
Gene Co-Expression Network Analysis Reveals the Hub Genes and Key Pathways Associated with Resistance to Salmonella Enteritidis Colonization in Chicken
 ,
,
Abstract
:1. Introduction
2. Results
2.1. Phenotypic, Immune, and Microbiome Diversity
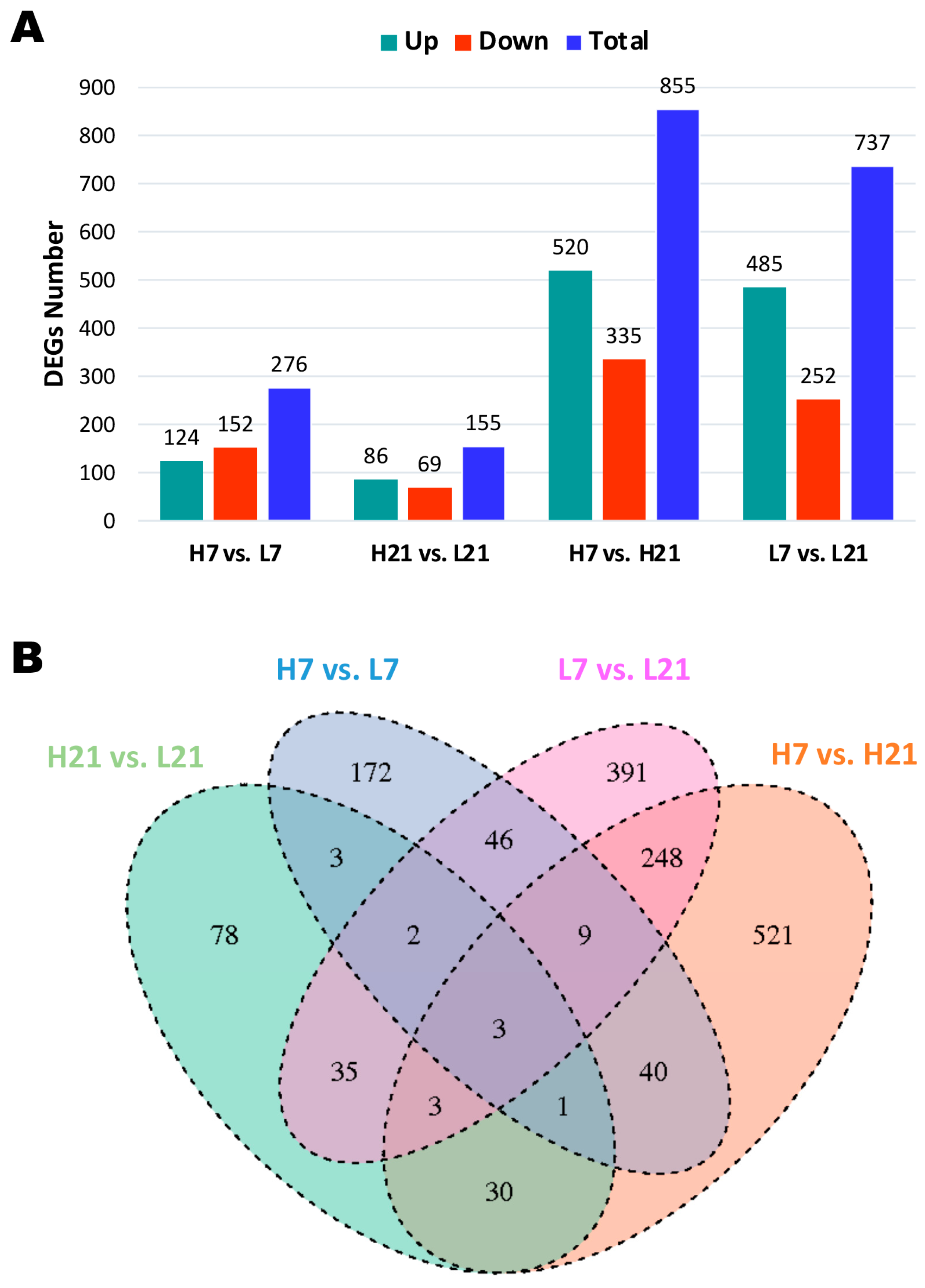
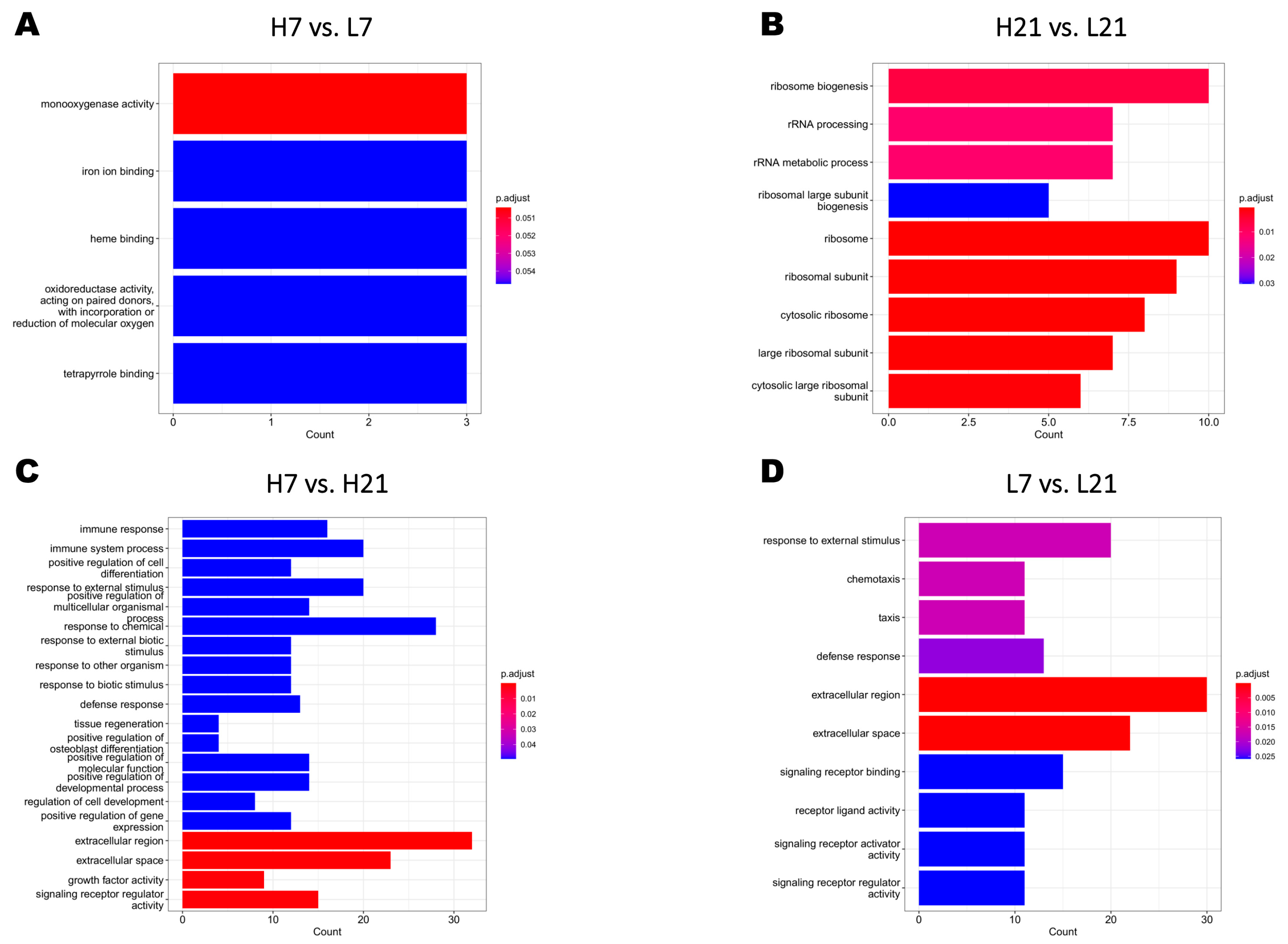
2.2. Transcriptome Profiling and Differentially Expressed Genes (DEGs)
2.3. Developmental Dynamics Genes and Gene Expression Patterns in the Cecum
2.4. Weighted Gene Co-Expression Network Analysis (WGCNA)
2.5. Screening of Hub Genes Associated with Resistance to Salmonella
2.6. Identification of Major Driver Genes by the Overlapping Method
2.7. Verification of Selected Candidate Genes Involvement in the Process of Salmonella Infection in Chicken
3. Discussion
4. Materials and Methods
4.1. Animal, Experimental Design, and Sample Collection
4.2. Phenotype and Microbiome Relative Abundance Determination
4.3. RNA Isolation
4.4. Transcriptome Profiling and Differentially Expressed Genes
4.5. Dynamic Developmental Genes Identification and Genes Expression Pattern
4.6. Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) Pathway Analyses
4.7. Weighted Gene Co-Expression Network Analysis
4.8. Quantitative Real-Time PCR
4.9. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Barrow, P.A.; Jones, M.A.; Smith, A.L.; Wigley, P. The long view: Salmonella--the last forty years. Avian Pathol. 2012, 41, 413–420. [Google Scholar] [CrossRef]
- Calenge, F.; Beaumont, C. Toward integrative genomics study of genetic resistance to Salmonella and Campylobacter intestinal colonization in fowl. Front. Genet. 2012, 3, 261. [Google Scholar] [CrossRef] [Green Version]
- Calenge, F.; Kaiser, P.; Vignal, A.; Beaumont, C. Genetic control of resistance to salmonellosis and to Salmonella carrier-state in fowl: A review. Genet. Sel. Evol. 2010, 42, 11. [Google Scholar] [CrossRef] [Green Version]
- Li, P.; Fan, W.; Li, Q.; Wang, J.; Liu, R.; Everaert, N.; Liu, J.; Zhang, Y.; Zheng, M.; Cui, H.; et al. Splenic microRNA Expression Profiles and Integration Analyses Involved in Host Responses to Salmonella enteritidis Infection in Chickens. Front. Cell. Infect. Microbiol. 2017, 7, 377. [Google Scholar] [CrossRef] [Green Version]
- Atterbury, R.J.; Gigante, A.M.; Rubio Lozano, M.d.l.S.; Méndez Medina, R.D.; Robinson, G.; Alloush, H.; Barrow, P.A.; Allen, V.M. Reduction of Salmonella contamination on the surface of chicken skin using bacteriophage. Virol. J. 2020, 17, 98. [Google Scholar] [CrossRef]
- Li, P.; Wang, H.; Zhao, X.; Gou, Z.; Liu, R.; Song, Y.; Li, Q.; Zheng, M.; Cui, H.; Everaert, N.; et al. Allelic variation in TLR4 is linked to resistance to Salmonella Enteritidis infection in chickens. Poult. Sci. 2017, 96, 2040–2048. [Google Scholar] [CrossRef]
- Kempf, F.; La Ragione, R.; Chirullo, B.; Schouler, C.; Velge, P. Super Shedding in Enteric Pathogens: A Review. Microorganisms 2022, 10, 2101. [Google Scholar] [CrossRef]
- Yegani, M.; Korver, D.R. Factors affecting intestinal health in poultry. Poult. Sci. 2008, 87, 2052–2063. [Google Scholar] [CrossRef]
- Apajalahti, J. Comparative gut microflora, metabolic challenges, and potential opportunities. J. Appl. Poult. Res. 2005, 14, 444–453. [Google Scholar] [CrossRef]
- Jeurissen, S.H.; Lewis, F.; van der Klis, J.D.; Mroz, Z.; Rebel, J.M.; ter Huurne, A.A. Parameters and techniques to determine intestinal health of poultry as constituted by immunity, integrity, and functionality. Curr. Issues Intest. Microbiol. 2002, 3, 1–14. [Google Scholar]
- Gaskins, H.R.; Collier, C.T.; Anderson, D.B. Antibiotics as growth promotants:mode of action. Anim. Biotechnol. 2002, 13, 29–42. [Google Scholar] [CrossRef]
