. Modeling the course of amyotrophic lateral sclerosis. Nat. Biotechnol. 2015, 33, 45–47. [Google Scholar] [CrossRef]
- Picher-Martel, V.; Valdmanis, P.N.; Gould, P.V.; Julien, J.-P.; Dupré, N. From animal models to human disease: A genetic approach for personalized medicine in ALS. Acta Neuropathol. Commun. 2016, 4, 1–29. [Google Scholar] [CrossRef]
- Pupillo, E.; Messina, P.; Logroscino, G.; Beghi, E.; the SLALOM Group. Long-term survival in amyotrophic lateral sclerosis: A population-based study. Ann. Neurol. 2014, 75, 287–297. [Google Scholar] [CrossRef]
- Swinnen, B.; Robberecht, W. The phenotypic variability of amyotrophic lateral sclerosis. Nat. Rev. Neurol. 2014, 10, 661–670. [Google Scholar] [CrossRef] [Green Version]
- Logroscino, G. Classifying change and heterogeneity in amyotrophic lateral sclerosis. Lancet Neurol. 2016, 15, 1111–1112. [Google Scholar] [CrossRef]
- Sabatelli, M.; Conte, A.; Zollino, M. Clinical and genetic heterogeneity of amyotrophic lateral sclerosis. Clin. Genet. 2013, 83, 408–416. [Google Scholar] [CrossRef]
- Zoccolella, S.; Beghi, E.; Palagano, G.; Fraddosio, A.; Guerra, V.; Samarelli, V.; Lepore, V.; Simone, I.L.; Lamberti, P.; Serlenga, L.; et al. Analysis of survival and prognostic factors in amyotrophic lateral sclerosis: A population based study. J. Neurol. Neurosurg. Psychiatry 2008, 79, 33–37. [Google Scholar] [CrossRef]
- Kollewe, K.; Mauss, U.; Krampfl, K.; Petri, S.; Dengler, R.; Mohammadi, B. ALSFRS-R score and its ratio: A useful predictor for ALS-progression. J. Neurol. Sci. 2008, 275, 69–73. [Google Scholar] [CrossRef] [PubMed]
- Steyn, F.J.; Ioannides, Z.A.; Van Eijk, R.P.A.; Heggie, S.; Thorpe, K.A.; Ceslis, A.; Heshmat, S.; Henders, A.K.; Wray, N.R.; Berg, L.H.V.D.; et al. Hypermetabolism in ALS is associated with greater functional decline and shorter survival. J. Neurol. Neurosurg. Psychiatry 2018, 89, 1016–1023. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kasarskis, E.J.; Mendiondo, M.S.; Matthews, D.E.; Mitsumoto, H.; Tandan, R.; Simmons, Z.; Bromberg, M.B.; Kryscio, R.J. Estimating daily energy expenditure in individuals with amyotrophic lateral sclerosis. Am. J. Clin. Nutr. 2014, 99, 792–803. [Google Scholar] [CrossRef] [Green Version]
- Fernandez-Martos, C.M.; Atkinson, R.A.K.; Chuah, M.I.; King, A.E.; Vickers, J.C. Combination treatment with leptin and pioglitazone in a mouse model of Alzheimer’s disease. Alzheimer’s Dement. 2017, 3, 92–106. [Google Scholar] [CrossRef]
- Fernández-Martos, C.M.; González, P.; Rodriguez, F.J. Acute leptin treatment enhances functional recovery after spinal cord injury. PLoS ONE 2012, 7, e35594. [Google Scholar] [CrossRef] [PubMed]
- Farr, S.A.; Banks, W.A.; Morley, J.E. Effects of leptin on memory processing. Peptides 2006, 27, 1420–1425. [Google Scholar] [CrossRef]
- Santos-Alvarez, J.; Goberna, R.; Sánchez-Margalet, V. Human leptin stimulates proliferation and activation of human circulating monocytes. Cell. Immunol. 1999, 194, 6–11. [Google Scholar] [CrossRef]
- Gabay, C.; Dreyer, M.G.; Pellegrinelli, N.; Chicheportiche, R.; Meier, C.A. Leptin directly induces the secretion of interleukin 1 receptor antagonist in human monocytes. J. Clin. Endocrinol. Metab. 2001, 86, 783–791. [Google Scholar] [CrossRef]
