
Latest Knowledge on the Role of Vitamin D in Hypertension

Abstract
:1. Introduction
2. Methods
3. Results and Discussion
3.1. Arterial Hypertension
3.2. Forms of Hypertension
3.3. Health Risks and Mortality
3.4. Normal Treatment of Hypertension
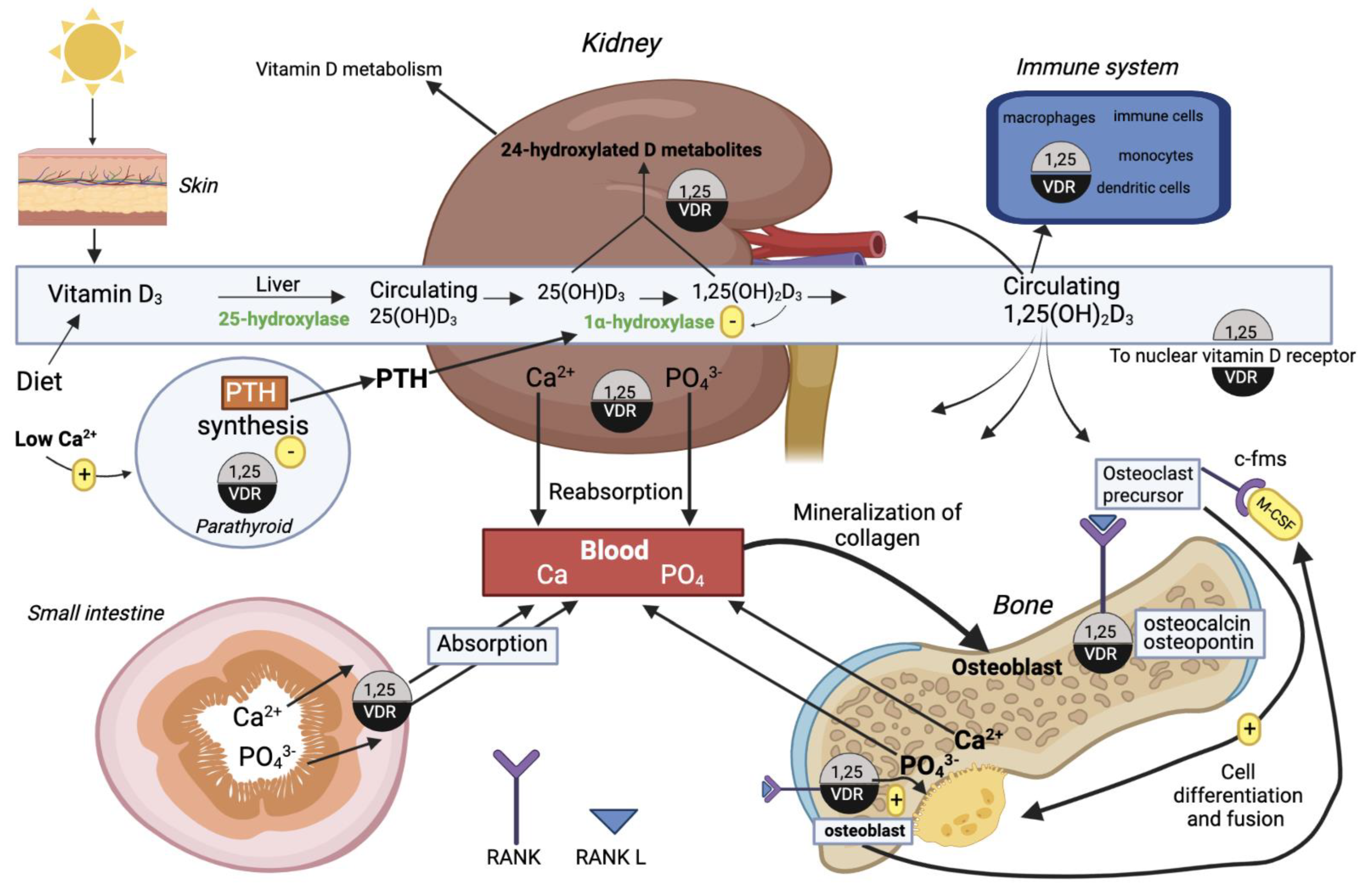
3.5. Vitamin D Metabolism
3.6. Genomic and Non-Genomic Response of the Vitamin D Receptor
3.7. Functions of Vitamin D
3.8. Vitamin D and Hypertension
3.9. Clinical Trials with Vitamin D and Hypertension
3.10. Discussion
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| 1,25(OH)2D3 | 1,25-dihydroxyvitamin D3 |
| 1,25-D-MARRS | 1,25D-membrane-associated rapid response steroid-binding protein |
| 7-DHC | 7-dehydrocholesterol |
| AB | Antibody |
| ABPM | Ambulatory Blood pressure measurement |
| ACE | Angiotensin converting enzyme |
| AE | Adverse effects |
| AH | Arterial hypertension |
| Ang I | Angiotensin I |
| Ang II | Angiotensin II |
| ARB | Angiotensin receptor blocker |
| AT1R | Angiotensin II type 1 receptor |
| B-AR | Beta-adrenergic receptor |
| BARB | Beta-adrenergic receptor blockers |
| C39 | C. elegans miR-39 |
| CCB | Calcium channel blocker |
| CTGF | Connective Tissue Growth Factor |
| CVD | Cardiovascular disease |
| DBP | Diastolic blood pressure |
| EMT | Epithelial to Mesenchymal Transition |
| FGF23 | Fibroblast growth factor |
| FMD | Flow mediated dilatation |
| HR | Heart rate |
| HT | Hypertension |
| ICAM1 | Intracellular adhesion molecule 1 |
| O-LDL | Oxidized LDL |
| Pi | Serum inorganic phosphorus |
| PKC | Protein kinase C |
| PRA | Plasma renin activity |
| PRC | Plasma renin concentration |
| Pre-D3 | Pre Vitamin D3 |
| PTH | Parathyroid hormone |
| RAAS | Renin-angiotensin-aldosterone-system |
| RXR | Retinoid X receptor |
| SBP | Systolic blood pressure |
| SH | Secondary hypertension |
| SNP | Single nucleotide polymorphism |
| T2DM | Type 2 diabetes |
| VD3 | Vitamin D3 |
| VDBP | Vitamin D-binding protein |
| VDR | Vitamin D receptor |
| VitD | Vitamin D |
References
- Legarth, C.; Grimm, D.; Krüger, M.; Infanger, M.; Wehland, M. Potential Beneficial Effects of Vitamin D in Coronary Artery Disease. Nutrients 2019, 12, 99. [Google Scholar] [CrossRef] [Green Version]
- Wimalawansa, S.J. Vitamin D and cardiovascular diseases: Causality. J. Steroid Biochem. Mol. Biol. 2018, 175, 29–43. [Google Scholar] [CrossRef]
- Rai, V.; Agrawal, D.K. Role of Vitamin D in Cardiovascular Diseases. Endocrinol. Metab. Clin. N. Am. 2017, 46, 1039–1059. [Google Scholar] [CrossRef]
- Delmi, M.; Rapin, C.H.; Bengoa, J.M.; Delmas, P.D.; Vasey, H.; Bonjour, J.P. Dietary supplementation in elderly patients with fractured neck of the femur. Lancet 1990, 335, 1013–1016. [Google Scholar] [CrossRef]
- Zhou, Y.; Zhao, L.; Meng, X.; Cai, Q.J.; Zhao, X.L.; Zhou, X.L.; Hu, A.H. Seasonal variation of ambulatory blood pressure in Chinese hypertensive adolescents. Front. Pediatr. 2022, 10, 1022865. [Google Scholar] [CrossRef]
