
Phosphorylated Peptide Derived from the Myosin Phosphatase Target Subunit Is a Novel Inhibitor of Protein Phosphatase-1
 , , , and
, , , and
Abstract
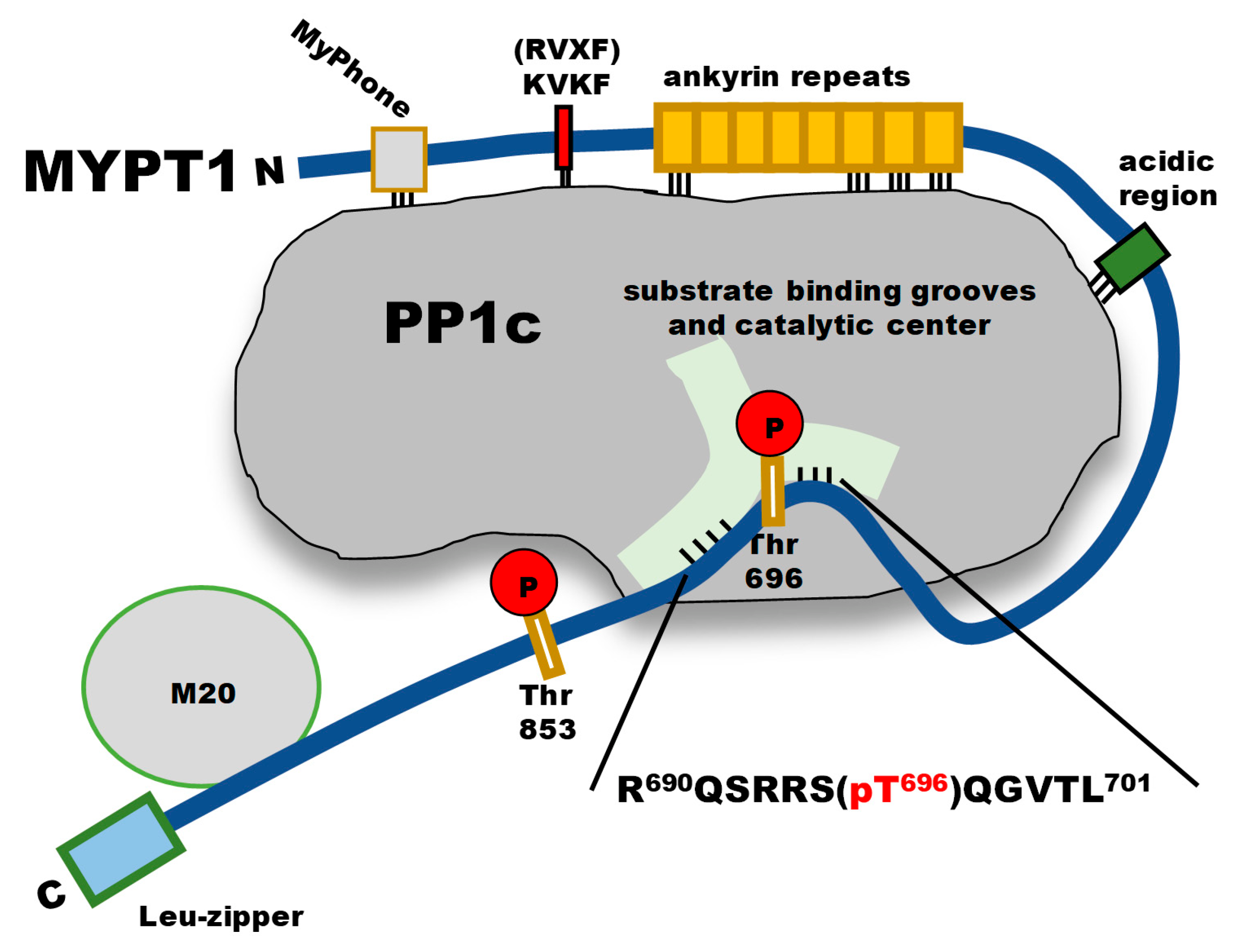
:1. Introduction
2. Results
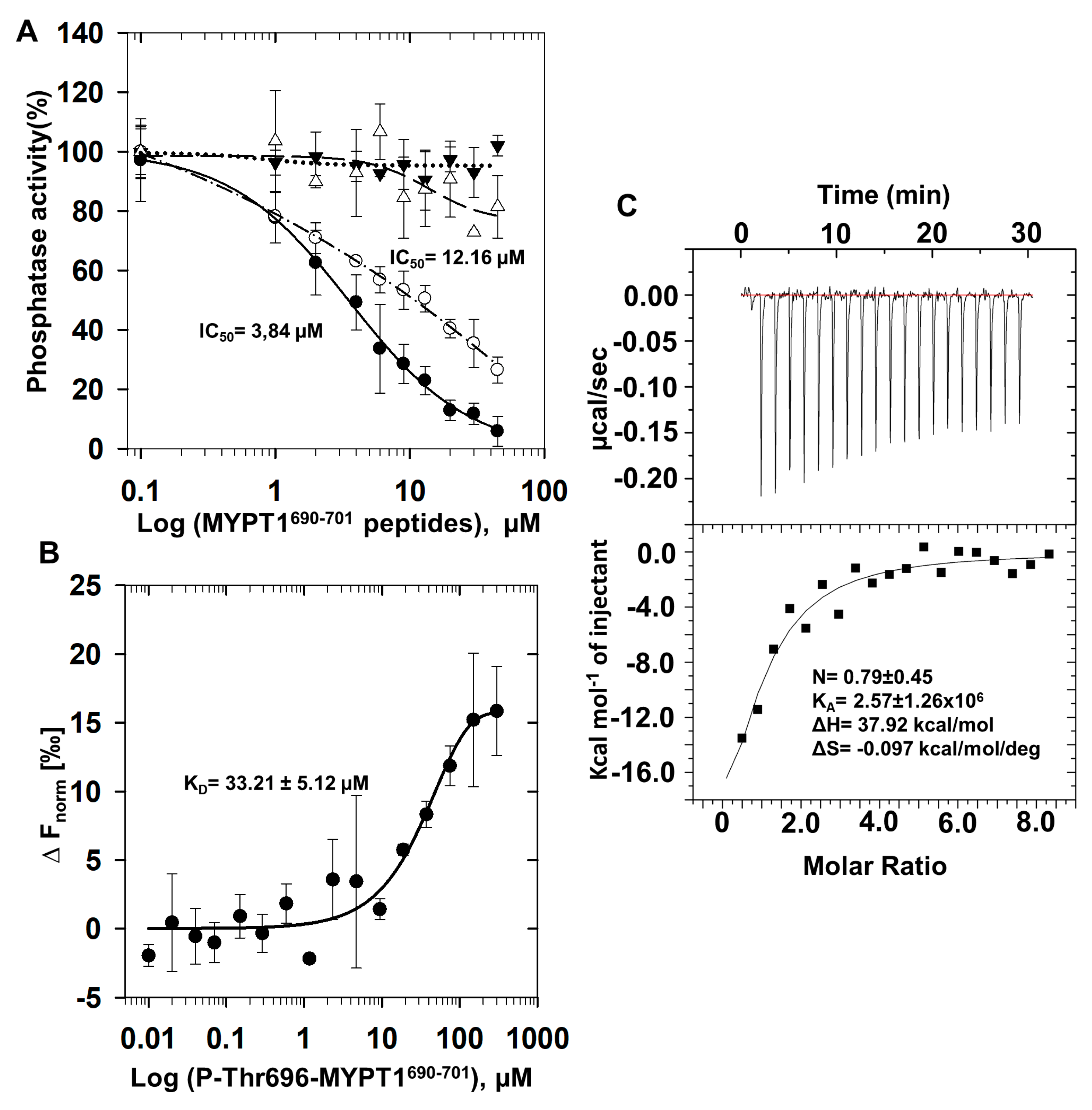
2.1. Inhibition and Interaction of PP1 with P-Thr696-MYPT1690−701 Peptide
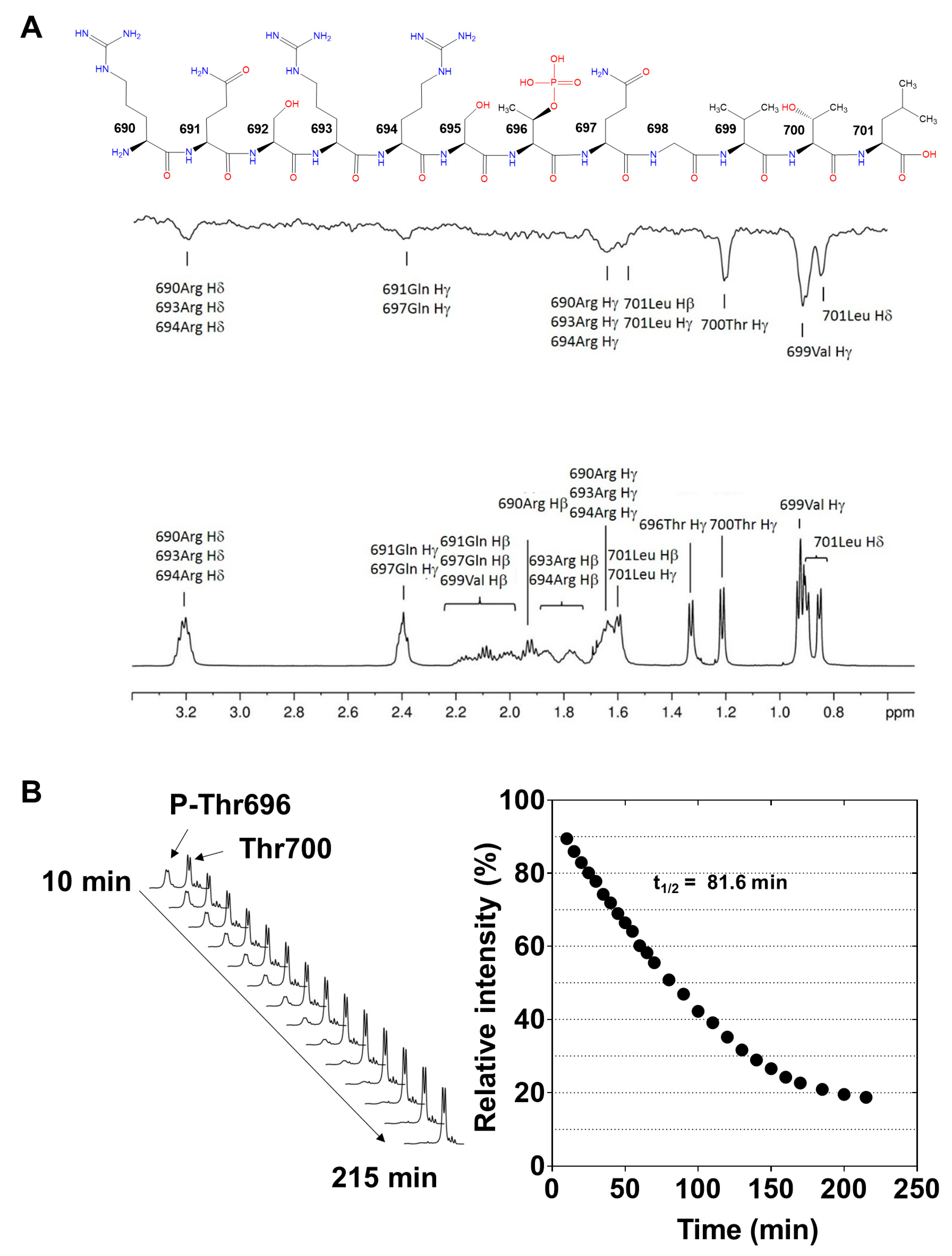
2.2. Interaction of P-Thr696-MYPT1690−701 with PP1c Revealed by Saturation Transfer Difference NMR Measurements
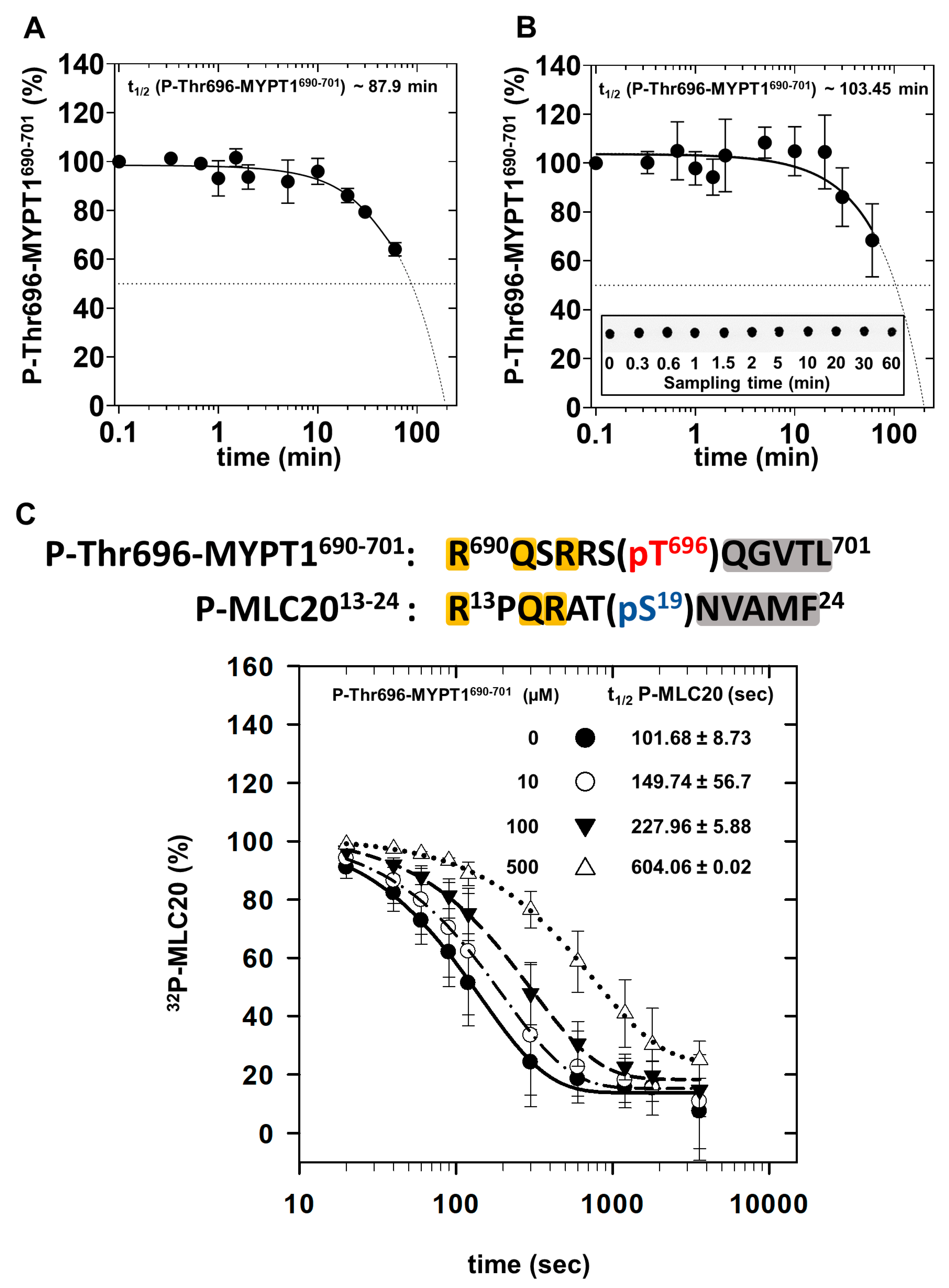
2.3. Dephosphorylation of P-Thr696-MYPT1690−701, 32P-MLC20 and Their Combination by PP1c
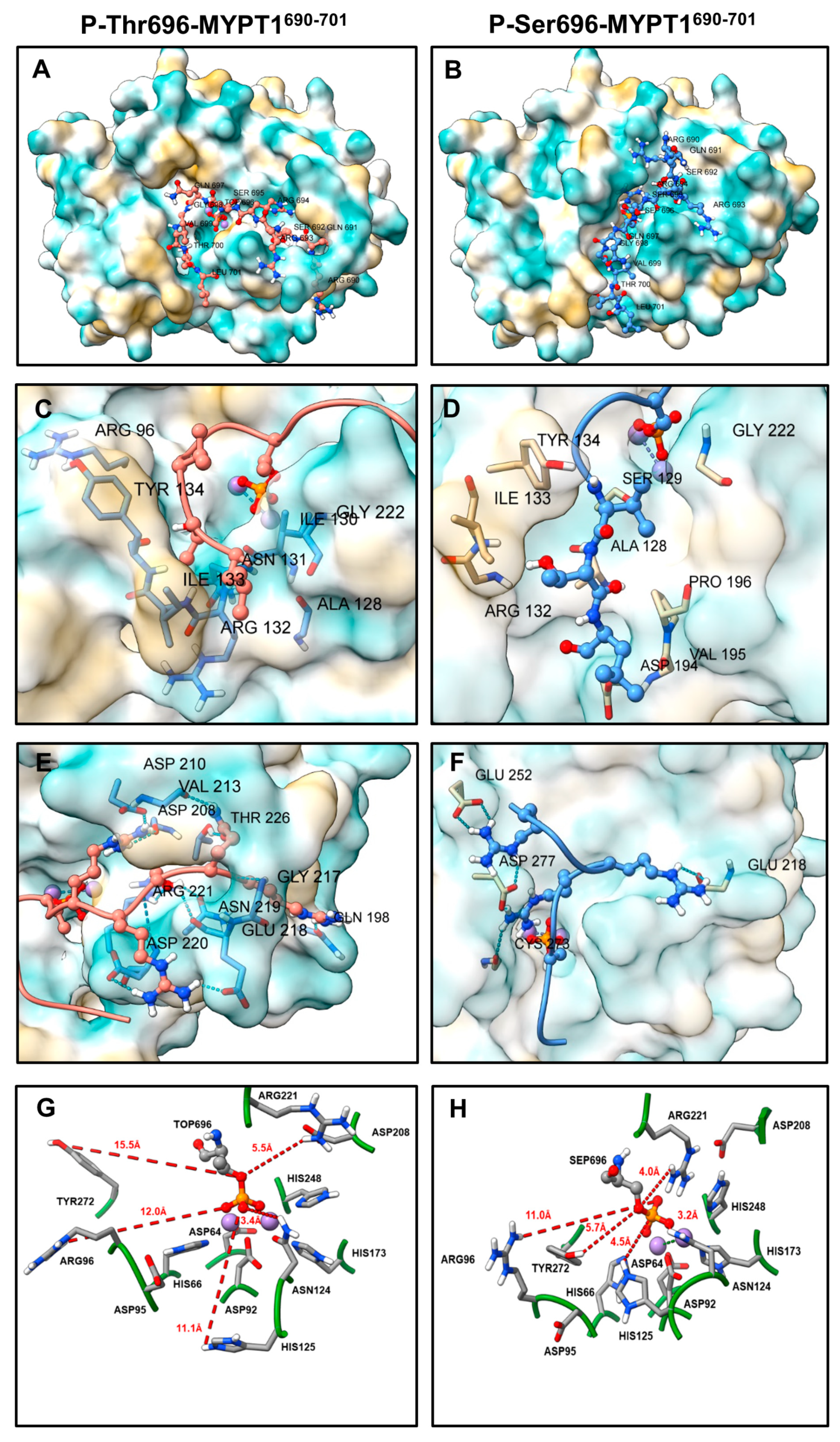
2.4. PP1c-Peptide Docking
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Proteins
4.3. Phosphatase Assays
4.4. Microscale Thermophoresis (MST) Experiments
4.5. Isothermal Titration Calorimetry (ITC)
4.6. Saturation Transfer Difference NMR
4.7. Malachite Green Assay
4.8. Dot Blot Assay
4.9. Peptide Docking
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sharma, K.; D’Souza, R.C.; Tyanova, S.; Schaab, C.; Wisniewski, J.R.; Cox, J.; Mann, M. Ultradeep human phosphoproteome reveals a distinct regulatory nature of Tyr and Ser/Thr-based signaling. Cell Rep. 2014, 8, 1583–1594. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brautigan, D.L.; Shenolikar, S. Protein Serine/Threonine Phosphatases: Keys to Unlocking Regulators and Substrates. Annu. Rev. Biochem. 2018, 87, 921–964. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chatterjee, J.; Kohn, M. Targeting the untargetable: Recent advances in the selective chemical modulation of protein phosphatase-1 activity. Curr. Opin. Chem. Biol. 2013, 17, 361–368. [Google Scholar] [CrossRef] [PubMed]