- Nagel, G.; Peter, R.S.; Rosenbohm, A.; Koenig, W.; Dupuis, L.; Rothenbacher, D.; Ludolph, A.C. Adipokines, C-reactive protein and Amyotrophic Lateral Sclerosis—Results from a population-based ALS registry in Germany. Sci. Rep. 2017, 7, 4374. [Google Scholar] [CrossRef] [Green Version]
- Ngo, S.T.; Steyn, F.J.; Huang, L.; Mantovani, S.; Pfluger, C.M.M.; Woodruff, T.M.; O’Sullivan, J.; Henderson, R.; McCombe, P. Altered expression of metabolic proteins and adipokines in patients with amyotrophic lateral sclerosis. J. Neurol. Sci. 2015, 357, 22–27. [Google Scholar] [CrossRef] [Green Version]
- Ferrer-Donato, A.; Contreras, A.; Frago, L.M.; Chowen, J.A.; Fernandez-Martos, C.M. Alterations in Leptin Signaling in Amyotrophic Lateral Sclerosis (ALS). Int. J. Mol. Sci. 2021, 22, 10305. [Google Scholar] [CrossRef] [PubMed]
- Ferrer-Donato, A.; Contreras, A.; Fernandez, P.; Fernandez-Martos, C.M. The potential benefit of leptin therapy against amyotrophic lateral sclerosis (ALS). Brain Behav. 2022, 12, e2465. [Google Scholar] [CrossRef] [PubMed]
- Béland, L.-C.; Markovinovic, A.; Jakovac, H.; De Marchi, F.; Bilic, E.; Mazzini, L.; Kriz, J.; Munitic, I. Immunity in amyotrophic lateral sclerosis: Blurred lines between excessive inflammation and inefficient immune responses. Brain Commun. 2020, 2, fcaa124. [Google Scholar] [CrossRef] [PubMed]
- Beers, D.R.; Appel, S.H. Immune dysregulation in amyotrophic lateral sclerosis: Mechanisms and emerging therapies. Lancet Neurol. 2019, 18, 211–220. [Google Scholar] [CrossRef]
- Abe, K.; Aoki, M.; Tsuji, S.; Itoyama, Y.; Sobue, G.; Togo, M.; Hamada, C.; Tanaka, M.; Akimoto, M.; Nakamura, K.; et al. Safety and efficacy of edaravone in well defined patients with amyotrophic lateral sclerosis: A randomised, double-blind, placebo-controlled trial. Lancet Neurol. 2017, 16, 505–512. [Google Scholar] [CrossRef]
- Gurney, M.E.; Pu, H.; Chiu, A.Y.; Dal Canto, M.C.; Polchow, C.Y.; Alexander, D.D.; Caliendo, J.; Hentati, A.; Kwon, Y.W.; Deng, H.X.; et al. Motor neuron degeneration in mice that express a human Cu,Zn superoxide dismutase mutation. Science 1994, 264, 1772–1775. [Google Scholar] [CrossRef] [PubMed]
- Picher-Martel, V.; Renaud, L.; Bareil, C.; Julien, J.-P. Neuronal Expression of UBQLN2P497H Exacerbates TDP-43 Pathology in TDP-43G348C Mice through Interaction with Ubiquitin. Mol. Neurobiol. 2019, 56, 4680–4696. [Google Scholar] [CrossRef] [Green Version]
- Inoki, K.; Kim, J.; Guan, K.-L. AMPK and mTOR in Cellular Energy Homeostasis and Drug Targets. Annu. Rev. Pharmacol. Toxicol. 2012, 52, 381–400. [Google Scholar] [CrossRef]
- Lee, M.J.; Wang, Y.; Ricci, M.R.; Sullivan, S.; Russell, C.D.; Fried, S.K. Acute and chronic regulation of leptin synthesis, storage, and secretion by insulin and dexamethasone in human adipose tissue. Am. J. Physiol. Endocrinol. Metab. 2007, 292, E858–E864. [Google Scholar] [CrossRef] [Green Version]
- Marques-Oliveira, G.H.; Silva, T.M.; Lima, W.G.; Valadares, H.M.S.; Chaves, V.E. Insulin as a hormone regulator of the synthesis and release of leptin by white adipose tissue. Peptides 2018, 106, 49–58. [Google Scholar] [CrossRef]
