
Simvastatin Improves Benign Prostatic Hyperplasia: Role of Peroxisome-Proliferator-Activated Receptor-γ and Classic WNT/β-Catenin Pathway
 , , and
, , and
Abstract
:1. Introduction
2. Results
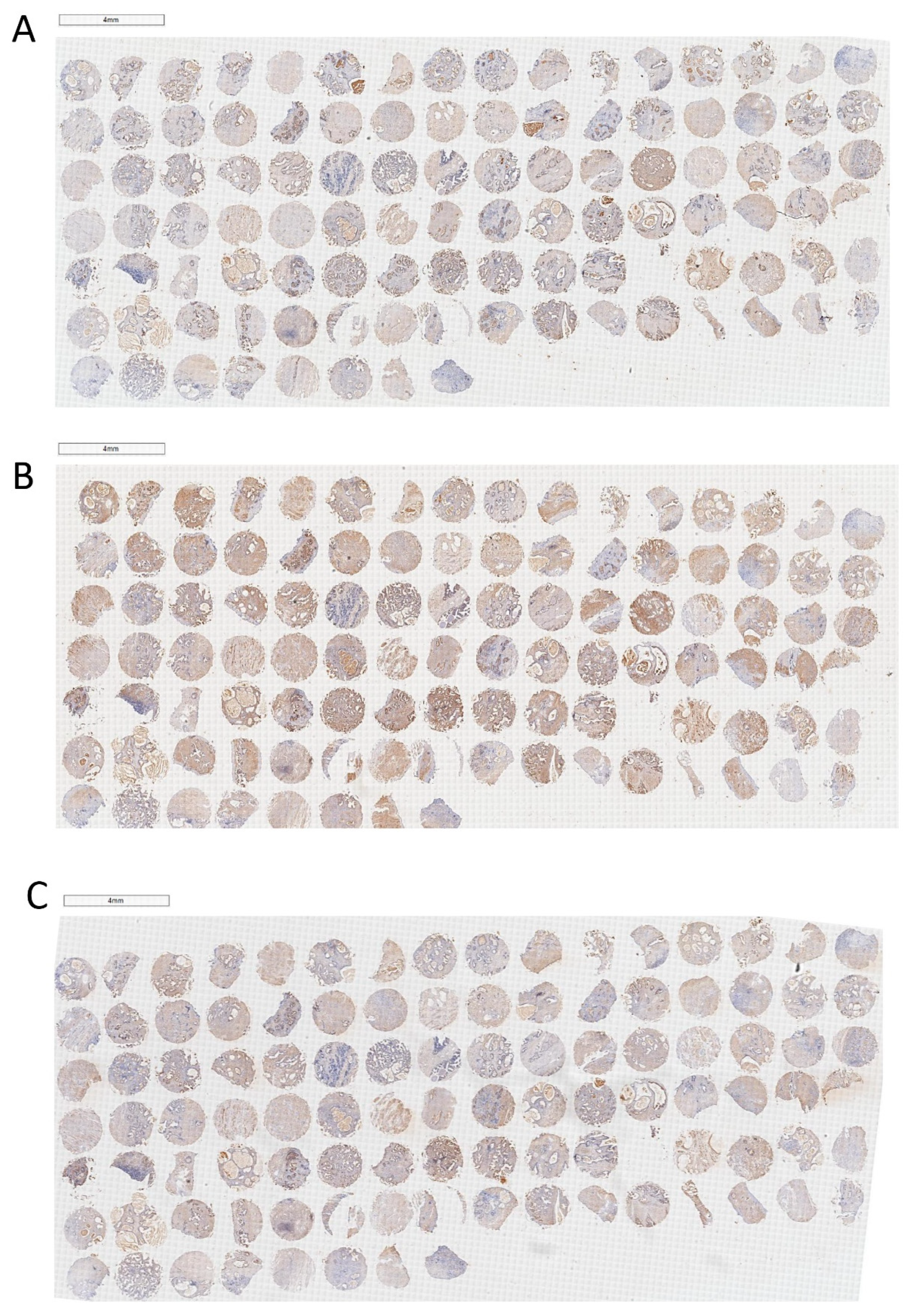
2.1. The Expression and Localization of PPARγ in Human Prostate Tissues and Cell Lines
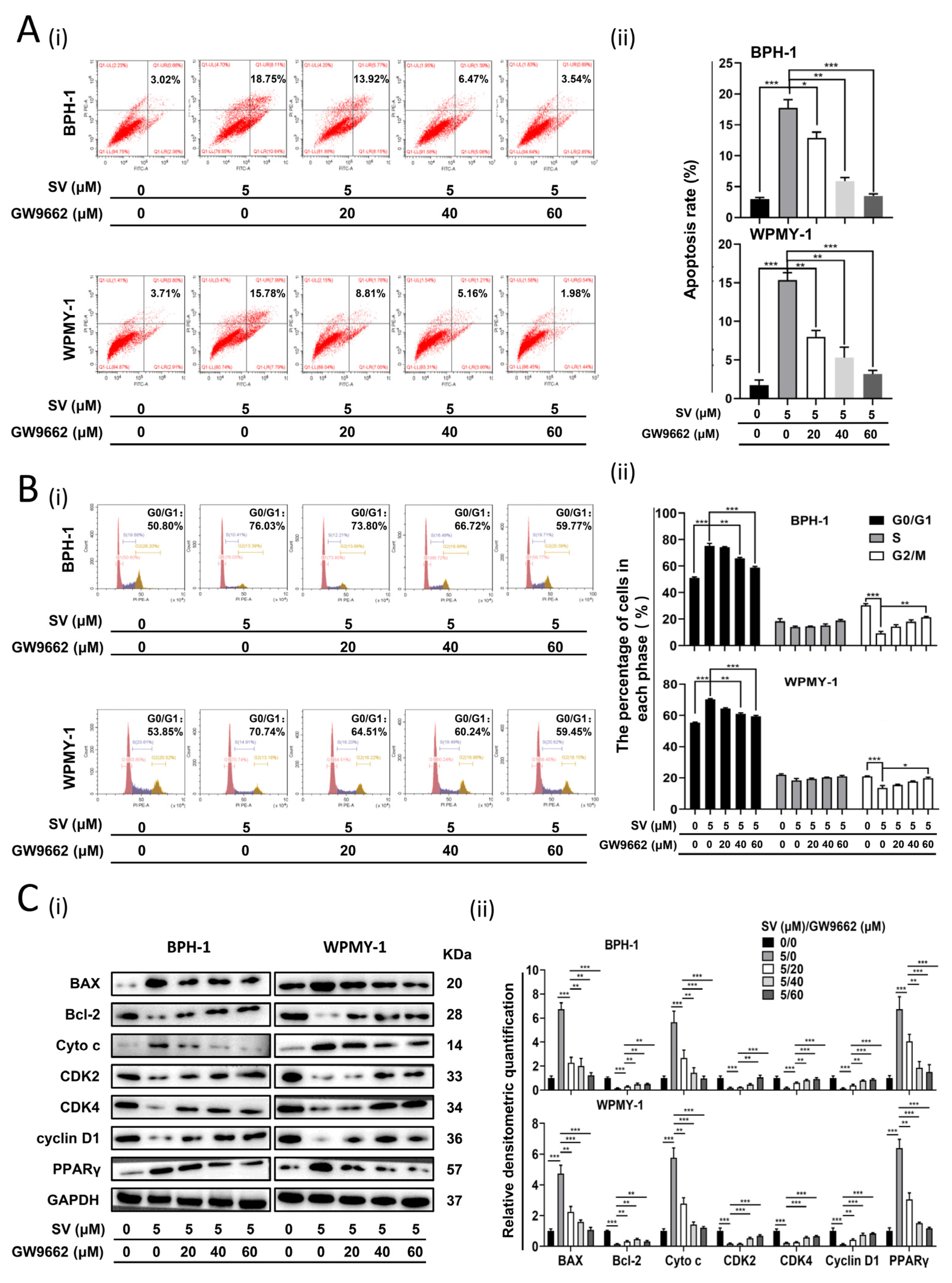
2.2. SV Inhibits Cell Survival by Promoting Cell Apoptosis and Inducing G0/G1 Phase Arrest through PPARγ
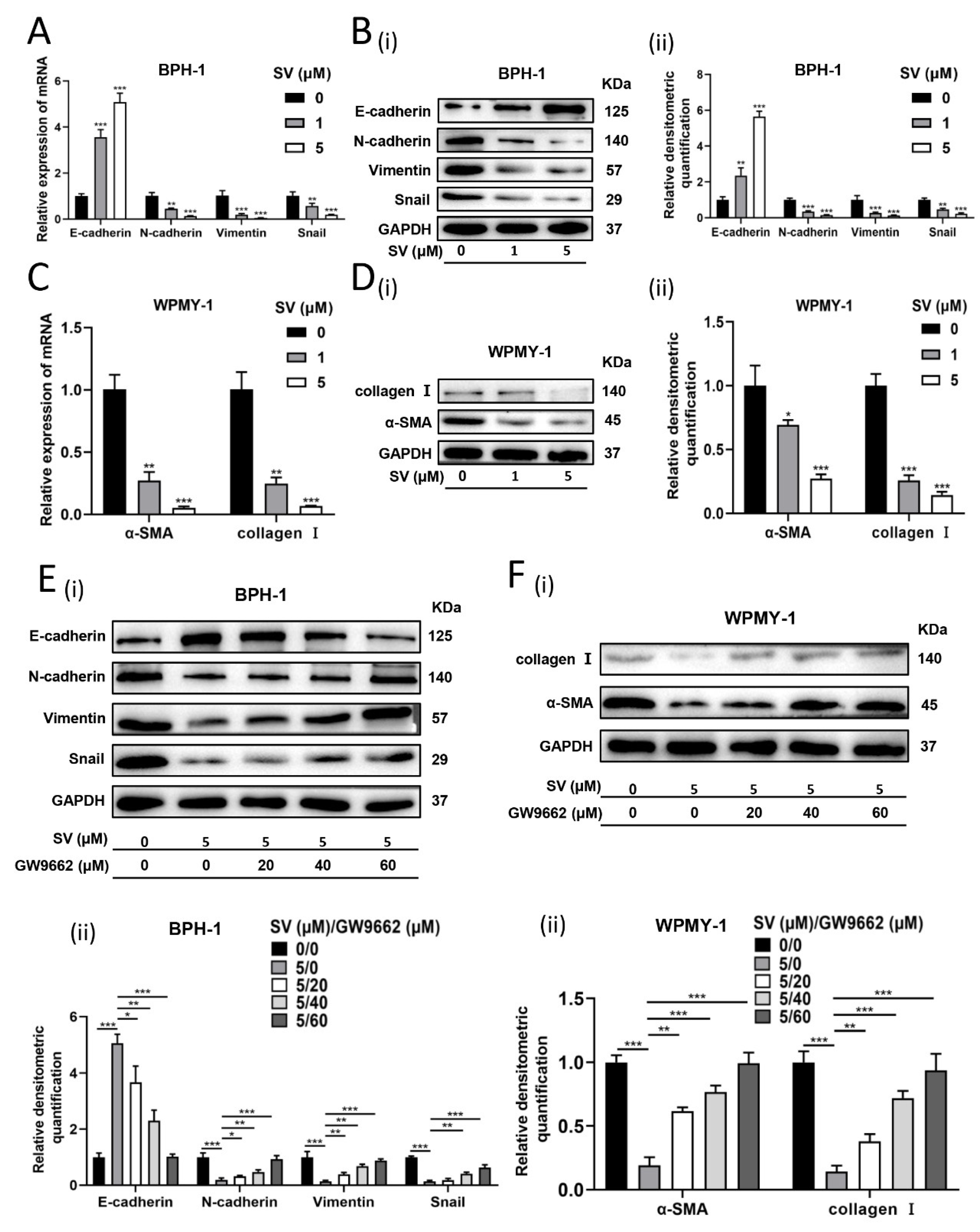
2.3. SV Attenuates Fibrosis and EMT Process in Prostate Cells through PPARγ Pathway
2.4. WNT/β-Catenin Pathway Crosstalks with PPARγ in Prostate Cells
2.5. SV Suppressed BPH by Increasing PPARγ In Vivo
2.6. PPARγ/WNT-1/β-Catenin Is Associated with Several Clinical Parameters in Patients with BPH
2.7. Overview of SV-PPARγ-WNT/β-Catenin Pathway in BPH
3. Discussion
4. Materials and Methods
4.1. Overview of Common Methods
4.2. Animals and Tissues
4.3. The Construction and Immunohistochemical Analysis of TMA
4.4. Drug Treatment of Cells
4.4.1. SV Treatment
4.4.2. GW9662 Treatment
4.5. Enzyme-Linked Immunosorbent Assay (ELISA)
4.6. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Berry, S.J.; Coffey, D.S.; Walsh, P.C.; Ewing, L.L. The development of human benign prostatic hyperplasia with age. J. Urol. 1984, 132, 474–479. [Google Scholar] [CrossRef] [PubMed]
- Chughtai, B.; Forde, J.C.; Thomas, D.D.; Laor, L.; Hossack, T.; Woo, H.H.; Te, A.E.; Kaplan, S.A. Benign prostatic hyperplasia. Nat. Rev. Dis. Prim. 2016, 2, 16031. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roehrborn, C.G.; Marks, L.; Harkaway, R. Enlarged prostate: A landmark national survey of its prevalence and impact on US men and their partners. Prostate Cancer Prostatic Dis. 2006, 9, 30–34. [Google Scholar] [CrossRef] [Green Version]
- Speakman, M.; Kirby, R.; Doyle, S.; Ioannou, C. Burden of male lower urinary tract symptoms (LUTS) suggestive of benign prostatic hyperplasia (BPH)-ocus on the UK. BJU Int. 2015, 115, 508–519. [Google Scholar] [CrossRef] [PubMed]
- van Exel, N.J.; Koopmanschap, M.A.; McDonnell, J.; Chapple, C.R.; Berges, R.; Rutten, F.F.; Panel, T.P.-E.E. Medical consumption and costs during a one-year follow-up of patients with LUTS suggestive of BPH in six european countries: Report of the TRIUMPH study. Eur Urol. 2006, 49, 92–102. [Google Scholar] [CrossRef]
- Timms, B.G.; Hofkamp, L.E. Prostate development and growth in benign prostatic hyperplasia. Differentiation 2011, 82, 173–183. [Google Scholar] [CrossRef]
- Robert, G.; Descazeaud, A.; Nicolaiew, N.; Terry, S.; Sirab, N.; Vacherot, F.; Maille, P.; Allory, Y.; de la Taille, A. Inflammation in benign prostatic hyperplasia: A 282 patients’ immunohistochemical analysis. Prostate 2009, 69, 1774–1780. [Google Scholar] [CrossRef] [Green Version]
- Carson, C., 3rd; Rittmaster, R. The role of dihydrotestosterone in benign prostatic hyperplasia. Urology 2003, 61, 2–7. [Google Scholar] [CrossRef]
- Pashootan, P.; Ploussard, G.; Cocaul, A.; de Gouvello, A.; Desgrandchamps, F. Association between metabolic syndrome and severity of lower urinary tract symptoms (LUTS): An observational study in a 4666 European men cohort. BJU Int. 2015, 116, 124–130. [Google Scholar] [CrossRef] [Green Version]