- Ceulemans, H.; Bollen, M. Functional diversity of protein phosphatase-1, a cellular economizer and reset button. Physiol. Rev. 2004, 84, 1–39. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cohen, P.T. Protein phosphatase 1—Targeted in many directions. J. Cell Sci. 2002, 115, 241–256. [Google Scholar] [CrossRef]
- Egloff, M.P.; Cohen, P.T.; Reinemer, P.; Barford, D. Crystal structure of the catalytic subunit of human protein phosphatase 1 and its complex with tungstate. J. Mol. Biol. 1995, 254, 942–959. [Google Scholar] [CrossRef]
- Goldberg, J.; Huang, H.B.; Kwon, Y.G.; Greengard, P.; Nairn, A.C.; Kuriyan, J. Three-dimensional structure of the catalytic subunit of protein serine/threonine phosphatase-1. Nature 1995, 376, 745–753. [Google Scholar] [CrossRef]
- Maynes, J.T.; Bateman, K.S.; Cherney, M.M.; Das, A.K.; Luu, H.A.; Holmes, C.F.; James, M.N. Crystal structure of the tumor-promoter okadaic acid bound to protein phosphatase-1. J. Biol. Chem. 2001, 276, 44078–44082. [Google Scholar] [CrossRef] [Green Version]
- Choy, M.S.; Swingle, M.; D’Arcy, B.; Abney, K.; Rusin, S.F.; Kettenbach, A.N.; Page, R.; Honkanen, R.E.; Peti, W. PP1:Tautomycetin Complex Reveals a Path toward the Development of PP1-Specific Inhibitors. J. Am. Chem. Soc. 2017, 139, 17703–17706. [Google Scholar] [CrossRef]
- Egloff, M.P.; Johnson, D.F.; Moorhead, G.; Cohen, P.T.; Cohen, P.; Barford, D. Structural basis for the recognition of regulatory subunits by the catalytic subunit of protein phosphatase 1. EMBO J. 1997, 16, 1876–1887. [Google Scholar] [CrossRef]