- Levy, M.; Thaiss, C.A.; Elinav, E. Metabolites: Messengers between the microbiota and the immune system. Genes Dev. 2016, 30, 1589–1597. [Google Scholar] [CrossRef] [Green Version]
- Al-Murrani, W.K.; Al-Rawi, A.J.; Al-Hadithi, M.F.; Al-Tikriti, B. Association between heterophil/lymphocyte ratio, a marker of ′resistance′ to stress, and some production and fitness traits in chickens. Br. Poult. Sci. 2006, 47, 443–448. [Google Scholar] [CrossRef]
- Al-Murrani, W.K.; Al-Rawi, I.K.; Raof, N.M. Genetic resistance to Salmonella typhimurium in two lines of chickens selected as resistant and sensitive on the basis of heterophil/lymphocyte ratio. Br. Poult. Sci. 2002, 43, 501–507. [Google Scholar] [CrossRef]
- Minias, P.; Włodarczyk, R.; Meissner, W.; Husak, J. Leukocyte profiles are associated with longevity and survival, but not migratory effort: A comparative analysis of shorebirds. Funct. Ecol. 2017, 32, 369–378. [Google Scholar] [CrossRef] [Green Version]
- Thiam, M.; Barreto Sánchez, A.L.; Zhang, J.; Wen, J.; Zhao, G.; Wang, Q. Investigation of the Potential of Heterophil/Lymphocyte Ratio as a Biomarker to Predict Colonization Resistance and Inflammatory Response to Salmonella enteritidis Infection in Chicken. Pathogens 2022, 11, 72. [Google Scholar] [CrossRef]
- Thiam, M.; Wang, Q.; Barreto Sánchez, A.L.; Zhang, J.; Ding, J.; Wang, H.; Zhang, Q.; Zhang, N.; Wang, J.; Li, Q.; et al. Heterophil/Lymphocyte Ratio Level Modulates Salmonella Resistance, Cecal Microbiota Composition and Functional Capacity in Infected Chicken. Front. Immunol. 2022, 13, 816689. [Google Scholar] [CrossRef]
- Minias, P. Evolution of heterophil/lymphocyte ratios in response to ecological and life-history traits: A comparative analysis across the avian tree of life. J. Anim. Ecol. 2019, 88, 554–565. [Google Scholar] [CrossRef]
- Thiam, M.; Barreto Sanchez, A.L.; Zhang, J.; Zheng, M.; Wen, J.; Zhao, G.; Wang, Q. Association of Heterophil/Lymphocyte Ratio with Intestinal Barrier Function and Immune Response to Salmonella enteritidis Infection in Chicken. Animals (Basel) 2021, 11, 3498. [Google Scholar] [CrossRef]
- Chalghoumi, R.; Marcq, C.; Thewis, A.; Portetelle, D.; Beckers, Y. Effects of feed supplementation with specific hen egg yolk antibody (immunoglobin Y) on Salmonella species cecal colonization and growth performances of challenged broiler chickens. Poult. Sci. 2009, 88, 2081–2092. [Google Scholar] [CrossRef]
- Beal, R.K.; Powers, C.; Wigley, P.; Barrow, P.A.; Smith, A.L. Temporal dynamics of the cellular, humoral and cytokine responses in chickens during primary and secondary infection with Salmonella enterica serovar Typhimurium. Avian. Pathol. 2004, 33, 25–33. [Google Scholar] [CrossRef] [PubMed]
- Beal, R.K.; Wigley, P.; Powers, C.; Hulme, S.D.; Barrow, P.A.; Smith, A.L. Age at primary infection with Salmonella enterica serovar Typhimurium in the chicken influences persistence of infection and subsequent immunity to re-challenge. Vet. Immunol. Immunopathol. 2004, 100, 151–164. [Google Scholar] [CrossRef] [PubMed]
- Belkaid, Y.; Hand, T.W. Role of the microbiota in immunity and inflammation. Cell 2014, 157, 121–141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spiljar, M.; Merkler, D.; Trajkovski, M. The Immune System Bridges the Gut Microbiota with Systemic Energy Homeostasis: Focus on TLRs, Mucosal Barrier, and SCFAs. Front. Immunol. 2017, 8, 1353. [Google Scholar] [CrossRef] [Green Version]
- Turnbaugh, P.J.; Ley, R.E.; Hamady, M.; Fraser-Liggett, C.M.; Knight, R.; Gordon, J.I. The human microbiome project. Nature 2007, 449, 804–810. [Google Scholar] [CrossRef] [Green Version]