- Fu, Y.; Zhou, Z.; Yang, B.; Zhang, K.; He, L.; Zhang, X. The Relationship between the Clinical Progression of Benign Prostatic Hyperplasia and Metabolic Syndrome: A Prospective Study. Urol. Int. 2016, 97, 330–335. [Google Scholar] [CrossRef]
- Mongiu, A.K.; McVary, K.T. Lower urinary tract symptoms, benign prostatic hyperplasia, and obesity. Curr. Urol. Rep. 2009, 10, 247–253. [Google Scholar] [CrossRef]
- O’Neill, S.; O’Driscoll, L. Metabolic syndrome: A closer look at the growing epidemic and its associated pathologies. Obes. Rev. 2015, 16, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Dogan, Y.; Uruc, F.; Aras, B.; Sahin, A.; Kivrak, M.; Urkmez, A.; Guner, N.D.; Aydin, S. The relationships between metabolic syndrome, erectile dysfunction and lower urinary tract symptoms associated with benign prostatic hyperplasia. Turk. J. Urol. 2015, 41, 7–12. [Google Scholar] [CrossRef] [PubMed]
- Telli, O.; Demirbas, A.; Kabar, M.; Karagoz, M.A.; Sarici, H.; Resorlu, B. Does metabolic syndrome or its components correlate with lower urinary tract symptoms in benign prostatic hyperplasia patients? Nephrourol. Mon. 2015, 7, e27253. [Google Scholar] [CrossRef] [Green Version]
- Xiong, Y.; Zhang, Y.; Tan, J.; Qin, F.; Yuan, J. The association between metabolic syndrome and lower urinary tract symptoms suggestive of benign prostatic hyperplasia in aging males: Evidence based on propensity score matching. Transl. Androl. Urol. 2021, 10, 384–396. [Google Scholar] [CrossRef]
- Hammarsten, J.; Peeker, R. Urological aspects of the metabolic syndrome. Nat. Rev. Urol. 2011, 8, 483–494. [Google Scholar] [CrossRef] [PubMed]
- Gacci, M.; Corona, G.; Vignozzi, L.; Salvi, M.; Serni, S.; De Nunzio, C.; Tubaro, A.; Oelke, M.; Carini, M.; Maggi, M. Metabolic syndrome and benign prostatic enlargement: A systematic review and meta-analysis. BJU Int. 2015, 115, 24–31. [Google Scholar] [CrossRef] [PubMed]
- Pan, J.G.; Jiang, C.; Luo, R.; Zhou, X. Association of metabolic syndrome and benign prostatic hyperplasia in Chinese patients of different age decades. Urol. Int. 2014, 93, 10–16. [Google Scholar] [CrossRef]
- Vignozzi, L.; Morelli, A.; Sarchielli, E.; Comeglio, P.; Filippi, S.; Cellai, I.; Maneschi, E.; Serni, S.; Gacci, M.; Carini, M.; et al. Testosterone protects from metabolic syndrome-associated prostate inflammation: An experimental study in rabbit. J. Endocrinol. 2012, 212, 71–84. [Google Scholar] [CrossRef] [Green Version]
- Vignozzi, L.; Rastrelli, G.; Corona, G.; Gacci, M.; Forti, G.; Maggi, M. Benign prostatic hyperplasia: A new metabolic disease? J. Endocrinol. Investig. 2014, 37, 313–322. [Google Scholar] [CrossRef]
- Nandeesha, H.; Koner, B.C.; Dorairajan, L.N.; Sen, S.K. Hyperinsulinemia and dyslipidemia in non-diabetic benign prostatic hyperplasia. Clin. Chim. Acta 2006, 370, 89–93. [Google Scholar] [CrossRef] [PubMed]
- Hammarsten, J.; Hogstedt, B.; Holthuis, N.; Mellstrom, D. Components of the metabolic syndrome-risk factors for the development of benign prostatic hyperplasia. Prostate Cancer Prostatic Dis. 1998, 1, 157–162. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parsons, J.K.; Bergstrom, J.; Barrett-Connor, E. Lipids, lipoproteins and the risk of benign prostatic hyperplasia in community-dwelling men. BJU Int. 2008, 101, 313–318. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eckel, R.H.; Grundy, S.M.; Zimmet, P.Z. The metabolic syndrome. Lancet 2005, 365, 1415–1428. [Google Scholar] [CrossRef]