- Terrak, M.; Kerff, F.; Langsetmo, K.; Tao, T.; Dominguez, R. Structural basis of protein phosphatase 1 regulation. Nature 2004, 429, 780–784. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eto, M. Regulation of cellular protein phosphatase-1 (PP1) by phosphorylation of the CPI-17 family, C-kinase-activated PP1 inhibitors. J. Biol. Chem. 2009, 284, 35273–35277. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Filter, J.J.; Williams, B.C.; Eto, M.; Shalloway, D.; Goldberg, M.L. Unfair competition governs the interaction of pCPI-17 with myosin phosphatase (PP1-MYPT1). Elife 2017, 6, e24665. [Google Scholar] [CrossRef] [PubMed]
- Bollen, M. Combinatorial control of protein phosphatase-1. Trends Biochem. Sci. 2001, 26, 426–431. [Google Scholar] [CrossRef] [PubMed]
- Bollen, M.; Peti, W.; Ragusa, M.J.; Beullens, M. The extended PP1 toolkit: Designed to create specificity. Trends Biochem. Sci. 2010, 35, 450–458. [Google Scholar] [CrossRef] [Green Version]
- Kiss, A.; Lontay, B.; Bécsi, B.; Márkász, L.; Oláh, E.; Gergely, P.; Erdődi, F. Myosin phosphatase interacts with and dephosphorylates the retinoblastoma protein in THP-1 leukemic cells: Its inhibition is involved in the attenuation of daunorubicin-induced cell death by calyculin-A. Cell. Signal. 2008, 20, 2059–2070. [Google Scholar] [CrossRef] [PubMed]
- Tóth, E.; Erdődi, F.; Kiss, A. Activation of Myosin Phosphatase by Epigallocatechin-Gallate Sensitizes THP-1 Leukemic Cells to Daunorubicin. Anticancer Agents Med. Chem. 2021, 21, 1092–1098. [Google Scholar] [CrossRef] [PubMed]
- Kiss, A.; Bécsi, B.; Kolozsvári, B.; Komáromi, I.; Kövér, K.E.; Erdődi, F. Epigallocatechin-3-gallate and penta-O-galloyl-beta-D-glucose inhibit protein phosphatase-1. FEBS J. 2013, 280, 612–626. [Google Scholar] [CrossRef]