- Ricke, S.C. Perspectives on the use of organic acids and short chain fatty acids as antimicrobials. Poult. Sci. 2003, 82, 632–639. [Google Scholar] [CrossRef]
- Wada, K.; Misaka, T.; Yokokawa, T.; Kimishima, Y.; Kaneshiro, T.; Oikawa, M.; Yoshihisa, A.; Takeishi, Y. Blood-Based Epigenetic Markers of FKBP5 Gene Methylation in Patients With Dilated Cardiomyopathy. J. Am. Heart Assoc. 2021, 10, e021101. [Google Scholar] [CrossRef]
- Zannas, A.S.; Jia, M.; Hafner, K.; Baumert, J.; Wiechmann, T.; Pape, J.C.; Arloth, J.; Ködel, M.; Martinelli, S.; Roitman, M.; et al. Epigenetic upregulation of FKBP5 by aging and stress contributes to NF-κB–driven inflammation and cardiovascular risk. PNAS 2019, 116, 11370–11379. [Google Scholar] [CrossRef] [Green Version]
- Grabacka, M.; Płonka, P.M.; Pierzchalska, M. The PPAR & alpha; Regulation of the Gut Physiology in Regard to Interaction with Microbiota, Intestinal Immunity, Metabolism, and Permeability. Int. J. Mol. Sci. 2022, 23, 14156. [Google Scholar]
- Reddy, A.T.; Lakshmi, S.P.; Reddy, R.C. PPARγ in Bacterial Infections: A Friend or Foe? PPAR Res. 2016, 2016, 7963540. [Google Scholar] [CrossRef] [Green Version]
- Kelly, D.; Campbell, J.I.; King, T.P.; Grant, G.; Jansson, E.A.; Coutts, A.G.; Pettersson, S.; Conway, S. Commensal anaerobic gut bacteria attenuate inflammation by regulating nuclear-cytoplasmic shuttling of PPAR-gamma and RelA. Nat. Immunol. 2004, 5, 104–112. [Google Scholar] [CrossRef] [PubMed]
- Manoharan, I.; Suryawanshi, A.; Hong, Y.; Ranganathan, P.; Shanmugam, A.; Ahmad, S.; Swafford, D.; Manicassamy, B.; Ramesh, G.; Koni, P.A.; et al. Homeostatic PPARα Signaling Limits Inflammatory Responses to Commensal Microbiota in the Intestine. J. Immunol. 2016, 196, 4739–4749. [Google Scholar] [CrossRef] [Green Version]
- Tsai, P.Y.; Zhang, B.; He, W.Q.; Zha, J.M.; Odenwald, M.A.; Singh, G.; Tamura, A.; Shen, L.; Sailer, A.; Yeruva, S.; et al. IL-22 Upregulates Epithelial Claudin-2 to Drive Diarrhea and Enteric Pathogen Clearance. Cell Host Microbe 2017, 21, 671–681.e674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, Y.; Valdez, P.A.; Danilenko, D.M.; Hu, Y.; Sa, S.M.; Gong, Q.; Abbas, A.R.; Modrusan, Z.; Ghilardi, N.; de Sauvage, F.J.; et al. Interleukin-22 mediates early host defense against attaching and effacing bacterial pathogens. Nat. Med. 2008, 14, 282–289. [Google Scholar] [CrossRef] [PubMed]
- Di Paola, M.; Bonechi, E.; Provensi, G.; Costa, A.; Clarke, G.; Ballerini, C.; De Filippo, C.; Passani, M.B. Oleoylethanolamide treatment affects gut microbiota composition and the expression of intestinal cytokines in Peyer’s patches of mice. Sci. Rep. 2018, 8, 14881. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Yu, C.; Liu, X.; Yang, J.; Feng, Y.; Wu, Y.; Xu, Y.; Zhu, Y.; Li, W. Fenofibrate Ameliorated Systemic and Retinal Inflammation and Modulated Gut Microbiota in High-Fat Diet-Induced Mice. Front. Cell. Infect. Microbiol. 2022, 12, 839592. [Google Scholar] [CrossRef] [PubMed]
- Meijer, B.; Gearry, R.B.; Day, A.S. The role of S100A12 as a systemic marker of inflammation. Int. J. Inflam. 2012, 2012, 907078. [Google Scholar] [CrossRef] [Green Version]
- Lira-Junior, R.; Holmström, S.B.; Clark, R.; Zwicker, S.; Majster, M.; Johannsen, G.; Axtelius, B.; Åkerman, S.; Svensson, M.; Klinge, B.; et al. S100A12 Expression Is Modulated During Monocyte Differentiation and Reflects Periodontitis Severity. Front. Immunol. 2020, 11. [Google Scholar] [CrossRef]
- Yang, Z.; Tao, T.; Raftery, M.J.; Youssef, P.; Di Girolamo, N.; Geczy, C.L. Proinflammatory properties of the human S100 protein S100A12. J. Leukoc. Biol. 2001, 69, 986–994. [Google Scholar] [CrossRef]