- Mitchell, R.M.; Freeman, W.M.; Randazzo, W.T.; Stephens, H.E.; Beard, J.L.; Simmons, Z.; Connor, J.R. A CSF biomarker panel for identification of patients with amyotrophic lateral sclerosis. Neurology 2009, 72, 14–19. [Google Scholar] [PubMed]
- Guo, J.; Yang, X.; Gao, L.; Zang, D. Evaluating the levels of CSF and serum factors in ALS. Brain Behav. 2017, 7, e00637. [Google Scholar] [CrossRef]
- Kuhle, J.; Lindberg, R.L.P.; Regeniter, A.; Mehling, M.; Steck, A.J.; Kappos, L.; Czaplinski, A. Increased levels of inflammatory chemokines in amyotrophic lateral sclerosis. Eur. J. Neurol. 2009, 16, 771–774. [Google Scholar] [PubMed]
- Italiani, P.; Carlesi, C.; Giungato, P.; Puxeddu, I.; Borroni, B.; Bossù, P.; Migliorini, P.; Siciliano, G.; Boraschi, D. Evaluating the levels of interleukin-1 family cytokines in sporadic amyotrophic lateral sclerosis. J. Neuroinflamm. 2014, 11, 94. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, J.; Gao, L.; Zang, D. Elevated levels of IFN-γ in CSF and serum of patients with amyotrophic lateral sclerosis. PLoS ONE 2015, 10, e0136937. [Google Scholar] [CrossRef] [Green Version]
- Babu, G.N.; Kumar, A.; Chandra, R.; Puri, S.K.; Kalita, J.; Misra, U.K. Elevated inflammatory markers in a group of amyotrophic lateral sclerosis patients from northern India. Neurochem. Res. 2008, 33, 1145–1149. [Google Scholar] [CrossRef]
- Tortelli, R.; Zecca, C.; Piccininni, M.; Benmahamed, S.; Dell’Abate, M.T.; Barulli, M.R.; Capozzo, R.; Battista, P.; Logroscino, G. Plasma Inflammatory Cytokines Are Elevated in ALS. Front. Neurol. 2020, 11, 552295. [Google Scholar] [CrossRef]
- Lu, C.-H.; Allen, K.; Oei, F.; Leoni, E.; Kuhle, J.; Tree, T.; Fratta, P.; Sharma, N.; Sidle, K.; Howard, R.; et al. Systemic inflammatory response and neuromuscular involvement in amyotrophic lateral sclerosis. Neurol. Neuroimmunol. Neuroinflamm. 2016, 3, e244. [Google Scholar] [CrossRef] [Green Version]
- Polverino, A.; Rucco, R.; Stillitano, I.; Bonavita, S.; Grimaldi, M.; Minino, R.; Pesoli, M.; Trojsi, F.; D’Ursi, A.M.; Sorrentino, G.; et al. In Amyotrophic Lateral Sclerosis Blood Cytokines Are Altered, but Do Not Correlate with Changes in Brain Topology. Brain Connect. 2020, 10, 411–421. [Google Scholar]
- Jin, M.; Günther, R.; Akgün, K.; Hermann, A.; Ziemssen, T. Peripheral proinflammatory Th1/Th17 immune cell shift is linked to disease severity in amyotrophic lateral sclerosis. Sci. Rep. 2020, 10, 5941. [Google Scholar]
- Pronto-Laborinho, A.; Pinto, S.; Gromicho, M.; Pereira, M.; Swash, M.; de Carvalho, M. Interleukin-6 and amyotrophic lateral sclerosis. J. Neurol. Sci. 2019, 398, 50–53. [Google Scholar] [CrossRef] [PubMed]
- Prado, L.d.G.R.; Rocha, N.P.; de Souza, L.C.; Bicalho, I.C.S.; Gomez, R.S.; Vidigal-Lopes, M.; Braz, N.F.T.; Vieira, É.L.M.; Teixeira, A.L. Longitudinal assessment of clinical and inflammatory markers in patients with amyotrophic lateral sclerosis. J. Neurol. Sci. 2018, 394, 69–74. [Google Scholar] [CrossRef] [PubMed]
- Andrés-Benito, P.; Moreno, J.; Domínguez, R.; Aso, E.; Povedano, M.; Ferrer, I. Inflammatory Gene Expression in Whole Peripheral Blood at Early Stages of Sporadic Amyotrophic Lateral Sclerosis. Front. Neurol. 2017, 8, 546. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Staats, K.A.; Borchelt, D.R.; Tansey, M.G.; Wymer, J. Blood-based biomarkers of inflammation in amyotrophic lateral sclerosis. Mol. Neurodegener. 2022, 17, 11. [Google Scholar] [CrossRef] [PubMed]