- Kristal-Boneh, E.; Harari, G.; Green, M.S.; Ribak, J. Summer-winter variation in 24 h ambulatory blood pressure. Blood. Press. Monit. 1996, 1, 87–94. [Google Scholar]
- Hanazawa, T.; Asayama, K.; Watabe, D.; Hosaka, M.; Satoh, M.; Yasui, D.; Obara, T.; Inoue, R.; Metoki, H.; Kikuya, M.; et al. Seasonal variation in self-measured home blood pressure among patients on antihypertensive medications: HOMED-BP study. Hypertens. Res. 2017, 40, 284–290. [Google Scholar] [CrossRef]
- High Blood Pressure (Hypertension). Available online: https://www.mayoclinic.org/diseases-conditions/high-blood-pressure/symptoms-causes/syc-20373410 (accessed on 4 November 2022).
- Elliott, W.J. Systemic hypertension. Curr. Probl. Cardiol. 2007, 32, 201–259. [Google Scholar] [CrossRef]
- O’Shea, P.M.; Griffin, T.P.; Fitzgibbon, M. Hypertension: The role of biochemistry in the diagnosis and management. Clin. Chim. Acta 2017, 465, 131–143. [Google Scholar] [CrossRef]
- Williams, B. Resistant hypertension: An unmet treatment need. Lancet 2009, 374, 1396–1398. [Google Scholar] [CrossRef]
- Anstey, D.E.; Pugliese, D.; Abdalla, M.; Bello, N.A.; Givens, R.; Shimbo, D. An Update on Masked Hypertension. Curr. Hypertens. Rep. 2017, 19, 94. [Google Scholar] [CrossRef]
- Nuredini, G.; Saunders, A.; Rajkumar, C.; Okorie, M. Current status of white coat hypertension: Where are we? Ther. Adv. Cardiovasc. Dis. 2020, 14, 1753944720931637. [Google Scholar] [CrossRef]
- Oliveras, A.; de la Sierra, A. Resistant hypertension: Patient characteristics, risk factors, co-morbidities and outcomes. J. Hum. Hypertens. 2014, 28, 213–217. [Google Scholar] [CrossRef]
- Unger, T.; Borghi, C.; Charchar, F.; Khan, N.A.; Poulter, N.R.; Prabhakaran, D.; Ramirez, A.; Schlaich, M.; Stergiou, G.S.; Tomaszewski, M.; et al. 2020 International Society of Hypertension Global Hypertension Practice Guidelines. Hypertension 2020, 75, 1334–1357. [Google Scholar] [CrossRef]
- Ezzati, M.; Lopez, A.D.; Rodgers, A.; Vander Hoorn, S.; Murray, C.J. Selected major risk factors and global and regional burden of disease. Lancet 2002, 360, 1347–1360. [Google Scholar] [CrossRef]
- Kearney, P.M.; Whelton, M.; Reynolds, K.; Muntner, P.; Whelton, P.K.; He, J. Global burden of hypertension: Analysis of worldwide data. Lancet 2005, 365, 217–223. [Google Scholar] [CrossRef]
- Chow, C.K.; Teo, K.K.; Rangarajan, S.; Islam, S.; Gupta, R.; Avezum, A.; Bahonar, A.; Chifamba, J.; Dagenais, G.; Diaz, R.; et al. Prevalence, Awareness, Treatment, and Control of Hypertension in Rural and Urban Communities in High-, Middle-, and Low-Income Countries. JAMA 2013, 310, 959–968. [Google Scholar] [CrossRef] [Green Version]
- WHO. Hypertension—Key Facts. Available online: https://www.who.int/news-room/fact-sheets/detail/hypertension (accessed on 24 February 2023).