- Kónya, Z.; Bécsi, B.; Kiss, A.; Horváth, D.; Raics, M.; Kövér, K.E.; Lontay, B.; Erdődi, F. Inhibition of protein phosphatase-1 and -2A by ellagitannins: Structure-inhibitory potency relationships and influences on cellular systems. J. Enzym. Inhib. Med. Chem. 2019, 34, 500–509. [Google Scholar] [CrossRef] [Green Version]
- Kónya, Z.; Bécsi, B.; Kiss, A.; Tamás, I.; Lontay, B.; Szilágyi, L.; Kövér, K.E.; Erdődi, F. Aralkyl selenoglycosides and related selenosugars in acetylated form activate protein phosphatase-1 and -2A. Bioorg. Med. Chem. 2018, 26, 1875–1884. [Google Scholar] [CrossRef]
- Tappan, E.; Chamberlin, A.R. Activation of protein phosphatase 1 by a small molecule designed to bind to the enzyme’s regulatory site. Chem. Biol. 2008, 15, 167–174. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chatterjee, J.; Beullens, M.; Sukackaite, R.; Qian, J.; Lesage, B.; Hart, D.J.; Bollen, M.; Köhn, M. Development of a peptide that selectively activates protein phosphatase-1 in living cells. Angew. Chem. Int. Ed. Engl. 2012, 51, 10054–10059. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, H.B.; Horiuchi, A.; Watanabe, T.; Shih, S.R.; Tsay, H.J.; Li, H.C.; Greengard, P.; Nairn, A.C. Characterization of the inhibition of protein phosphatase-1 by DARPP-32 and inhibitor-2. J. Biol. Chem. 1999, 274, 7870–7878. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hartshorne, D.J.; Ito, M.; Erdődi, F. Role of protein phosphatase type 1 in contractile functions: Myosin phosphatase. J. Biol. Chem. 2004, 279, 37211–37214. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kiss, A.; Erdődi, F.; Lontay, B. Myosin phosphatase: Unexpected functions of a long-known enzyme. Biochim. Biophys. Acta Mol. Cell Res. 2019, 1866, 2–15. [Google Scholar] [CrossRef]