- Russo, G.I.; Regis, F.; Spatafora, P.; Frizzi, J.; Urzi, D.; Cimino, S.; Serni, S.; Carini, M.; Gacci, M.; Morgia, G. Association between metabolic syndrome and intravesical prostatic protrusion in patients with benign prostatic enlargement and lower urinary tract symptoms (MIPS Study). BJU Int. 2018, 121, 799–804. [Google Scholar] [CrossRef] [PubMed]
- Davis, M.E.; Korn, M.A.; Gumucio, J.P.; Harning, J.A.; Saripalli, A.L.; Bedi, A.; Mendias, C.L. Simvastatin reduces fibrosis and protects against muscle weakness after massive rotator cuff tear. J. Shoulder Elb. Surg. 2015, 24, 280–287. [Google Scholar] [CrossRef] [Green Version]
- Nakashima, Y.; Miyagi-Shiohira, C.; Noguchi, H.; Omasa, T. Atorvastatin inhibits the HIF1alpha-PPAR axis, which is essential for maintaining the function of human induced pluripotent stem cells. Mol. Ther. 2018, 26, 1715–1734. [Google Scholar] [CrossRef] [Green Version]
- Pose, E.; Trebicka, J.; Mookerjee, R.P.; Angeli, P.; Gines, P. Statins: Old drugs as new therapy for liver diseases? J. Hepatol. 2019, 70, 194–202. [Google Scholar] [CrossRef] [Green Version]
- Rzouq, F.; Alahdab, F.; Olyaee, M. Statins and hepatitis C virus infection: An old therapy with new scope. Am. J. Med. Sci 2014, 348, 426–430. [Google Scholar] [CrossRef]
- Stopsack, K.H. Statins and prostate cancer: Bias, precision medicine, or population health? Eur. Urol. 2021, 79, 453–455. [Google Scholar] [CrossRef]
- Zhang, X.; Zeng, X.; Dong, L.; Zhao, X.; Qu, X. The effects of statins on benign prostatic hyperplasia in elderly patients with metabolic syndrome. World J. Urol. 2015, 33, 2071–2077. [Google Scholar] [CrossRef] [PubMed]
- Zou, C.; Qi, H.; Liu, Z.H.; Han, L.; Zhao, C.; Yang, X. Simvastatin activates the PPARgamma-dependent pathway to prevent left ventricular hypertrophy associated with inhibition of RhoA signaling. Tex. Heart Inst. J. 2013, 40, 140–147. [Google Scholar] [PubMed]
- Du, H.; Hu, H.; Zheng, H.; Hao, J.; Yang, J.; Cui, W. Effects of peroxisome proliferator-activated receptor gamma in simvastatin antiplatelet activity: Influences on cAMP and mitogen-activated protein kinases. Thromb. Res. 2014, 134, 111–120. [Google Scholar] [CrossRef] [PubMed]
- Walther, U.; Emmrich, K.; Ramer, R.; Mittag, N.; Hinz, B. Lovastatin lactone elicits human lung cancer cell apoptosis via a COX-2/PPARgamma-dependent pathway. Oncotarget 2016, 7, 10345–10362. [Google Scholar] [CrossRef] [Green Version]
- Rosen, E.D.; Spiegelman, B.M. PPARgamma: A nuclear regulator of metabolism, differentiation, and cell growth. J. Biol. Chem. 2001, 276, 37731–37734. [Google Scholar] [CrossRef] [Green Version]
- Cheng, H.S.; Tan, W.R.; Low, Z.S.; Marvalim, C.; Lee, J.Y.H.; Tan, N.S. Exploration and development of PPAR modulators in health and disease: An update of clinical evidence. Int. J. Mol. Sci. 2019, 20, 5055. [Google Scholar] [CrossRef] [Green Version]
- Francis, G.A.; Fayard, E.; Picard, F.; Auwerx, J. Nuclear receptors and the control of metabolism. Annu. Rev. Physiol. 2003, 65, 261–311. [Google Scholar] [CrossRef]
- Forman, B.M.; Tontonoz, P.; Chen, J.; Brun, R.P.; Spiegelman, B.M.; Evans, R.M. 15-Deoxy-delta 12, 14-prostaglandin J2 is a ligand for the adipocyte determination factor PPAR gamma. Cell 1995, 83, 803–812. [Google Scholar] [CrossRef] [Green Version]