- Hasegawa, T.; Kosaki, A.; Kimura, T.; Matsubara, H.; Mori, Y.; Okigaki, M.; Masaki, H.; Toyoda, N.; Inoue-Shibata, M.; Kimura, Y.; et al. The regulation of EN-RAGE (S100A12) gene expression in human THP-1 macrophages. Atherosclerosis 2003, 171, 211–218. [Google Scholar] [CrossRef]
- Realegeno, S.; Kelly-Scumpia, K.M.; Dang, A.T.; Lu, J.; Teles, R.; Liu, P.T.; Schenk, M.; Lee, E.Y.; Schmidt, N.W.; Wong, G.C.; et al. S100A12 Is Part of the Antimicrobial Network against Mycobacterium leprae in Human Macrophages. PLoS Pathog. 2016, 12, e1005705. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brightbill, H.D.; Libraty, D.H.; Krutzik, S.R.; Yang, R.B.; Belisle, J.T.; Bleharski, J.R.; Maitland, M.; Norgard, M.V.; Plevy, S.E.; Smale, S.T.; et al. Host defense mechanisms triggered by microbial lipoproteins through toll-like receptors. Science 1999, 285, 732–736. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thoma-Uszynski, S.; Kiertscher, S.M.; Ochoa, M.T.; Bouis, D.A.; Norgard, M.V.; Miyake, K.; Godowski, P.J.; Roth, M.D.; Modlin, R.L. Activation of toll-like receptor 2 on human dendritic cells triggers induction of IL-12, but not IL-10. J. Immunol. 2000, 165, 3804–3810. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kommadath, A.; Bao, H.; Arantes, A.S.; Plastow, G.S.; Tuggle, C.K.; Bearson, S.M.; Guan le, L.; Stothard, P. Gene co-expression network analysis identifies porcine genes associated with variation in Salmonella shedding. BMC Genomics 2014, 15, 452. [Google Scholar] [CrossRef] [Green Version]
- Iwasaki, Y.; Takeshima, Y.; Fujio, K. Basic mechanism of immune system activation by mitochondria. Immunol. Med. 2020, 43, 142–147. [Google Scholar] [CrossRef]
- Kelly, B.; O’Neill, L.A. Metabolic reprogramming in macrophages and dendritic cells in innate immunity. Cell Res. 2015, 25, 771–784. [Google Scholar] [CrossRef] [Green Version]
- Park, S.; Juliana, C.; Hong, S.; Datta, P.; Hwang, I.; Fernandes-Alnemri, T.; Yu, J.W.; Alnemri, E.S. The mitochondrial antiviral protein MAVS associates with NLRP3 and regulates its inflammasome activity. J. Immunol. 2013, 191, 4358–4366. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Jia, A.; Wang, Y.; Dong, L.; Wang, Y.; He, Y.; Wang, S.; Cao, Y.; Yang, H.; Bi, Y.; et al. Immune effects of glycolysis or oxidative phosphorylation metabolic pathway in protecting against bacterial infection. J. Cell. Physiol. 2019, 234, 20298–20309. [Google Scholar] [CrossRef]
- Wang, Y.; McLean, A.S. The Role of Mitochondria in the Immune Response in Critical Illness. Critical. Care 2022, 26, 80. [Google Scholar] [CrossRef]
- Hu, G.; Liu, L.; Miao, X.; Zhao, Y.; Peng, Y.; Liu, L.; Li, X. The response of cecal microbiota to inflammatory state induced by Salmonella enterica serovar Enteritidis. Front. Microbiol. 2022, 13. [Google Scholar] [CrossRef]
- Litvak, Y.; Mon, K.K.Z.; Nguyen, H.; Chanthavixay, G.; Liou, M.; Velazquez, E.M.; Kutter, L.; Alcantara, M.A.; Byndloss, M.X.; Tiffany, C.R.; et al. Commensal Enterobacteriaceae Protect against Salmonella Colonization through Oxygen Competition. Cell Host Microbe 2019, 25, 128–139.e125. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cazals, A.; Rau, A.; Estellé, J.; Bruneau, N.; Coville, J.L.; Menanteau, P.; Rossignol, M.N.; Jardet, D.; Bevilacqua, C.; Bed’Hom, B.; et al. Comparative analysis of the caecal tonsil transcriptome in two chicken lines experimentally infected with Salmonella Enteritidis. PLoS ONE 2022, 17, e0270012. [Google Scholar] [CrossRef] [PubMed]