- Tóth, A.; Kiss, E.; Herberg, F.W.; Gergely, P.; Hartshorne, D.J.; Erdődi, F. Study of the subunit interactions in myosin phosphatase by surface plasmon resonance. Eur. J. Biochem. 2000, 267, 1687–1697. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tóth, A.; Kiss, E.; Gergely, P.; Walsh, M.P.; Hartshorne, D.J.; Erdődi, F. Phosphorylation of MYPT1 by protein kinase C attenuates interaction with PP1 catalytic subunit and the 20 kDa light chain of myosin. FEBS Lett. 2000, 484, 113–117. [Google Scholar] [CrossRef] [Green Version]
- Velasco, G.; Armstrong, C.; Morrice, N.; Frame, S.; Cohen, P. Phosphorylation of the regulatory subunit of smooth muscle protein phosphatase 1M at Thr850 induces its dissociation from myosin. FEBS Lett. 2002, 527, 101–104. [Google Scholar] [CrossRef]
- Murányi, A.; Derkach, D.; Erdődi, F.; Kiss, A.; Ito, M.; Hartshorne, D.J. Phosphorylation of Thr695 and Thr850 on the myosin phosphatase target subunit: Inhibitory effects and occurrence in A7r5 cells. FEBS Lett. 2005, 579, 6611–6615. [Google Scholar] [CrossRef] [Green Version]
- Murányi, A.; MacDonald, J.A.; Deng, J.T.; Wilson, D.P.; Haystead, T.A.; Walsh, M.P.; Erdődi, F.; Kiss, E.; Wu, Y.; Hartshorne, D.J. Phosphorylation of the myosin phosphatase target subunit by integrin-linked kinase. Biochem. J. 2002, 366, 211–216. [Google Scholar] [CrossRef] [Green Version]