- Olsen, M.H.; Angell, S.Y.; Asma, S.; Boutouyrie, P.; Burger, D.; Chirinos, J.A.; Damasceno, A.; Delles, C.; Gimenez-Roqueplo, A.P.; Hering, D.; et al. A call to action and a lifecourse strategy to address the global burden of raised blood pressure on current and future generations: The Lancet Commission on hypertension. Lancet 2016, 388, 2665–2712. [Google Scholar] [CrossRef]
- Forouzanfar, M.H.; Liu, P.; Roth, G.A.; Ng, M.; Biryukov, S.; Marczak, L.; Alexander, L.; Estep, K.; Hassen Abate, K.; Akinyemiju, T.F.; et al. Global Burden of Hypertension and Systolic Blood Pressure of at Least 110 to 115 mm Hg, 1990–2015. JAMA 2017, 317, 165–182. [Google Scholar] [CrossRef] [Green Version]
- Williams, B.; Mancia, G.; Spiering, W.; Agabiti Rosei, E.; Azizi, M.; Burnier, M.; Clement, D.L.; Coca, A.; de Simone, G.; Dominiczak, A.; et al. 2018 ESC/ESH Guidelines for the management of arterial hypertension. Eur. Heart J. 2018, 39, 3021–3104. [Google Scholar] [CrossRef] [Green Version]
- Sacks, F.M.; Svetkey, L.P.; Vollmer, W.M.; Appel, L.J.; Bray, G.A.; Harsha, D.; Obarzanek, E.; Conlin, P.R.; Miller, E.R., 3rd; Simons-Morton, D.G.; et al. Effects on blood pressure of reduced dietary sodium and the Dietary Approaches to Stop Hypertension (DASH) diet. DASH-Sodium Collaborative Research Group. N. Engl. J. Med. 2001, 344, 3–10. [Google Scholar] [CrossRef] [PubMed]
- Faselis, C.; Doumas, M.; Kokkinos, J.P.; Panagiotakos, D.; Kheirbek, R.; Sheriff, H.M.; Hare, K.; Papademetriou, V.; Fletcher, R.; Kokkinos, P. Exercise Capacity and Progression From Prehypertension to Hypertension. Hypertension 2012, 60, 333–338. [Google Scholar] [CrossRef] [Green Version]
- Zanchetti, A.; Grassi, G.; Mancia, G. When should antihypertensive drug treatment be initiated and to what levels should systolic blood pressure be lowered? A critical reappraisal. J. Hypertens. 2009, 27, 923–934. [Google Scholar] [CrossRef]
- Dustan, H.R.; Tarazi, R.C.; Bravo, E.L. Diuretic and Diet Treatment of Hypertension. Arch. Intern. Med. 1974, 133, 1007–1013. [Google Scholar] [CrossRef]
- Somasekharan, S.; Tanis, J.; Forbush, B. Loop diuretic and ion-binding residues revealed by scanning mutagenesis of transmembrane helix 3 (TM3) of Na-K-Cl cotransporter (NKCC1). J. Biol. Chem. 2012, 287, 17308–17317. [Google Scholar] [CrossRef] [Green Version]
- Mroczek, W.J. The rational use of diuretics in the treatment of arterial hypertension. Angiology 1976, 27, 358–369. [Google Scholar] [CrossRef]
- Gamba, G.; Miyanoshita, A.; Lombardi, M.; Lytton, J.; Lee, W.S.; Hediger, M.A.; Hebert, S.C. Molecular cloning, primary structure, and characterization of two members of the mammalian electroneutral sodium-(potassium)-chloride cotransporter family expressed in kidney. J. Biol. Chem. 1994, 269, 17713–17722. [Google Scholar] [CrossRef] [PubMed]
- Bezerra, E.M.; de Alvarenga É, C.; Dos Santos, R.P.; de Sousa, J.S.; Fulco, U.L.; Freire, V.N.; Albuquerque, E.L.; da Costa, R.F. Losartan as an ACE inhibitor: A description of the mechanism of action through quantum biochemistry. RSC Adv. 2022, 12, 28395–28404. [Google Scholar] [CrossRef]
- Takezako, T.; Unal, H.; Karnik, S.S.; Node, K. Structure-Function Basis of Attenuated Inverse Agonism of Angiotensin II Type 1 Receptor Blockers for Active-State Angiotensin II Type 1 Receptor. Mol. Pharmacol. 2015, 88, 488–501. [Google Scholar] [CrossRef] [Green Version]
- Post, S.R.; Hammond, H.K.; Insel, P.A. Beta-adrenergic receptors and receptor signaling in heart failure. Annu. Rev. Pharmacol. Toxicol. 1999, 39, 343–360. [Google Scholar] [CrossRef]
- López-Sendón, J.; Swedberg, K.; McMurray, J.; Tamargo, J.; Maggioni, A.P.; Dargie, H.; Tendera, M.; Waagstein, F.; Kjekshus, J.; Lechat, P.; et al. Expert consensus document on beta-adrenergic receptor blockers. Eur. Heart J. 2004, 25, 1341–1362. [Google Scholar] [CrossRef] [Green Version]
- Wehland, M.; Grosse, J.; Simonsen, U.; Infanger, M.; Bauer, J.; Grimm, D. The effects of newer beta-adrenoceptor antagonists on vascular function in cardiovascular disease. Curr. Vasc. Pharmacol. 2012, 10, 378–390. [Google Scholar] [CrossRef]
- Elliott, W.J.; Ram, C.V. Calcium channel blockers. J. Clin. Hypertens. 2011, 13, 687–689. [Google Scholar] [CrossRef]