- Dokoshi, T.; Zhang, L.J.; Li, F.; Nakatsuji, T.; Butcher, A.; Yoshida, H.; Shimoda, M.; Okada, Y.; Gallo, R.L. Hyaluronan Degradation by Cemip Regulates Host Defense against Staphylococcus aureus Skin Infection. Cell Rep. 2020, 30, 61–68.e64. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, P.; Xia, P.; Wen, J.; Zheng, M.; Chen, J.; Zhao, J.; Jiang, R.; Liu, R.; Zhao, G. Up-regulation of the MyD88-dependent pathway of TLR signaling in spleen and caecum of young chickens infected with Salmonella serovar Pullorum. Vet. Microbiol. 2010, 143, 346–351. [Google Scholar] [CrossRef]
- Fidan, E.D.; Nazlıgül, A.; Türkyılmaz, M.K.; Aypak, S.Ü.; Kilimci, F.S.; Karaarslan, S.; Kaya, M. Effect of photoperiod length and light intensity on some welfare criteria, carcass, and meat quality characteristics in broilers. Rev. Bras. De Zootec. 2017, 46, 202–210. [Google Scholar] [CrossRef] [Green Version]
- Tan, Z.; Luo, L.; Wang, X.; Wen, Q.; Zhou, L.; Wu, K. Characterization of the cecal microbiome composition of Wenchang chickens before and after fattening. PLoS ONE 2019, 14, e0225692. [Google Scholar] [CrossRef] [Green Version]
- Andrews, S. FastQC: A quality control tool for high throughput sequence data. In Babraham Bioinformatics; Babraham Institute: Cambridge, UK, 2010. [Google Scholar]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [Green Version]
- Benjamini, Y.; Drai, D.; Elmer, G.; Kafkafi, N.; Golani, I. Controlling the false discovery rate in behavior genetics research. Behav. Brain Res. 2001, 125, 279–284. [Google Scholar] [CrossRef] [Green Version]
- Conesa, A.; Nueda, M.J.; Ferrer, A.; Talón, M. maSigPro: A method to identify significantly differential expression profiles in time-course microarray experiments. Bioinformatics 2006, 22, 1096–1102. [Google Scholar] [CrossRef] [Green Version]
- Nueda, M.J.; Tarazona, S.; Conesa, A. Next maSigPro: Updating maSigPro bioconductor package for RNA-seq time series. Bioinformatics 2014, 30, 2598–2602. [Google Scholar] [CrossRef] [Green Version]
- Yu, G.; Wang, L.G.; Han, Y.; He, Q.Y. clusterProfiler: An R package for comparing biological themes among gene clusters. Omics 2012, 16, 284–287. [Google Scholar] [CrossRef] [PubMed]
- Carlson, M. Genome Wide Annotation for Chicken org.Gg.eg.db R Version 3.8.2. 2019. Available online: https://bioconductor.org/packages/release/data/annotation/html/org.Gg.eg.db.html (accessed on 15 January 2023).
- Langfelder, P.; Horvath, S. WGCNA: An R package for weighted correlation network analysis. BMC Bioinform. 2008, 9, 559. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barreto Sánchez, A.L.; Wang, Q.; Thiam, M.; Wang, Z.; Zhang, J.; Zhang, Q.; Zhang, N.; Li, Q.; Wen, J.; Zhao, G. Liver Transcriptome Response to Heat Stress in Beijing You Chickens and Guang Ming Broilers. Genes (Basel) 2022, 13, 416. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Saelao, P.; Kern, C.; Jin, S.; Gallardo, R.A.; Kelly, T.; Dekkers, J.M.; Lamont, S.J.; Zhou, H. Liver Transcriptome Responses to Heat Stress and Newcastle Disease Virus Infection in Genetically Distinct Chicken Inbred Lines. Genes 2020, 11, 1067. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Kaiser, M.G.; Deist, M.S.; Gallardo, R.A.; Bunn, D.A.; Kelly, T.R.; Dekkers, J.C.M.; Zhou, H.; Lamont, S.J. Transcriptome Analysis in Spleen Reveals Differential Regulation of Response to Newcastle Disease Virus in Two Chicken Lines. Sci. Rep. 2018, 8, 1278. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Horvath, S.; Dong, J. Geometric Interpretation of Gene Coexpression Network Analysis. PLoS Comput. Biol. 2008, 4, e1000117. [Google Scholar] [CrossRef] [PubMed]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef]
- Xing, S.; Liu, R.; Zhao, G.; Groenen, M.A.M.; Madsen, O.; Liu, L.; Zheng, M.; Wang, Q.; Wu, Z.; Crooijmans, R.P.M.A.; et al. Time Course Transcriptomic Study Reveals the Gene Regulation During Liver Development and the Correlation With Abdominal Fat Weight in Chicken. Front. Genet. 2021, 12, 723519. [Google Scholar] [CrossRef]