- Haakonsson, A.K.; Stahl Madsen, M.; Nielsen, R.; Sandelin, A.; Mandrup, S. Acute genome-wide effects of rosiglitazone on PPARgamma transcriptional networks in adipocytes. Mol. Endocrinol. 2013, 27, 1536–1549. [Google Scholar] [CrossRef]
- Lehrke, M.; Lazar, M.A. The many faces of PPARgamma. Cell 2005, 123, 993–999. [Google Scholar] [CrossRef] [Green Version]
- Rogue, A.; Spire, C.; Brun, M.; Claude, N.; Guillouzo, A. Gene expression changes induced by ppar gamma agonists in animal and human liver. PPAR Res. 2010, 2010, 325183. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schupp, M.; Cristancho, A.G.; Lefterova, M.I.; Hanniman, E.A.; Briggs, E.R.; Steger, D.J.; Qatanani, M.; Curtin, J.C.; Schug, J.; Ochsner, S.A.; et al. Re-expression of GATA2 cooperates with peroxisome proliferator-activated receptor-gamma depletion to revert the adipocyte phenotype. J. Biol. Chem. 2009, 284, 9458–9464. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Farshbaf, M.J.; Ghaedi, K.; Shirani, M.; Nasr-Esfahani, M.H. Peroxisome proliferator activated receptor gamma (PPARgamma) as a therapeutic target for improvement of cognitive performance in Fragile-X. Med. Hypotheses 2014, 82, 291–294. [Google Scholar] [CrossRef] [PubMed]
- Ghoochani, A.; Shabani, K.; Peymani, M.; Ghaedi, K.; Karamali, F.; Karbalaei, K.; Tanhaie, S.; Salamian, A.; Esmaeili, A.; Valian-Borujeni, S.; et al. The influence of peroxisome proliferator-activated receptor gamma(1) during differentiation of mouse embryonic stem cells to neural cells. Differentiation 2012, 83, 60–67. [Google Scholar] [CrossRef] [PubMed]
- Shu, L.; Huang, R.; Wu, S.; Chen, Z.; Sun, K.; Jiang, Y.; Cai, X. PPARgamma and Its Ligands: Potential Antitumor Agents in the Digestive System. Curr. Stem Cell Res. 2016, 11, 274–281. [Google Scholar] [CrossRef]
- Olokpa, E.; Bolden, A.; Stewart, L.V. The androgen receptor regulates ppargamma expression and activity in human prostate cancer cells. J. Cell Physiol. 2016, 231, 2664–2672. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hisatake, J.I.; Ikezoe, T.; Carey, M.; Holden, S.; Tomoyasu, S.; Koeffler, H.P. Down-Regulation of prostate-specific antigen expression by ligands for peroxisome proliferator-activated receptor gamma in human prostate cancer. Cancer Res. 2000, 60, 5494–5498. [Google Scholar]
- Kubota, T.; Koshizuka, K.; Williamson, E.A.; Asou, H.; Said, J.W.; Holden, S.; Miyoshi, I.; Koeffler, H.P. Ligand for peroxisome proliferator-activated receptor gamma (troglitazone) has potent antitumor effect against human prostate cancer both in vitro and in vivo. Cancer Res. 1998, 58, 3344–3352. [Google Scholar]
- Mueller, E.; Smith, M.; Sarraf, P.; Kroll, T.; Aiyer, A.; Kaufman, D.S.; Oh, W.; Demetri, G.; Figg, W.D.; Zhou, X.P.; et al. Effects of ligand activation of peroxisome proliferator-activated receptor gamma in human prostate cancer. Proc. Natl. Acad. Sci. USA 2000, 97, 10990–10995. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Zhang, X.; Wang, J.; Shen, Y.; Tang, X.; Yu, F.; Wang, R. Expression and function of PPARs in cancer stem cells. Curr. Stem Cell Res. 2016, 11, 226–234. [Google Scholar] [CrossRef]