- Clemens, T.L.; Adams, J.S.; Henderson, S.L.; Holick, M.F. Increased skin pigment reduces the capacity of skin to synthesise vitamin D3. Lancet 1982, 1, 74–76. [Google Scholar] [CrossRef]
- Holick, M.F.; MacLaughlin, J.A.; Clark, M.B.; Holick, S.A.; Potts, J.T., Jr.; Anderson, R.R.; Blank, I.H.; Parrish, J.A.; Elias, P. Photosynthesis of previtamin D3 in human skin and the physiologic consequences. Science 1980, 210, 203–205. [Google Scholar] [CrossRef]
- Houghton, L.A.; Vieth, R. The case against ergocalciferol (vitamin D2) as a vitamin supplement. Am. J. Clin. Nutr. 2006, 84, 694–697. [Google Scholar] [CrossRef] [Green Version]
- Armas, L.A.G.; Hollis, B.W.; Heaney, R.P. Vitamin D2 Is Much Less Effective than Vitamin D3 in Humans. J. Clin. Endocrinol. Metab. 2004, 89, 5387–5391. [Google Scholar] [CrossRef] [Green Version]
- Smith, J.E.; Goodman, D.S. The turnover and transport of vitamin D and of a polar metabolite with the properties of 25-hydroxycholecalciferol in human plasma. J. Clin. Investig. 1971, 50, 2159–2167. [Google Scholar] [CrossRef]
- Di Nisio, A.; De Toni, L.; Sabovic, I.; Rocca, M.S.; De Filippis, V.; Opocher, G.; Azzena, B.; Vettor, R.; Plebani, M.; Foresta, C. Impaired Release of Vitamin D in Dysfunctional Adipose Tissue: New Cues on Vitamin D Supplementation in Obesity. J. Clin. Endocrinol. Metab. 2017, 102, 2564–2574. [Google Scholar] [CrossRef]
- ZEHNDER, D.; BLAND, R.; WALKER, E.A.; BRADWELL, A.R.; HOWIE, A.J.; HEWISON, M.; STEWART, P.M. Expression of 25-Hydroxyvitamin D<sub>3</sub>-1α-Hydroxylase in the Human Kidney. J. Am. Soc. Nephrol. 1999, 10, 2465–2473. [Google Scholar] [CrossRef]
- Midgett, R.J.; Spielvogel, A.M.; Coburn, J.W.; Norman, A.W. Studies on calciferol metabolism. VII. The renal production of the biologically active form of vitamin D, 1,25-dihydroxycholecalciferol; species, tissue and subcellular distribution. J. Clin. Endocrinol. Metab. 1973, 36, 1153–1161. [Google Scholar] [CrossRef]
- Brumbaugh, P.F.; Haussler, M.R. 1 Alpha,25-dihydroxycholecalciferol receptors in intestine. I. Association of 1 alpha,25-dihydroxycholecalciferol with intestinal mucosa chromatin. J. Biol. Chem. 1974, 249, 1251–1257. [Google Scholar] [CrossRef]
- Pike, J.W.; Haussler, M.R. Purification of chicken intestinal receptor for 1,25-dihydroxyvitamin D. Proc. Natl. Acad. Sci. USA 1979, 76, 5485–5489. [Google Scholar] [CrossRef] [Green Version]
- Haussler, M.R.; Jurutka, P.W.; Mizwicki, M.; Norman, A.W. Vitamin D receptor (VDR)-mediated actions of 1α,25(OH)₂vitamin D₃: Genomic and non-genomic mechanisms. Best. Pract. Res. Clin. Endocrinol. Metab. 2011, 25, 543–559. [Google Scholar] [CrossRef]
- Kurokawa, R.; Yu, V.C.; Näär, A.; Kyakumoto, S.; Han, Z.; Silverman, S.; Rosenfeld, M.G.; Glass, C.K. Differential orientations of the DNA-binding domain and carboxy-terminal dimerization interface regulate binding site selection by nuclear receptor heterodimers. Genes Dev. 1993, 7, 1423–1435. [Google Scholar] [CrossRef]
- Prüfer, K.; Racz, A.; Lin, G.C.; Barsony, J. Dimerization with retinoid X receptors promotes nuclear localization and subnuclear targeting of vitamin D receptors. J. Biol. Chem. 2000, 275, 41114–41123. [Google Scholar] [CrossRef] [Green Version]
- Dwivedi, P.P.; Hii, C.S.; Ferrante, A.; Tan, J.; Der, C.J.; Omdahl, J.L.; Morris, H.A.; May, B.K. Role of MAP kinases in the 1,25-dihydroxyvitamin D3-induced transactivation of the rat cytochrome P450C24 (CYP24) promoter. Specific functions for ERK1/ERK2 and ERK5. J. Biol. Chem. 2002, 277, 29643–29653. [Google Scholar] [CrossRef] [Green Version]
- Brumbaugh, P.F.; Haussler, M.R. 1 Alpha,25-dihydroxycholecalciferol receptors in intestine. II. Temperature-dependent transfer of the hormone to chromatin via a specific cytosol receptor. J. Biol. Chem. 1974, 249, 1258–1262. [Google Scholar] [CrossRef]
- Nemere, I.; Safford, S.E.; Rohe, B.; DeSouza, M.M.; Farach-Carson, M.C. Identification and characterization of 1,25D3-membrane-associated rapid response, steroid (1,25D3-MARRS) binding protein. J. Steroid Biochem. Mol. Biol. 2004, 89–90, 281–285. [Google Scholar] [CrossRef]
- Hii, C.S.; Ferrante, A. The Non-Genomic Actions of Vitamin D. Nutrients 2016, 8, 135. [Google Scholar] [CrossRef] [Green Version]