- Khromov, A.; Choudhury, N.; Stevenson, A.S.; Somlyo, A.V.; Eto, M. Phosphorylation-dependent autoinhibition of myosin light chain phosphatase accounts for Ca2+ sensitization force of smooth muscle contraction. J. Biol. Chem. 2009, 284, 21569–21579. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bátori, R.; Bécsi, B.; Nagy, D.; Kónya, Z.; Hegedűs, C.; Bordán, Z.; Verin, A.; Lontay, B.; Erdődi, F. Interplay of myosin phosphatase and protein phosphatase-2A in the regulation of endothelial nitric-oxide synthase phosphorylation and nitric oxide production. Sci. Rep. 2017, 7, 44698. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mayer, M.; Meyer, B. Group epitope mapping by saturation transfer difference NMR to identify segments of a ligand in direct contact with a protein receptor. J. Am. Chem. Soc. 2001, 123, 6108–6117. [Google Scholar] [CrossRef] [PubMed]
- Groves, P.; Kövér, K.E.; André, S.; Bandorowicz-Pikula, J.; Batta, G.; Bruix, M.; Buchet, R.; Canales, A.; Canada, F.J.; Gabius, H.J.; et al. Temperature dependence of ligand-protein complex formation as reflected by saturation transfer difference NMR experiments. Magn. Reson. Chem. 2007, 45, 745–748. [Google Scholar] [CrossRef] [Green Version]
- Itaya, K.; Ui, M. A new micromethod for the colorimetric determination of inorganic phosphate. Clin. Chim. Acta 1966, 14, 361–366. [Google Scholar] [CrossRef]
- Veloria, J.; Shin, M.; Devkota, A.K.; Payne, S.M.; Cho, E.J.; Dalby, K.N. Developing Colorimetric and Luminescence-Based High-Throughput Screening Platforms for Monitoring the GTPase Activity of Ferrous Iron Transport Protein B (FeoB). SLAS Discov. 2019, 24, 597–605. [Google Scholar] [CrossRef]