- Rehan, V.K.; Torday, J.S. PPARgamma Signaling Mediates the Evolution, Development, Homeostasis, and Repair of the Lung. PPAR Res. 2012, 2012, 289867. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reddy, A.T.; Lakshmi, S.P.; Zhang, Y.; Reddy, R.C. Nitrated fatty acids reverse pulmonary fibrosis by dedifferentiating myofibroblasts and promoting collagen uptake by alveolar macrophages. FASEB J. 2014, 28, 5299–5310. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lecarpentier, Y.; Claes, V.; Duthoit, G.; Hebert, J.L. Circadian rhythms, Wnt/beta-catenin pathway and PPAR alpha/gamma profiles in diseases with primary or secondary cardiac dysfunction. Front. Physiol. 2014, 5, 429. [Google Scholar] [CrossRef] [Green Version]
- MacDonald, B.T.; Tamai, K.; He, X. Wnt/beta-catenin signaling: Components, mechanisms, and diseases. Dev. Cell 2009, 17, 9–26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, S.Y.; Du, Y.; Song, J. MicroRNA-340 inhibits epithelial-mesenchymal transition by impairing rock-1-dependent wnt/beta-catenin signaling pathway in epithelial cells from human benign prostatic hyperplasia. Chin. Med. J. 2018, 131, 2008–2012. [Google Scholar] [CrossRef]
- Katoh, M. Multilayered prevention and treatment of chronic inflammation, organ fibrosis and cancer associated with canonical WNT/betacatenin signaling activation (Review). Int. J. Mol. Med. 2018, 42, 713–725. [Google Scholar] [CrossRef] [Green Version]
- Keil, K.P.; Mehta, V.; Branam, A.M.; Abler, L.L.; Buresh-Stiemke, R.A.; Joshi, P.S.; Schmitz, C.T.; Marker, P.C.; Vezina, C.M. Wnt inhibitory factor 1 (Wif1) is regulated by androgens and enhances androgen-dependent prostate development. Endocrinology 2012, 153, 6091–6103. [Google Scholar] [CrossRef] [Green Version]
- Pan, C.; Chen, Y.; Xu, T.; Wang, J.; Li, D.; Han, X. Chronic exposure to microcystin-leucine-arginine promoted proliferation of prostate epithelial cells resulting in benign prostatic hyperplasia. Environ. Pollut. 2018, 242, 1535–1545. [Google Scholar] [CrossRef]
- Wei, X.; Zhang, L.; Zhou, Z.; Kwon, O.J.; Zhang, Y.; Nguyen, H.; Dumpit, R.; True, L.; Nelson, P.; Dong, B.; et al. Spatially restricted stromal wnt signaling restrains prostate epithelial progenitor growth through direct and indirect mechanisms. Cell Stem Cell 2019, 24, 753–768. [Google Scholar] [CrossRef]
- Fagerberg, L.; Hallstrom, B.M.; Oksvold, P.; Kampf, C.; Djureinovic, D.; Odeberg, J.; Habuka, M.; Tahmasebpoor, S.; Danielsson, A.; Edlund, K.; et al. Analysis of the human tissue-specific expression by genome-wide integration of transcriptomics and antibody-based proteomics. Mol. Cell Proteom. 2014, 13, 397–406. [Google Scholar] [CrossRef] [Green Version]
- Vidal-Puig, A.; Jimenez-Linan, M.; Lowell, B.B.; Hamann, A.; Hu, E.; Spiegelman, B.; Flier, J.S.; Moller, D.E. Regulation of PPAR gamma gene expression by nutrition and obesity in rodents. J. Clin. Investig. 1996, 97, 2553–2561. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Werman, A.; Hollenberg, A.; Solanes, G.; Bjorbaek, C.; Vidal-Puig, A.J.; Flier, J.S. Ligand-independent activation domain in the N terminus of peroxisome proliferator-activated receptor gamma (PPARgamma). Differential activity of PPARgamma1 and -2 isoforms and influence of insulin. J. Biol. Chem. 1997, 272, 20230–20235. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, D.; Zhang, F.; Zhang, X.; Xue, C.; Namwanje, M.; Fan, L.; Reilly, M.P.; Hu, F.; Qiang, L. Distinct functions of PPARgamma isoforms in regulating adipocyte plasticity. Biochem. Biophys. Res. Commun 2016, 481, 132–138. [Google Scholar] [CrossRef] [Green Version]