- Dwivedi, P.P.; Gao, X.H.; Tan, J.C.; Evdokiou, A.; Ferrante, A.; Morris, H.A.; May, B.K.; Hii, C.S. A role for the phosphatidylinositol 3-kinase--protein kinase C zeta--Sp1 pathway in the 1,25-dihydroxyvitamin D3 induction of the 25-hydroxyvitamin D3 24-hydroxylase gene in human kidney cells. Cell Signal 2010, 22, 543–552. [Google Scholar] [CrossRef]
- Armbrecht, H.J.; Boltz, M.A.; Hodam, T.L.; Kumar, V.B. Differential responsiveness of intestinal epithelial cells to 1,25-dihydroxyvitamin D3--role of protein kinase C. J. Endocrinol. 2001, 169, 145–151. [Google Scholar] [CrossRef] [Green Version]
- Whitfield, G.K.; Jurutka, P.W.; Haussler, C.A.; Hsieh, J.C.; Barthel, T.K.; Jacobs, E.T.; Dominguez, C.E.; Thatcher, M.L.; Haussler, M.R. Nuclear Vitamin D Receptor: Structure-Function, Molecular Control of Gene Transcription, and Novel Bioactions. Vitamin D 2005, 1, 219–261. [Google Scholar]
- Haussler, M.R.; Haussler, C.A.; Whitfield, G.K.; Hsieh, J.C.; Thompson, P.D.; Barthel, T.K.; Bartik, L.; Egan, J.B.; Wu, Y.; Kubicek, J.L.; et al. The nuclear vitamin D receptor controls the expression of genes encoding factors which feed the "Fountain of Youth" to mediate healthful aging. J. Steroid Biochem. Mol. Biol. 2010, 121, 88–97. [Google Scholar] [CrossRef] [Green Version]
- Meyer, M.B.; Watanuki, M.; Kim, S.; Shevde, N.K.; Pike, J.W. The human transient receptor potential vanilloid type 6 distal promoter contains multiple vitamin D receptor binding sites that mediate activation by 1,25-dihydroxyvitamin D3 in intestinal cells. Mol. Endocrinol. 2006, 20, 1447–1461. [Google Scholar] [CrossRef] [Green Version]
- Kolek, O.I.; Hines, E.R.; Jones, M.D.; LeSueur, L.K.; Lipko, M.A.; Kiela, P.R.; Collins, J.F.; Haussler, M.R.; Ghishan, F.K. 1alpha,25-Dihydroxyvitamin D3 upregulates FGF23 gene expression in bone: The final link in a renal-gastrointestinal-skeletal axis that controls phosphate transport. Am. J. Physiol. Gastrointest. Liver Physiol. 2005, 289, G1036–G1042. [Google Scholar] [CrossRef] [Green Version]
- Silver, J.; Russell, J.; Sherwood, L.M. Regulation by vitamin D metabolites of messenger ribonucleic acid for preproparathyroid hormone in isolated bovine parathyroid cells. Proc. Natl. Acad. Sci. USA 1985, 82, 4270–4273. [Google Scholar] [CrossRef] [Green Version]
- Hernando, N.; Pastor-Arroyo, E.M.; Marks, J.; Schnitzbauer, U.; Knöpfel, T.; Bürki, M.; Bettoni, C.; Wagner, C.A. 1,25(OH)(2) vitamin D(3) stimulates active phosphate transport but not paracellular phosphate absorption in mouse intestine. J. Physiol. 2021, 599, 1131–1150. [Google Scholar] [CrossRef]
- Barthel, T.K.; Mathern, D.R.; Whitfield, G.K.; Haussler, C.A.; Hopper, H.A.t.; Hsieh, J.C.; Slater, S.A.; Hsieh, G.; Kaczmarska, M.; Jurutka, P.W.; et al. 1,25-Dihydroxyvitamin D3/VDR-mediated induction of FGF23 as well as transcriptional control of other bone anabolic and catabolic genes that orchestrate the regulation of phosphate and calcium mineral metabolism. J. Steroid Biochem. Mol. Biol. 2007, 103, 381–388. [Google Scholar] [CrossRef]
- Kitazawa, R.; Kitazawa, S. Vitamin D(3) augments osteoclastogenesis via vitamin D-responsive element of mouse RANKL gene promoter. Biochem. Biophys. Res. Commun. 2002, 290, 650–655. [Google Scholar] [CrossRef]
- Noda, M.; Vogel, R.L.; Craig, A.M.; Prahl, J.; DeLuca, H.F.; Denhardt, D.T. Identification of a DNA sequence responsible for binding of the 1,25-dihydroxyvitamin D3 receptor and 1,25-dihydroxyvitamin D3 enhancement of mouse secreted phosphoprotein 1 (SPP-1 or osteopontin) gene expression. Proc. Natl. Acad. Sci. USA 1990, 87, 9995–9999. [Google Scholar] [CrossRef] [Green Version]
- Haussler, M.R.; Whitfield, G.K.; Haussler, C.A.; Hsieh, J.C.; Thompson, P.D.; Selznick, S.H.; Dominguez, C.E.; Jurutka, P.W. The nuclear vitamin D receptor: Biological and molecular regulatory properties revealed. J. Bone Miner. Res. 1998, 13, 325–349. [Google Scholar] [CrossRef]
- Fernandez, G.J.; Ramírez-Mejía, J.M.; Urcuqui-Inchima, S. Vitamin D boosts immune response of macrophages through a regulatory network of microRNAs and mRNAs. J. Nutr. Biochem. 2022, 109, 109105. [Google Scholar] [CrossRef]