- Zhao, W.; Beers, D.R.; Hooten, K.G.; Sieglaff, D.H.; Zhang, A.; Kalyana-Sundaram, S.; Traini, C.M.; Halsey, W.S.; Hughes, A.M.; Sathe, G.M.; et al. Characterization of Gene Expression Phenotype in Amyotrophic Lateral Sclerosis Monocytes. JAMA Neurol. 2017, 74, 677–685. [Google Scholar] [CrossRef] [Green Version]
- Saad, M.F.; Damani, S.; Gingerich, R.L.; Riad-Gabriel, M.G.; Khan, A.; Boyadjian, R.; Jinagouda, S.D.; El-Tawil, K.; Rude, R.K.; Kamdar, V. Sexual dimorphism in plasma leptin concentration. J. Clin. Endocrinol. Metab. 1997, 82, 579–584. [Google Scholar] [CrossRef]
- Vitery, R.J.; Ortega, G.; Salazar, C.B.C. Leptin sexual dimorphism, insulin resistance, and body composition in normal weight prepubescent. Rev. Chil. Pediatr. 2020, 91, 924–929. [Google Scholar]
- Martin, L.J.; Mahaney, M.C.; Almasy, L.; MacCluer, J.W.; Blangero, J.; Jaquish, C.E.; Comuzzie, A.G. Leptin’s sexual dimorphism results from genotype by sex interactions mediated by testosterone. Obes. Res. 2002, 10, 14–21. [Google Scholar] [CrossRef]
- Zhang, F.; Wang, S.; Signore, A.P.; Chen, J. Neuroprotective effects of leptin against ischemic injury induced by oxygen-glucose deprivation and transient cerebral ischemia. Stroke 2007, 38, 2329–2336. [Google Scholar] [CrossRef] [Green Version]
- Fewlass, D.C.; Noboa, K.; Pi-Sunyer, F.X.; Johnston, J.M.; Yan, S.D.; Tezapsidis, N. Obesity-related leptin regulates Alzheimer’s Abeta. FASEB J. 2004, 18, 1870–1878. [Google Scholar] [CrossRef]
- Greco, S.J.; Sarkar, S.; Casadesus, G.; Zhu, X.; Smith, M.A.; Ashford, J.W.; Johnston, J.M.; Tezapsidis, N. Leptin inhibits glycogen synthase kinase-3beta to prevent tau phosphorylation in neuronal cells. Neurosci. Lett. 2009, 455, 191–194. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Greco, S.J.; Sarkar, S.; Johnston, J.M.; Tezapsidis, N. Leptin regulates tau phosphorylation and amyloid through AMPK in neuronal cells. Biochem. Biophys. Res. Commun. 2009, 380, 98–104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weng, Z.; Signore, A.P.; Gao, Y.; Wang, S.; Zhang, F.; Hastings, T.; Yin, X.-M.; Chen, J. Leptin protects against 6-hydroxydopamine-induced dopaminergic cell death via mitogen-activated protein kinase signaling. J. Biol. Chem. 2007, 282, 34479–34491. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bouret, S.G. Neurodevelopmental actions of leptin. Brain Res. 2010, 1350, 2–9. [Google Scholar] [CrossRef] [Green Version]
- Fujita, Y.; Yamashita, T. The Effects of Leptin on Glial Cells in Neurological Diseases. Front. Neurosci. 2019, 13, 828. [Google Scholar] [CrossRef]
- Ahmed, R.M.; Phan, K.; Highton-Williamson, E.; Strikwerda-Brown, C.; Caga, J.; Ramsey, E.; Zoing, M.; Devenney, E.; Kim, W.S.; Hodges, J.R.; et al. Eating peptides: Biomarkers of neurodegeneration in amyotrophic lateral sclerosis and frontotemporal dementia. Ann. Clin. Transl. Neurol. 2019, 6, 486–495. [Google Scholar] [CrossRef] [Green Version]