- Hu, W.; Jiang, C.; Kim, M.; Xiao, Y.; Richter, H.J.; Guan, D.; Zhu, K.; Krusen, B.M.; Roberts, A.N.; Miller, J.; et al. Isoform-specific functions of PPARgamma in gene regulation and metabolism. Genes Dev. 2022, 36, 300–312. [Google Scholar] [CrossRef]
- Pang, X.; Shu, Y.; Niu, Z.; Zheng, W.; Wu, H.; Lu, Y.; Shen, P. PPARgamma1 phosphorylation enhances proliferation and drug resistance in human fibrosarcoma cells. Exp. Cell Res. 2014, 322, 30–38. [Google Scholar] [CrossRef]
- Koga, H.; Sakisaka, S.; Harada, M.; Takagi, T.; Hanada, S.; Taniguchi, E.; Kawaguchi, T.; Sasatomi, K.; Kimura, R.; Hashimoto, O.; et al. Involvement of p21(WAF1/Cip1), p27(Kip1), and p18(INK4c) in troglitazone-induced cell-cycle arrest in human hepatoma cell lines. Hepatology 2001, 33, 1087–1097. [Google Scholar] [CrossRef]
- Jiao, X.; Tian, L.; Zhang, Z.; Balcerek, J.; Kossenkov, A.V.; Casimiro, M.C.; Wang, C.; Liu, Y.; Ertel, A.; Soccio, R.E.; et al. Ppargamma1 Facilitates ErbB2-Mammary Adenocarcinoma in Mice. Cancers 2021, 13, 2171. [Google Scholar] [CrossRef] [PubMed]
- Mu, F.; Jing, Y.; Ning, B.; Huang, J.; Cui, T.; Guo, Y.; You, X.; Yan, X.; Li, H.; Wang, N. Peroxisome proliferator-activated receptor gamma isoforms differentially regulate preadipocyte proliferation, apoptosis, and differentiation in chickens. Poult. Sci. 2020, 99, 6410–6421. [Google Scholar] [CrossRef]
- Forootan, F.S.; Forootan, S.S.; Malki, M.I.; Chen, D.; Li, G.; Lin, K.; Rudland, P.S.; Foster, C.S.; Ke, Y. The expression of C-FABP and PPARgamma and their prognostic significance in prostate cancer. Int. J. Oncol. 2014, 44, 265–275. [Google Scholar] [CrossRef] [Green Version]
- Mehrbod, P.; Ande, S.R.; Alizadeh, J.; Rahimizadeh, S.; Shariati, A.; Malek, H.; Hashemi, M.; Glover, K.K.M.; Sher, A.A.; Coombs, K.M.; et al. The roles of apoptosis, autophagy and unfolded protein response in arbovirus, influenza virus, and HIV infections. Virulence 2019, 10, 376–413. [Google Scholar] [CrossRef] [Green Version]
- Sochalska, M.; Tuzlak, S.; Egle, A.; Villunger, A. Lessons from gain- and loss-of-function models of pro-survival Bcl2 family proteins: Implications for targeted therapy. FEBS J. 2015, 282, 834–849. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maggiora, M.; Oraldi, M.; Muzio, G.; Canuto, R.A. Involvement of PPARalpha and PPARgamma in apoptosis and proliferation of human hepatocarcinoma HepG2 cells. Cell Biochem. Funct. 2010, 28, 571–577. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Cao, R.; Wang, Y.; Qian, G.; Dan, H.C.; Jiang, W.; Ju, L.; Wu, M.; Xiao, Y.; Wang, X. Simvastatin induces cell cycle arrest and inhibits proliferation of bladder cancer cells via PPARgamma signalling pathway. Sci. Rep. 2016, 6, 35783. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Broster, S.A.; Kyprianou, N. Epithelial-mesenchymal transition in prostatic disease. Future Oncol. 2015, 11, 3197–3206. [Google Scholar] [CrossRef]
- Chen, W.; Pascal, L.E.; Wang, K.; Dhir, R.; Sims, A.M.; Campbell, R.; Gasper, G.; DeFranco, D.B.; Yoshimura, N.; Wang, Z. Differential impact of paired patient-derived BPH and normal adjacent stromal cells on benign prostatic epithelial cell growth in 3D culture. Prostate 2020, 80, 1177–1187. [Google Scholar] [CrossRef]