- Grübler, M.R.; Gaksch, M.; Kienreich, K.; Verheyen, N.; Schmid, J.; BW, Ó.H.; Richtig, G.; Scharnagl, H.; Meinitzer, A.; Pieske, B.; et al. Effects of Vitamin D Supplementation on Plasma Aldosterone and Renin-A Randomized Placebo-Controlled Trial. J. Clin. Hypertens. 2016, 18, 608–613. [Google Scholar] [CrossRef]
- Resnick, L.M.; Müller, F.B.; Laragh, J.H. Calcium-regulating hormones in essential hypertension. Relation to plasma renin activity and sodium metabolism. Ann. Intern. Med. 1986, 105, 649–654. [Google Scholar] [CrossRef]
- Zhang, Y.; Kong, J.; Deb, D.K.; Chang, A.; Li, Y.C. Vitamin D receptor attenuates renal fibrosis by suppressing the renin-angiotensin system. J. Am. Soc. Nephrol. 2010, 21, 966–973. [Google Scholar] [CrossRef] [Green Version]
- Karadeniz, Y.; Özpamuk-Karadeniz, F.; Ahbab, S.; Ataoğlu, E.; Can, G. Vitamin D Deficiency Is a Potential Risk for Blood Pressure Elevation and the Development of Hypertension. Medicina 2021, 57, 1297. [Google Scholar] [CrossRef]
- McMullan, C.J.; Borgi, L.; Curhan, G.C.; Fisher, N.; Forman, J.P. The effect of vitamin D on renin-angiotensin system activation and blood pressure: A randomized control trial. J. Hypertens. 2017, 35, 822–829. [Google Scholar] [CrossRef] [Green Version]
- Tomaschitz, A.; Pilz, S.; Ritz, E.; Grammer, T.; Drechsler, C.; Boehm, B.O.; März, W. Independent association between 1,25-dihydroxyvitamin D, 25-hydroxyvitamin D and the renin-angiotensin system: The Ludwigshafen Risk and Cardiovascular Health (LURIC) study. Clin. Chim. Acta 2010, 411, 1354–1360. [Google Scholar] [CrossRef]
- Li, Y.C.; Kong, J.; Wei, M.; Chen, Z.F.; Liu, S.Q.; Cao, L.P. 1,25-Dihydroxyvitamin D(3) is a negative endocrine regulator of the renin-angiotensin system. J. Clin. Investig. 2002, 110, 229–238. [Google Scholar] [CrossRef]
- Zhou, C.; Lu, F.; Cao, K.; Xu, D.; Goltzman, D.; Miao, D. Calcium-independent and 1,25(OH)2D3-dependent regulation of the renin-angiotensin system in 1alpha-hydroxylase knockout mice. Kidney Int. 2008, 74, 170–179. [Google Scholar] [CrossRef] [Green Version]
- Barbarawi, M.; Kheiri, B.; Zayed, Y.; Barbarawi, O.; Dhillon, H.; Swaid, B.; Yelangi, A.; Sundus, S.; Bachuwa, G.; Alkotob, M.L.; et al. Vitamin D Supplementation and Cardiovascular Disease Risks in More Than 83 000 Individuals in 21 Randomized Clinical Trials: A Meta-analysis. JAMA Cardiol. 2019, 4, 765–776. [Google Scholar] [CrossRef]
- Jorde, R.; Figenschau, Y.; Emaus, N.; Hutchinson, M.; Grimnes, G. Serum 25-hydroxyvitamin D levels are strongly related to systolic blood pressure but do not predict future hypertension. Hypertension 2010, 55, 792–798. [Google Scholar] [CrossRef] [Green Version]
- Peterlik, M.; Cross, H.S. Vitamin D and calcium deficits predispose for multiple chronic diseases. Eur. J. Clin. Investig. 2005, 35, 290–304. [Google Scholar] [CrossRef]
- Holick, M.F. Vitamin D deficiency. N. Engl. J. Med. 2007, 357, 266–281. [Google Scholar] [CrossRef]
- Snijder, M.B.; Lips, P.; Seidell, J.C.; Visser, M.; Deeg, D.J.; Dekker, J.M.; van Dam, R.M. Vitamin D status and parathyroid hormone levels in relation to blood pressure: A population-based study in older men and women. J. Intern. Med. 2007, 261, 558–565. [Google Scholar] [CrossRef]
- Jorde, R.; Sundsfjord, J.; Haug, E.; Bonaa, K.H. Relation between low calcium intake, parathyroid hormone, and blood pressure. Hypertension 2000, 35, 1154–1159. [Google Scholar] [CrossRef] [Green Version]
- Morfis, L.; Smerdely, P.; Howes, L.G. Relationship between serum parathyroid hormone levels in the elderly and 24 h ambulatory blood pressures. J. Hypertens. 1997, 15, 1271–1276. [Google Scholar] [CrossRef]
- Hulter, H.N.; Melby, J.C.; Peterson, J.C.; Cooke, C.R. Chronic continuous PTH infusion results in hypertension in normal subjects. J. Clin. Hypertens. 1986, 2, 360–370. [Google Scholar]
- Fliser, D.; Franek, E.; Fode, P.; Stefanski, A.; Schmitt, C.P.; Lyons, M.; Ritz, E. Subacute infusion of physiological doses of parathyroid hormone raises blood pressure in humans. Nephrol. Dial. Transplant. 1997, 12, 933–938. [Google Scholar] [CrossRef] [Green Version]
- Nilsson, I.L.; Rastad, J.; Johansson, K.; Lind, L. Endothelial vasodilatory function and blood pressure response to local and systemic hypercalcemia. Surgery 2001, 130, 986–990. [Google Scholar] [CrossRef]