- Xing, S.; Liu, R.; Zhao, G.; Liu, L.; Groenen, M.A.M.; Madsen, O.; Zheng, M.; Yang, X.; Crooijmans, R.P.M.A.; Wen, J. RNA-Seq Analysis Reveals Hub Genes Involved in Chicken Intramuscular Fat and Abdominal Fat Deposition During Development. Front. Genet. 2020, 11, 1009. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- Bardou, P.; Mariette, J.; Escudié, F.; Djemiel, C.; Klopp, C. jvenn: An interactive Venn diagram viewer. BMC Bioinform. 2014, 15, 293. [Google Scholar] [CrossRef] [PubMed] [Green Version]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Groups | Ensemble ID | Gene Symbol | Log2FC | pval | padj. |
|---|---|---|---|---|---|
| H7 vs. L7 | ENSGALG00000043754 | GLUL | −1.15 | 2.24E−06 | 0.0171 |
| ENSGALG00000038540 | ZBTB16 | −1.88 | 1.46E−05 | 0.0346 | |
| ENSGALG00000041202 | FBXO32 | −1.19 | 1.32E−05 | 0.0346 | |
| ENSGALG00000042148 | FKBP5 | −1.87 | 1.89E−05 | 0.0359 | |
| ENSGALG00000009700 | PDK4 | −1.34 | 3.55E−05 | 0.0490 | |
| ENSGALG00000016448 | KCNF1 | −1.04 | 3.27E−05 | 0.0490 | |
| ENSGALG00000044940 | GCNT3 | 1.01 | 0.0002 | 0.1347 | |
| H21 vs. L21 | ENSGALG00000029006 | SYPL1 | 1.55 | 9.26E−06 | 0.1911 |
| ENSGALG00000041921 | CWH43 | 1.40 | 5.39E−05 | 0.5567 | |
| ENSGALG00000002605 | MRPL17 | 1.04 | 0.0008 | 0.9946 | |
| ENSGALG00000020827 | ATP13A4 | 4.17 | 0.0015 | 0.9946 | |
| ENSGALG00000030502 | ADAMTS4 | −1.65 | 0.0018 | 0.9946 | |
| ENSGALG00000009919 | KCNG3 | 1.38 | 0.0018 | 0.9946 | |
| ENSGALG00000006413 | CEMIP | −1.27 | 0.0018 | 0.9946 | |
| ENSGALG00000036659 | NDUFAF8 | 1.27 | 0.0025 | 0.9946 | |
| H7 vs. H21 | ENSGALG00000026098 | IL8 | 2.46 | 2.19E−19 | 1.67E−15 |
| ENSGALG00000003876 | TIMD4 | −1.67 | 3.00E−17 | 1.44E−13 | |
| ENSGALG00000015307 | - | −1.32 | 2.22E−15 | 6.78E−12 | |
| ENSGALG00000027786 | SOCS3 | 1.99 | 2.90E−15 | 7.38E−12 | |
| ENSGALG00000016186 | PDE9A | 1.59 | 6.76E−14 | 1.47E−10 | |
| ENSGALG00000015733 | MDN1 | −1.01 | 7.97E−14 | 1.52E−10 | |
| ENSGALG00000001252 | CREB3L3 | 3.45 | 9.26E−13 | 1.57E−09 | |
| ENSGALG00000008518 | - | 1.13 | 1.79E−12 | 2.49E−09 | |
| ENSGALG00000010961 | IGF2BP3 | 1.18 | 1.79E−12 | 2.49E−09 | |
| ENSGALG00000005521 | PER2 | −1.08 | 2.13E−12 | 2.72E−09 | |
| L7 vs. L21 | ENSGALG00000043064 | EXFABP | 4.23 | 7.98E−16 | 1.21E−11 |
| ENSGALG00000004228 | USP40 | −1.62 | 9.62E−15 | 6.00E−11 | |
| ENSGALG00000025945 | AVD | 5.60 | 1.19E−14 | 6.00E−11 | |
| ENSGALG00000033807 | TYSND1 | 1.19 | 6.23E−12 | 1.05E−08 | |
| ENSGALG00000002722 | MST1 | 2.03 | 1.30E−11 | 1.64E−08 | |
| ENSGALG00000053916 | ABCB1 | −1.22 | 2.74E−10 | 2.18E−07 | |
| ENSGALG00000033204 | PQLC2 | 1.00 | 5.27E−10 | 3.99E−07 | |
| ENSGALG00000024272 | S100A9 | 5.69 | 8.29E−10 | 5.71E−07 | |
| ENSGALG00000041403 | DENND4A | −1.02 | 1.82E−09 | 1.20E−06 |
| Trait | Correlation * | Module Color | Gene Names (GS, MM) |
|---|---|---|---|
| Dpi | Positive | Blue | PER2 (0.95, 0.86), ROCK2 (0.94, 0.84), USP8 (0.94, 0.83), GOLGA4 (0.93, 0.87), CDC42SE1 (−0.93, −0.86), ENSGALG00000029691 (−0.93, −0.80), PIGC (−0.91, −0.83), CKAP2L (−0.91, −0.78), H3F3B (−0.91, −0.79), C8H1orf52 (−0.90, −0.77) |
| Brown | CGNL1 (0.87, 0.77), TLN2 (0.85, 0.79), HRH1 (0.85, 0.77), ECM2 (0.85, 0.87), TMEM245 (0.84, 0.66), NFU1 (−0.91, −0.70), EMC6 (−0.88, −0.70), ENSGALG00000021686 (−0.81, −0.84), GNG5 (−0.81, −0.81), JAGN1 (−0.80, −0.72) | ||
| Negative | Yellow | FAM168B (0.89, −0.83), C2CD5 (0.88, −0.86), RAF1 (0.87, −0.78), HESX1 (0.83, −0.70), LYPLA1 (0.80, −0.83), DDTL (0.79, −0.74), ENSGALG00000053041 (−0.91, 0.87), ENSGALG00000043126 (−0.90, 0.82), MARCKSL1 (−0.89, 0.83), TMEM115 (−0.89, 0.84) | |