- Li, F.; Pascal, L.E.; Stolz, D.B.; Wang, K.; Zhou, Y.; Chen, W.; Xu, Y.; Chen, Y.; Dhir, R.; Parwani, A.V.; et al. E-cadherin is downregulated in benign prostatic hyperplasia and required for tight junction formation and permeability barrier in the prostatic epithelial cell monolayer. Prostate 2019, 79, 1226–1237. [Google Scholar] [CrossRef]
- Vallee, A.; Lecarpentier, Y.; Guillevin, R.; Vallee, J.N. Thermodynamics in gliomas: Interactions between the canonical WNT/Beta-catenin pathway and PPAR gamma. Front. Physiol. 2017, 8, 352. [Google Scholar] [CrossRef] [Green Version]
- Lecarpentier, Y.; Claes, V.; Vallee, A.; Hebert, J.L. Interactions between PPAR gamma and the canonical Wnt/Beta-catenin pathway in type 2 diabetes and colon cancer. PPAR Res. 2017, 2017, 5879090. [Google Scholar] [CrossRef] [Green Version]
- Takada, I.; Kouzmenko, A.P.; Kato, S. Wnt and PPARgamma signaling in osteoblastogenesis and adipogenesis. Nat. Rev. Rheumatol. 2009, 5, 442–447. [Google Scholar] [CrossRef]
- Lu, D.; Carson, D.A. Repression of beta-catenin signaling by PPAR gamma ligands. Eur. J. Pharm. 2010, 636, 198–202. [Google Scholar] [CrossRef] [Green Version]
- Grimes, C.A.; Jope, R.S. The multifaceted roles of glycogen synthase kinase 3beta in cellular signaling. Prog. Neurobiol. 2001, 65, 391–426. [Google Scholar] [CrossRef] [PubMed]
- Jeon, M.; Rahman, N.; Kim, Y.S. Wnt/beta-catenin signaling plays a distinct role in methyl gallate-mediated inhibition of adipogenesis. Biochem. Biophys. Res. Commun. 2016, 479, 22–27. [Google Scholar] [CrossRef] [PubMed]
- Gustafson, B.; Eliasson, B.; Smith, U. Thiazolidinediones increase the wingless-type MMTV integration site family (WNT) inhibitor Dickkopf-1 in adipocytes: A link with osteogenesis. Diabetologia 2010, 53, 536–540. [Google Scholar] [CrossRef] [Green Version]
- Jiang, H.; Zhang, Z.; Yu, Y.; Chu, H.Y.; Yu, S.; Yao, S.; Zhang, G.; Zhang, B.T. Drug discovery of DKK1 inhibitors. Front. Pharm. 2022, 13, 847387. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Zang, N.; Jiang, Y.; Chen, P.; Wang, X.; Zhang, X. Upregulation of Phosphodiesterase type 5 in the Hyperplastic Prostate. Sci. Rep. 2015, 5, 17888. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, M.Y.; Shin, I.S.; Seo, C.S.; Lee, N.H.; Ha, H.K.; Son, J.K.; Shin, H.K. Effects of Melandrium firmum methanolic extract on testosterone-induced benign prostatic hyperplasia in Wistar rats. Asian J. Androl. 2012, 14, 320–324. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gong, Y.M.; Wang, X.; Liu, S.; Hu, X.C.; Xu, Y.; Huang, T. Simvastatin inhibits prostatic hyperplasia in rats with metabolic syndrome. Int. Urol. Nephrol. 2022, 54, 2125–2131. [Google Scholar] [CrossRef]
- Ishola, I.O.; Tijani, H.K.; Dosumu, O.O.; Anunobi, C.C.; Oshodi, T.O. Atorvastatin attenuates testosterone-induced benign prostatic hyperplasia in rats: Role of peroxisome proliferator-activated receptor-gamma and cyclo-oxygenase-2. Fundam. Clin. Pharm. 2017, 31, 652–662. [Google Scholar] [CrossRef]
- Foo, K.T. What is a disease? What is the disease clinical benign prostatic hyperplasia (BPH)? World J. Urol. 2019, 37, 1293–1296. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.; Yin, J.; Chen, P.; Liu, D.; He, W.; Li, Y.; Li, M.; Fu, X.; Zeng, G.; Guo, Y.; et al. Smoothened inhibition leads to decreased cell proliferation and suppressed tissue fibrosis in the development of benign prostatic hyperplasia. Cell Death Discov. 2021, 7, 115. [Google Scholar] [CrossRef]