- de Boer, I.H.; Kestenbaum, B.; Shoben, A.B.; Michos, E.D.; Sarnak, M.J.; Siscovick, D.S. 25-hydroxyvitamin D levels inversely associate with risk for developing coronary artery calcification. J. Am. Soc. Nephrol. 2009, 20, 1805–1812. [Google Scholar] [CrossRef] [Green Version]
- Zehnder, D.; Bland, R.; Chana, R.S.; Wheeler, D.C.; Howie, A.J.; Williams, M.C.; Stewart, P.M.; Hewison, M. Synthesis of 1,25-dihydroxyvitamin D(3) by human endothelial cells is regulated by inflammatory cytokines: A novel autocrine determinant of vascular cell adhesion. J. Am. Soc. Nephrol. 2002, 13, 621–629. [Google Scholar] [CrossRef]
- Wong, M.S.; Delansorne, R.; Man, R.Y.; Vanhoutte, P.M. Vitamin D derivatives acutely reduce endothelium-dependent contractions in the aorta of the spontaneously hypertensive rat. Am. J. Physiol. Heart Circ. Physiol. 2008, 295, H289–H296. [Google Scholar] [CrossRef]
- Richart, T.; Li, Y.; Staessen, J.A. Renal versus extrarenal activation of vitamin D in relation to atherosclerosis, arterial stiffening, and hypertension. Am. J. Hypertens. 2007, 20, 1007–1015. [Google Scholar] [CrossRef] [Green Version]
- Dao, H.H.; Essalihi, R.; Bouvet, C.; Moreau, P. Evolution and modulation of age-related medial elastocalcinosis: Impact on large artery stiffness and isolated systolic hypertension. Cardiovasc. Res. 2005, 66, 307–317. [Google Scholar] [CrossRef] [Green Version]
- Jono, S.; Nishizawa, Y.; Shioi, A.; Morii, H. 1,25-Dihydroxyvitamin D3 increases in vitro vascular calcification by modulating secretion of endogenous parathyroid hormone-related peptide. Circulation 1998, 98, 1302–1306. [Google Scholar] [CrossRef] [Green Version]
- Yamamoto, T.; Kozawa, O.; Tanabe, K.; Akamatsu, S.; Matsuno, H.; Dohi, S.; Hirose, H.; Uematsu, T. 1,25-dihydroxyvitamin D3 stimulates vascular endothelial growth factor release in aortic smooth muscle cells: Role of p38 mitogen-activated protein kinase. Arch. Biochem. Biophys. 2002, 398, 1–6. [Google Scholar] [CrossRef]
- Kim, D.H.; Sabour, S.; Sagar, U.N.; Adams, S.; Whellan, D.J. Prevalence of hypovitaminosis D in cardiovascular diseases (from the National Health and Nutrition Examination Survey 2001 to 2004). Am. J. Cardiol. 2008, 102, 1540–1544. [Google Scholar] [CrossRef]
- Ke, L.; Mason, R.S.; Kariuki, M.; Mpofu, E.; Brock, K.E. Vitamin D status and hypertension: A review. Integr. Blood Press. Control. 2015, 8, 13–35. [Google Scholar] [CrossRef] [Green Version]
- Yilmaz, S.; Sen, F.; Ozeke, O.; Temizhan, A.; Topaloglu, S.; Aras, D.; Aydogdu, S. The relationship between vitamin D levels and nondipper hypertension. Blood Press. Monit. 2015, 20, 330–334. [Google Scholar] [CrossRef]
- Kunutsor, S.K.; Burgess, S.; Munroe, P.B.; Khan, H. Vitamin D and high blood pressure: Causal association or epiphenomenon? Eur. J. Epidemiol. 2014, 29, 1–14. [Google Scholar] [CrossRef]
- Del Pinto, R.; Wright, J.T.; Monaco, A.; Pietropaoli, D.; Ferri, C. Vitamin D and blood pressure control among hypertensive adults: Results from NHANES 2001–2014. J. Hypertens. 2020, 38, 150–158. [Google Scholar] [CrossRef]
- Rostand, S.G.; McClure, L.A.; Kent, S.T.; Judd, S.E.; Gutiérrez, O.M. Associations of blood pressure, sunlight, and vitamin D in community-dwelling adults. J. Hypertens. 2016, 34, 1704–1710. [Google Scholar] [CrossRef] [Green Version]
- Rostand, S.G. Ultraviolet light may contribute to geographic and racial blood pressure differences. Hypertension 1997, 30, 150–156. [Google Scholar] [CrossRef] [Green Version]
- Brennan, P.J.; Greenberg, G.; Miall, W.E.; Thompson, S.G. Seasonal variation in arterial blood pressure. Br. Med. J. (Clin. Res. Ed.) 1982, 285, 919–923. [Google Scholar] [CrossRef] [Green Version]
- Mokhtari, E.; Hajhashemy, Z.; Saneei, P. Serum Vitamin D Levels in Relation to Hypertension and Pre-hypertension in Adults: A Systematic Review and Dose-Response Meta-Analysis of Epidemiologic Studies. Front. Nutr. 2022, 9, 829307. [Google Scholar] [CrossRef]
- Wu, Z.; Wu, Y.; Rao, J.; Hu, H.; Wang, C.; Wu, J.; Shi, Y.; Fu, Y.; Cheng, X.; Li, P. Associations among vitamin D, tobacco smoke, and hypertension: A cross-sectional study of the NHANES 2001–2016. Hypertens. Res. 2022, 45, 1986–1996. [Google Scholar] [CrossRef]