| H/L ratio | Positive | Dark-green | ENSGALG00000038918 (0.62, 0.66), DNAJC17 (0.65, 0.52) |
| Body weight post-infection | Positive | Blue | GOLGA4 (0.92, 0.87), ENSGALG00000048303 (0.91, 0.74), ROCK2 (0.90, 0.84), USP8 (0.90, 0.83), PER2 (0.90, 0.86), ORMDL2 (−0.92, −0.86), CDC42SE1 (−0.92, −0.86), UBALD1 (−0.91, −0.91), ENSGALG00000029691 (−0.90, −0.80), PIGC (−0.90, −0.83) |
| Negative | Yellow | C2CD5 (0.89, −0.86), RAF1 (0.87, −0.78), DDTL (0.84, −0.74), FAM168B (0.84, −0.83), PLA2G4F (0.82, −0.89), HESX1 (0.82, −0.70), ENSGALG00000053041 (−0.90, 0.87), TMEM115 (−0.89, 0.84), UBE2M (−0.89, 0.90), ENSGALG00000052202 (−0.89, 0.91) | |
| Bacterial load (SE) | Positive | Dark-turquoise | SOWAHA (0.73, 0.70), ENSGALG00000007596 (0.73, 0.67), ENSGALG00000040718 (0.72, 0.69), STBD1 (0.71, 0.75), HMX3 (0.70, 0.57), ENSGALG00000050785 (0.58, 0.68), ENSGALG00000050054 (0.58, 0.80), RASSF10 (0.59, 0.70), ENSGALG00000049917 (0.59, 0.92), LYPD6 (0.60, 0.74) |
| Magenta | CREB3L3 (0.82, 0.69), ENSGALG00000045581 (0.81, 0.74), ENSGALG00000029381 (0.80, 0.78), WFDC2 (0.80, 0.73), ENSGALG00000007645 (0.78, 0.84), GXYLT2 (−0.63, −0.74), GTF2E2 (−0.59, −0.72), EEF1A2 (0.58, 0.83), RNF128 (0.58, 0.93), MLN (0.58, 0.97) | ||
| Negative | Blue | TMEM243 (0.89, −0.74), ENSGALG00000049966 (0.87, −0.83), FOXA1 (0.87, −0.80), OTP (0.86, −0.60), SDF2 (0.85, −0.93), PI4KA (−0.86, 0.87), INPP5K (−0.83, 0.87), ENSGALG00000004881 (−0.83, 0.91), TUBGCP4 (−0.83, 0.79), ENSGALG00000053997 (−0.83, 0.92) | |
| Pink | FUCA1 (0.86, −0.69), ENSGALG00000051392 (0.78, −0.69), ENSGALG00000047357 (0.67, −0.77), ENSA (0.65, −0.86), IRF2 (0.65, −0.78), NABP1 (−0.77, 0.80), NVL (−0.76, 0.79), LARS1 (−0.75, 0.86), NRDC (−0.74, 0.89), MKLN1 (−0.73, 0.84) | ||
| Brown | ENSGALG00000047515 (0.75, −0.66), EFNB1 (0.75, −0.76), ENSGALG00000048205 (0.73, −0.74), ENSGALG00000021686 (0.73, −0.84), ENSGALG00000048159 (0.72, −0.69), NFU1 (0.71, −0.70), EMC6 (0.69, −0.70), PCMT1 (−0.81, 0.77), TMEM245 (−0.77, 0.66), CD109 (−0.73, 0.83) | ||
| Propionate | Positive | Blue | PER2 (0.86, 0.86), USP8 (0.86, 0.83), SLC25A1 (0.86, 0.71), NEK1 (0.85, 0.88), TIMD4 (0.85, 0.84), C8H1orf52 (−0.79, −0.77), PIGC (−0.79, −0.83), CKAP2L (−0.79, −0.78), H3F3B (−0.79, −0.79), ENSGALG00000029691 (−0.79, −0.80) |
| Tan | HECW1 (0.81, 0.74), LGI2 (0.77, 0.70), ENSGALG00000035854 (0.75, 0.85), SLC6A15 (0.74, 0.56), TACSTD2 (0.72, 0.69), ENSGALG00000054224 (0.72, 0.80), ENSGALG00000044674 (0.58, 0.96), ENSGALG00000013762 (0.58, 0.80), CNDP1 (0.58, 0.77), NTNG1 (0.59, 0.76) | ||
| Brown | TLN2 (0.87, 0.79), ECM2 (0.86, 0.87), HRH1 (0.81, 0.77), CGNL1 (0.81, 0.77), ENSGALG00000048205 (−0.79, −0.74), EMC6 (−0.78, −0.70), ENSGALG00000021686 (−0.78, −0.84), NFU1 (−0.76, −0.70), JAGN1 (−0.71, −0.72), GNG5 (−0.71, −0.81) | ||
| Negative | Yellow | C2CD5 (0.80, −0.86), HESX1 (0.79, −0.70), ENSGALG00000017139 (0.78, −0.80), FAM168B (0.78, −0.83), SASH1 (0.78, −0.61), ENSGALG00000004725 (0.75, −0.75), RAF1 (0.75, −0.78), CYP2U1 (0.74, −0.75), ENSGALG00000043126 (−0.82, 0.82), ENSGALG00000053041 (−0.81, 0.87) | |
| Valerate | Positive | Blue | TIMD4 (0.91, 0.84), NEK1 (091, 0.88), USP8 (0.86, 0.83), ROCK2 (0.85, 0.84), ORMDL2 (−0.88, −0.86), CKAP2L (−0.87, −0.78), C8H1orf52 (−0.86, −0.77), PIGC (−0.85, −0.83), CDC42SE1 (−0.83, −0.86), H3F3B (−0.82, −0.79) |
| Tan | ENSGALG00000035854 (0.74, 0.85), NT5E (0.72, 0.80), LGI2 (0.71, 0.70), ENSGALG00000052840 (0.70, 0.65), CCDC148 (0.69, 0.61), SLC16A12 (0.67, 0.67), ASIP (0.58, 0.85), ITGA11 (0.58, 0.63), KCNV1 (0.59, 0.53), ENSGALG00000054224 (0.59, 0.80) | ||
| Brown | CGNL1 (0.90, 0.77), TLN2 (0.87, 0.79), HRH1 (0.85, 0.77), ECM2 (0.80, 0.87), ENSGALG00000021686 (−0.80, −0.84), NFU1 (−0.79, −0.70), EMC6 (−0.76, −0.70), ENSGALG00000048205 (−0.74, −0.74), GNG5 (−0.72, −0.81), JAGN1 (−0.68, −0.72) | ||