- Cereda, C.; Baiocchi, C.; Bongioanni, P.; Cova, E.; Guareschi, S.; Metelli, M.R.; Rossi, B.; Sbalsi, I.; Cuccia, M.C.; Ceroni, M. TNF and sTNFR1/2 plasma levels in ALS patients. J. Neuroimmunol. 2008, 194, 123–131. [Google Scholar] [CrossRef]
- Ronin, E.; Pouchy, C.; Khosravi, M.; Hilaire, M.; Grégoire, S.; Casrouge, A.; Kassem, S.; Sleurs, D.; Martin, G.H.; Chanson, N.; et al. Tissue-restricted control of established central nervous system autoimmunity by TNF receptor 2-expressing Treg cells. Proc. Natl. Acad. Sci. USA 2021, 118, e2014043118. [Google Scholar]
- Probert, L. TNF and its receptors in the CNS: The essential, the desirable and the deleterious effects. Neuroscience 2015, 302, 2–22. [Google Scholar] [CrossRef] [Green Version]
- Magliozzi, R.; Pezzini, F.; Pucci, M.; Rossi, S.; Facchiano, F.; Marastoni, D.; Montagnana, M.; Lippi, G.; Reynolds, R.; Calabrese, M. Changes in Cerebrospinal Fluid Balance of TNF and TNF Receptors in Naïve Multiple Sclerosis Patients: Early Involvement in Compartmentalised Intrathecal Inflammation. Cells 2021, 10, 1712. [Google Scholar] [CrossRef]
- Mendelson, M.M.; Johannes, R.; Liu, C.; Huan, T.; Yao, C.; Miao, X.; Murabito, J.M.; Dupuis, J.; Levy, D.; Benjamin, E.J.; et al. Epigenome-Wide Association Study of Soluble Tumor Necrosis Factor Receptor 2 Levels in the Framingham Heart Study. Front. Pharmacol. 2018, 9, 207. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shai, I.; Schulze, M.B.; Manson, J.E.; Rexrode, K.M.; Stampfer, M.J.; Mantzoros, C.; Hu, F.B. A prospective study of soluble tumor necrosis factor-alpha receptor II (sTNF-RII) and risk of coronary heart disease among women with type 2 diabetes. Diabetes Care 2005, 28, 1376–1382. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Medler, J.; Wajant, H. Tumor necrosis factor receptor-2 (TNFR2): An overview of an emerging drug target. Expert Opin. Ther. Targets 2019, 23, 295–307. [Google Scholar] [CrossRef]
- Lim, M.A.; Selak, M.A.; Xiang, Z.; Krainc, D.; Neve, R.L.; Kraemer, B.C.; Watts, J.L.; Kalb, R.G. Reduced activity of AMP-activated protein kinase protects against genetic models of motor neuron disease. J. Neurosci. 2012, 32, 1123–1141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perera, N.D.; Turner, B.J. AMPK Signalling and Defective Energy Metabolism in Amyotrophic Lateral Sclerosis. Neurochem. Res. 2016, 41, 544–553. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.J.; Lee, L.M.; Lai, H.L.; Chern, Y. Aberrant activation of AMP-activated protein kinase contributes to the abnormal distribution of HuR in amyotrophic lateral sclerosis. FEBS Lett. 2015, 589, 432–439. [Google Scholar] [CrossRef] [Green Version]
- Perera, N.D.; Sheean, R.K.; Scott, J.W.; Kemp, B.E.; Horne, M.K.; Turner, B.J. Mutant TDP-43 deregulates AMPK activation by PP2A in ALS models. PLoS ONE 2014, 9, e90449. [Google Scholar] [CrossRef]
- Kaneb, H.M.; Sharp, P.S.; Rahmani-Kondori, N.; Wells, D.J. Metformin treatment has no beneficial effect in a dose-response survival study in the SOD1 G93A mouse model of ALS and is harmful in female mice. PLoS ONE 2011, 6, e24189. [Google Scholar] [CrossRef] [Green Version]