- Van Zundert, G.C.P.; Rodrigues, J.; Trellet, M.; Schmitz, C.; Kastritis, P.L.; Karaca, E.; Melquiond, A.S.J.; van Dijk, M.; de Vries, S.J.; Bonvin, A. The HADDOCK2.2 Web Server: User-Friendly Integrative Modeling of Biomolecular Complexes. J. Mol. Biol. 2016, 428, 720–725. [Google Scholar] [CrossRef] [Green Version]
- Hoermann, B.; Kokot, T.; Helm, D.; Heinzlmeir, S.; Chojnacki, J.E.; Schubert, T.; Ludwig, C.; Berteotti, A.; Kurzawa, N.; Kuster, B.; et al. Dissecting the sequence determinants for dephosphorylation by the catalytic subunits of phosphatases PP1 and PP2A. Nat. Commun. 2020, 11, 3583. [Google Scholar] [CrossRef]
- Erdődi, F.; Tóth, B.; Hirano, K.; Hirano, M.; Hartshorne, D.J.; Gergely, P. Endothall thioanhydride inhibits protein phosphatases-1 and -2A in vivo. Am. J. Physiol. 1995, 269, C1176–C1184. [Google Scholar] [CrossRef]
- Hirschi, A.; Cecchini, M.; Steinhardt, R.C.; Schamber, M.R.; Dick, F.A.; Rubin, S.M. An overlapping kinase and phosphatase docking site regulates activity of the retinoblastoma protein. Nat. Struct. Mol. Biol. 2010, 17, 1051–1057. [Google Scholar] [CrossRef]
- Honorato, R.V.; Koukos, P.I.; Jimenez-Garcia, B.; Tsaregorodtsev, A.; Verlato, M.; Giachetti, A.; Rosato, A.; Bonvin, A. Structural Biology in the Clouds: The WeNMR-EOSC Ecosystem. Front. Mol. Biosci. 2021, 8, 729513. [Google Scholar] [CrossRef] [PubMed]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Meng, E.C.; Couch, G.S.; Croll, T.I.; Morris, J.H.; Ferrin, T.E. UCSF ChimeraX: Structure visualization for researchers, educators, and developers. Protein Sci. 2021, 30, 70–82. [Google Scholar] [CrossRef] [PubMed]
- Reissmann, S. Cell penetration: Scope and limitations by the application of cell-penetrating peptides. J. Pept. Sci. 2014, 20, 760–784. [Google Scholar] [CrossRef] [PubMed]