- Liu, J.; Liu, D.; Zhang, X.; Li, Y.; Fu, X.; He, W.; Li, M.; Chen, P.; Zeng, G.; DiSanto, M.E.; et al. NELL2 modulates cell proliferation and apoptosis via ERK pathway in the development of benign prostatic hyperplasia. Clin. Sci. 2021, 135, 1591–1608. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.; Liu, J.; Li, Y.; Liu, H.; Hassan, H.M.; He, W.; Li, M.; Zhou, Y.; Fu, X.; Zhan, J.; et al. Upregulated bone morphogenetic protein 5 enhances proliferation and epithelial-mesenchymal transition process in benign prostatic hyperplasia via BMP/Smad signaling pathway. Prostate 2021, 81, 1435–1449. [Google Scholar] [CrossRef] [PubMed]
- Gaspar, J.C.; Healy, C.; Ferdousi, M.I.; Roche, M.; Finn, D.P. Pharmacological blockade of pparalpha exacerbates inflammatory pain-related impairment of spatial memory in rats. Biomedicines 2021, 9, 610. [Google Scholar] [CrossRef] [PubMed]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Pearson Correlation Coefficient | p-Value | |
|---|---|---|
| PPARγ vs. WNT-1 PPARγ vs. β-catenin WNT-1 vs. β-catenin | 0.1848 −0.2193 0.6525 | 0.3919 0.0253 * <0.001 *** |
| PPARγ | WNT-1 | β-Catenin | ||||
|---|---|---|---|---|---|---|
| Pearson Correlation Coefficient | p-Value | Pearson Correlation Coefficient | p-Value | Pearson Correlation Coefficient | p-Value | |
| Age | 0.0177 | 0.8585 | −0.1286 | 0.1933 | 0.0307 | 0.7571 |
| BMI | 0.0753 | 0.4497 | −0.0329 | 0.7417 | 0.0278 | 0.7805 |
| PV | −0.2133 | 0.0297 * | 0.0354 | 0.7212 | 0.0568 | 0.5667 |
| tPSA | −0.0578 | 0.5800 | −0.1540 | 0.1383 | 0.0386 | 0.7121 |
| fPSA | −0.3053 | 0.0025 ** | −0.0753 | 0.4661 | 0.0024 | 0.9811 |
| Qmax | 0.3125 | 0.0496 * | −0.0405 | 0.8042 | 0.0210 | 0.8979 |
| RU | 0.0738 | 0.5854 | −0.0260 | 0.8479 | 0.0417 | 0.7586 |
| IPSS | 0.0649 | 0.5150 | 0.2211 | 0.0248 * | 0.1386 | 0.1626 |
| N | −0.0313 | 0.7538 | 0.0740 | 0.4576 | 0.2356 | 0.0166 * |
| Mean | SD | |
|---|---|---|
| Age (years) | 70.13 | 7.46 |
| Body mass index (kg/m2) | 22.78 | 2.76 |
| Prostate volume (cm3) | 60.82 | 36.73 |
| Total prostate specific antigen (ng/mL) | 7.00 | 5.89 |
| Free prostate specific antigen (ng/mL) | 1.62 | 1.41 |
| Maximum flow rate (mL/s) | 9.97 | 5.86 |
| Residual urine (mL) | 160.66 | 114.48 |
| International prostate symptom score | 21.74 | 7.37 |
| Nocturia (N) | 3.07 | 1.94 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, Z.; Yang, S.; Li, Y.; Zhou, Y.; Liu, D.; Liu, J.; DiSanto, M.E.; Zhang, X. Simvastatin Improves Benign Prostatic Hyperplasia: Role of Peroxisome-Proliferator-Activated Receptor-γ and Classic WNT/β-Catenin Pathway. Int. J. Mol. Sci. 2023, 24, 4911. https://doi.org/10.3390/ijms24054911
Wang Z, Yang S, Li Y, Zhou Y, Liu D, Liu J, DiSanto ME, Zhang X. Simvastatin Improves Benign Prostatic Hyperplasia: Role of Peroxisome-Proliferator-Activated Receptor-γ and Classic WNT/β-Catenin Pathway. International Journal of Molecular Sciences. 2023; 24(5):4911. https://doi.org/10.3390/ijms24054911
Chicago/Turabian StyleWang, Zhen, Shu Yang, Yan Li, Yongying Zhou, Daoquan Liu, Jianmin Liu, Michael E. DiSanto, and Xinhua Zhang. 2023. "Simvastatin Improves Benign Prostatic Hyperplasia: Role of Peroxisome-Proliferator-Activated Receptor-γ and Classic WNT/β-Catenin Pathway" International Journal of Molecular Sciences 24, no. 5: 4911. https://doi.org/10.3390/ijms24054911