- Yun, Y.C.; Jeong, S.G.; Kim, S.H.; Cho, G.W. Reduced sirtuin 1/adenosine monophosphate-activated protein kinase in amyotrophic lateral sclerosis patient-derived mesenchymal stem cells can be restored by resveratrol. J. Tissue Eng. Regen. Med. 2019, 13, 110–115. [Google Scholar] [CrossRef] [Green Version]
- Ruiz-Perera, L.M.; Höving, A.L.; Schmidt, K.E.; Cenan, S.; Wohllebe, M.; Greiner, J.F.W.; Kaltschmidt, C.; Simon, M.; Knabbe, C.; Kaltschmidt, B. Neuroprotection Mediated by Human Blood Plasma in Mouse Hippocampal Slice Cultures and in Oxidatively Stressed Human Neurons. Int. J. Mol. Sci. 2021, 22, 9567. [Google Scholar] [CrossRef]
- Wang, P.; Li, X.; Li, X.; Yang, W.; Yu, S. Blood Plasma of Patients with Parkinson’s Disease Increases Alpha-Synuclein Aggregation and Neurotoxicity. Park. Dis. 2016, 2016, 7596482. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsubai, T.; Noda, Y.; Ito, K.; Nakao, M.; Seino, Y.; Oiso, Y.; Hamada, Y. Insulin elevates leptin secretion and mRNA levels via cyclic AMP in 3T3-L1 adipocytes deprived of glucose. Heliyon 2016, 2, e00194. [Google Scholar] [CrossRef] [PubMed] [Green Version]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Characteristic | Age-Matched Controls (N = 38) | Sporadic ALS (N = 51) | p-Value |
|---|---|---|---|
| Age—yr. | 65.86 ± 8.67 | 62.62 ± 10.62 | 0.1358 |
| Female sex—no. (%) * | 23 (60%) | 20 (39.2%) | NA |
| Riluzole—no. (%) | NA | 27 (53%) | NA |
| Weight—kg | 73.80 ± 16.97 | 72.49 ± 16.84 | 0.7879 |
| Weight loss—kg | NA | −4.53 ± 11.79 | NA |
| BMI—kg/m2 | 26.83 ± 6.17 | 25.5 ± 6.13 | 0.3219 |
| Disease duration—months | NA | 33 ± 27.98 | NA |
| ALSFRS-R—(/48) | NA | 32.86 ± 9.29 | NA |
| CVF—liter | NA | 2.69 ± 0.98 | NA |
| Fast progressor—no (%) | NA | 11 (21.6%) | NA |
| Bulbar onset—no (%) | NA | 11 (21.6%) | NA |
| Characteristic | Slow ALS (N = 40) | Fast ALS (N = 11) | p-Value |
|---|---|---|---|
| Rate of progression (ALSFRS-R points/12 weeks) | −1.683 ± 2.25 | −7.884 ± 4.77 | <0.0001 |
| Age—yr. | 62.75 ± 10.89 | 62.18 ± 10.02 | 0.877 |
| Female—no. (%) * | 18 (45%) | 2 (18%) | NA |
| Weight—kg | 74.62 ± 17.29 | 67.42 ± 15.03 | 0.161 |
| Weight loss—kg | −2.456 ± 12.03 | −10.03 ± 10.51 | 0.0703 |
| BMI—kg/m2 | 26.18 ± 6.04 | 22.76 ± 6.53 | 0.1099 |
| Disease duration—months | 37.16 ± 30.40 | 16.07 ± 6.40 | 0.0276 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Picher-Martel, V.; Boutej, H.; Vézina, A.; Cordeau, P.; Kaneb, H.; Julien, J.-P.; Genge, A.; Dupré, N.; Kriz, J. Distinct Plasma Immune Profile in ALS Implicates sTNFR-II in pAMPK/Leptin Homeostasis. Int. J. Mol. Sci. 2023, 24, 5065. https://doi.org/10.3390/ijms24065065
Picher-Martel V, Boutej H, Vézina A, Cordeau P, Kaneb H, Julien J-P, Genge A, Dupré N, Kriz J. Distinct Plasma Immune Profile in ALS Implicates sTNFR-II in pAMPK/Leptin Homeostasis. International Journal of Molecular Sciences. 2023; 24(6):5065. https://doi.org/10.3390/ijms24065065
Chicago/Turabian StylePicher-Martel, Vincent, Hejer Boutej, Alexandre Vézina, Pierre Cordeau, Hannah Kaneb, Jean-Pierre Julien, Angela Genge, Nicolas Dupré, and Jasna Kriz. 2023. "Distinct Plasma Immune Profile in ALS Implicates sTNFR-II in pAMPK/Leptin Homeostasis" International Journal of Molecular Sciences 24, no. 6: 5065. https://doi.org/10.3390/ijms24065065