- Lucana, M.C.; Arruga, Y.; Petrachi, E.; Roig, A.; Lucchi, R.; Oller-Salvia, B. Protease-Resistant Peptides for Targeting and Intracellular Delivery of Therapeutics. Pharmaceutics 2021, 13, 2065. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Distance (Å) | ||||
|---|---|---|---|---|
| Metal Ions * | Coordinating Residues ** | PP1c Crystal | PP1c-P-Thr696-MYPT1690−701 | PP1c-P-Ser696-MYPT1690−701 |
| Mn12+ | Asp92 | 2.3 | 2.3 | 2.1 |
| Mn12+ | Asn124 | 2.1 | 1.9 | 2.0 |
| Mn12+ | His173 | 2.1 | 2.7 | 1.8 |
| Mn12+ | His248 | 2.3 | 2.4 | 2.9 |
| Mn22+ | Asp64 | 2.0 | 1.5 | 1.6 |
| Mn22+ | His66 | 2.3 | 2.4 | 3.0 |
| Mn22+ | Asp92 | 2.3 | 1.9 | 1.6 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kónya, Z.; Tamás, I.; Bécsi, B.; Lontay, B.; Raics, M.; Timári, I.; Kövér, K.E.; Erdődi, F. Phosphorylated Peptide Derived from the Myosin Phosphatase Target Subunit Is a Novel Inhibitor of Protein Phosphatase-1. Int. J. Mol. Sci. 2023, 24, 4789. https://doi.org/10.3390/ijms24054789
Kónya Z, Tamás I, Bécsi B, Lontay B, Raics M, Timári I, Kövér KE, Erdődi F. Phosphorylated Peptide Derived from the Myosin Phosphatase Target Subunit Is a Novel Inhibitor of Protein Phosphatase-1. International Journal of Molecular Sciences. 2023; 24(5):4789. https://doi.org/10.3390/ijms24054789
Chicago/Turabian StyleKónya, Zoltán, István Tamás, Bálint Bécsi, Beáta Lontay, Mária Raics, István Timári, Katalin E. Kövér, and Ferenc Erdődi. 2023. "Phosphorylated Peptide Derived from the Myosin Phosphatase Target Subunit Is a Novel Inhibitor of Protein Phosphatase-1" International Journal of Molecular Sciences 24, no. 5: 4789. https://doi.org/10.3390/ijms24054789