| Negative | Yellow | C2CD5 (0.82, −0.86), HESX1 (0.78, −0.70), FAM168B (0.77, −0.83), RAF1 (0.75, −0.78), DDTL (0.75, −0.74), CYP2U1 (0.73, −0.75), ENSGALG00000053041 (−0.84, 0.87), ENSGALG00000043126 (−0.84, 0.82), UBE2M (−0.83, 0.90), TMEM115 (−0.81, 0.84) | |
| Firmicutes | Positive | Gray | CERS5 (0.56, 0.59), FKBP3 (−0.53, −0.76), HPF1 (−0.56, −0.63) |
| Negative | Blue | CALML4 (0.77, −0.57), H-RAS (0.77, −0.83), ENSGALG00000046988 (0.74, −0.59), ACSL5 (0.73, −0.61), ENSGALG00000004503 (0.71, −0.69), CNGA3 (−0.77, 0.65), ENSGALG00000001972 (−0.75, 0.80), DOCK7 (−0.75, 0.61), DENND5B (−0.73, 0.63), ADCY2 (−0.73, 0.71) | |
| Pink | EHHADH (0.78, −0.73), AADAC (0.67, −0.74), FAM81A (0.66, −0.58), ZDHHC9 (0.65, −0.80), IRF2 (0.64, −0.78), GPR176 (−0.72, 0.53), GAR1 (−0.72, 0.72), URI1 (−0.70, 0.82), CXCR4 (−0.69, 0.73), SRSF2 (−0.69, 0.72) | ||
| Bacteroidetes | Positive | Blue | SLC25A16 (0.86, 0.71), BAG3 (0.88, 0.63), ENSGALG00000048303 (0.85, 0.74), MYBL1 (0.85, 0.69), CDC27 (0.84, 0.78), ORMDL2 (−0.83, −0.86), H-RAS (−0.83, −0.83), GPX1 (−0.80, −0.74), PIGC (−0.80, −0.83), NHEJ1 (−0.79, −0.82) |
| Pink | ENSGALG00000045350 (0.76, 0.74), DNAJA2 (0.70, 0.73), FAM98A (0.70, 0.80), NEIL3 (0.70, 0.74), MKLN1 (0.69, 0.84), FUCA1 (−0.60, −0.69), ENSA (−0.58, −0.86), SDAD1 (0.58, 0.91), CCT6A (0.58, 0.87), SIKE1 (0.58, 0.82) | ||
| Tan | ENSGALG00000035854 (0.79, 0.85), MTURN (0.71, 0.78), RPP14 (0.69, 0.82), ENSGALG00000005938 (0.58, 0.87), PCDH10 (0.59, 0.62), FKBP1B (0.59, 0.62), PDCL2 (0.59, 0.63), SULT (0.60, 0.71), ENSGALG00000051072 (0.60, 0.67) | ||
| Negative | Yellow | RAF1 (0.76, −0.78), HESX1 (0.73, −0.70), IKBKB (0.73, −0.77), FAM168B (0.72, −0.83), LYPLA1 (0.70, −0.83), PLA2G4F (0.70, −0.89), ENSGALG00000052202 (−0.79, 0.91), MARCKSL1 (−0.77, 0.83), ZNF142 (−0.85, 0.84), XKR8 (−0.84, 0.79) | |
| Proteobacteria | Positive | Black | ENSGALG00000052317 (0.89, 0.73), AMIGO3 (0.87, 0.86), RFT1 (0.87, 0.74), ENSGALG00000035539 (0.87, 0.72), MEPE (0.86, 0.87), UQCRQ (−0.68, −0.52), ANAPC5 (−0.63, −0.58) |
| Green | WNT5B (0.85, 0.85), SUN2 (0.83, 0.80), PCNX2 (0.82, 0.91), ITGA2B (0.80, 0.80), OLFM4 (0.80, 0.80), UGT1A1 9−0.76, −0.68), SFRP1 (−0.74, −0.56), ACAA1 (−0.66, −0.55), COMTD1 (−0.65, −0.60), PDLIM1 (−0.61, −0.65) | ||
| Yellow | JPH3 (0.91, 0.81), CCDC130 (0.90, 0.82), TAF6 (0.85, 0.85), ATP13A2 (0.84, 0.86), C2CD4C (0.83, 0.76), EPB41L3 (−0.75, −0.86), OCIAD1 (−0.69, −0.65), TSKU (−0.69, −0.73), WBP2 (−0.69, −0.71), ANK3 (−0.67, −0.92) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, Q.; Thiam, M.; Barreto Sánchez, A.L.; Wang, Z.; Zhang, J.; Li, Q.; Wen, J.; Zhao, G. Gene Co-Expression Network Analysis Reveals the Hub Genes and Key Pathways Associated with Resistance to Salmonella Enteritidis Colonization in Chicken. Int. J. Mol. Sci. 2023, 24, 4824. https://doi.org/10.3390/ijms24054824
Wang Q, Thiam M, Barreto Sánchez AL, Wang Z, Zhang J, Li Q, Wen J, Zhao G. Gene Co-Expression Network Analysis Reveals the Hub Genes and Key Pathways Associated with Resistance to Salmonella Enteritidis Colonization in Chicken. International Journal of Molecular Sciences. 2023; 24(5):4824. https://doi.org/10.3390/ijms24054824
Chicago/Turabian StyleWang, Qiao, Mamadou Thiam, Astrid Lissette Barreto Sánchez, Zixuan Wang, Jin Zhang, Qinghe Li, Jie Wen, and Guiping Zhao. 2023. "Gene Co-Expression Network Analysis Reveals the Hub Genes and Key Pathways Associated with Resistance to Salmonella Enteritidis Colonization in Chicken" International Journal of Molecular Sciences 24, no. 5: 4824. https://doi.org/10.3390/ijms24054824