- Krivošíková, K.; Krivošíková, Z.; Wsolová, L.; Seeman, T.; Podracká, Ľ. Hypertension in obese children is associated with vitamin D deficiency and serotonin dysregulation. BMC Pediatr. 2022, 22, 289. [Google Scholar] [CrossRef]
- Sheehy, S.; Palmer, J.R.; Cozier, Y.; Bertrand, K.A.; Rosenberg, L. Vitamin D and risk of hypertension among Black women. J. Clin. Hypertens. 2023, 25, 168–174. [Google Scholar] [CrossRef]
- Liu, Y.; Shi, L.; Lin, Y.; Zhang, M.; Chen, F.; Li, A.; Liu, Y. Relationship between serum 25-hydroxyvitamin D and target organ damage in children with essential hypertension. J. Hum. Hypertens. 2022, 36, 604–609. [Google Scholar] [CrossRef]
- Nganou-Gnindjio, C.N.; Ama Moor, V.J.; Pieme, C.A.; Sadeu Wafeu, G.; Ngandjeu Kamtchoum, I.; Yerema, R.; Menanga, A.P. Short-term Effects of Calcium and Vitamin D Supplementation in Postmenopausal Hypertensive Patients in sub-Saharan Africa: A Double Blinded Randomized Controlled Trial. Acta Sci. Women’s Health 2021, 1, 59–64. [Google Scholar] [CrossRef]
- Schroten, N.F.; Ruifrok, W.P.; Kleijn, L.; Dokter, M.M.; Silljé, H.H.; Lambers Heerspink, H.J.; Bakker, S.J.; Kema, I.P.; van Gilst, W.H.; van Veldhuisen, D.J.; et al. Short-term vitamin D3 supplementation lowers plasma renin activity in patients with stable chronic heart failure: An open-label, blinded end point, randomized prospective trial (VitD-CHF trial). Am. Heart J. 2013, 166, 357–364.e352. [Google Scholar] [CrossRef]
- Bislev, L.S.; Langagergaard Rødbro, L.; Nørgaard Bech, J.; Bjerregaard Pedersen, E.; Rolighed, L.; Sikjaer, T.; Rejnmark, L. Effects of treatment with an angiotensin 2 receptor blocker and/or vitamin D3 on parathyroid hormone and aldosterone: A randomized, placebo-controlled trial. Clin. Endocrinol. 2018, 89, 656–666. [Google Scholar] [CrossRef]
- Pilz, S.; Gaksch, M.; Kienreich, K.; Grübler, M.; Verheyen, N.; Fahrleitner-Pammer, A.; Treiber, G.; Drechsler, C.; B, Ó.H.; Obermayer-Pietsch, B.; et al. Effects of vitamin D on blood pressure and cardiovascular risk factors: A randomized controlled trial. Hypertension 2015, 65, 1195–1201. [Google Scholar] [CrossRef] [PubMed]
- Arora, P.; Song, Y.; Dusek, J.; Plotnikoff, G.; Sabatine, M.S.; Cheng, S.; Valcour, A.; Swales, H.; Taylor, B.; Carney, E.; et al. Vitamin D therapy in individuals with prehypertension or hypertension: The DAYLIGHT trial. Circulation 2015, 131, 254–262. [Google Scholar] [CrossRef] [PubMed]
- Zaheer, S.; Taquechel, K.; Brown, J.M.; Adler, G.K.; Williams, J.S.; Vaidya, A. A randomized intervention study to evaluate the effect of calcitriol therapy on the renin-angiotensin system in diabetes. J. Renin Angiotensin Aldosterone Syst. 2018, 19, 1470320317754178. [Google Scholar] [CrossRef] [Green Version]
- Kennel, K.A.; Drake, M.T.; Hurley, D.L. Vitamin D deficiency in adults: When to test and how to treat. Mayo Clin. Proc. 2010, 85, 752–758. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bouillon, R. Comparative analysis of nutritional guidelines for vitamin D. Nat. Rev. Endocrinol. 2017, 13, 466–479. [Google Scholar] [CrossRef]
- Medicine, I.O. Dietary Reference Intakes for Calcium and Vitamin D; The National Academies Press: Washington, DC, USA, 2011; p. 1132. [Google Scholar] [CrossRef]
- EFSA Panel on Dietetic Products, Nutrition and Allergies (NDA). Scientific Opinion on the Tolerable Upper Intake Level of vitamin D. EFSA J. 2012, 10, 2813. [Google Scholar] [CrossRef]
- Theiler-Schwetz, V.; Trummer, C.; Grübler, M.R.; Keppel, M.H.; Zittermann, A.; Tomaschitz, A.; Karras, S.N.; März, W.; Pilz, S.; Gängler, S. Effects of Vitamin D Supplementation on 24-Hour Blood Pressure in Patients with Low 25-Hydroxyvitamin D Levels: A Randomized Controlled Trial. Nutrients 2022, 14, 1360. [Google Scholar] [CrossRef] [PubMed]
- Caccamo, D.; Cannata, A.; Ricca, S.; Catalano, L.M.; Montalto, A.F.; Alibrandi, A.; Ercoli, A.; Granese, R. Role of Vitamin-D Receptor (VDR) single nucleotide polymorphisms in gestational hypertension development: A case-control study. PLoS ONE 2020, 15, e0239407. [Google Scholar] [CrossRef]
- Abouzid, M.; Kruszyna, M.; Burchardt, P.; Kruszyna, Ł.; Główka, F.K.; Karaźniewicz-Łada, M. Vitamin D Receptor Gene Polymorphism and Vitamin D Status in Population of Patients with Cardiovascular Disease—A Preliminary Study. Nutrients 2021, 13, 3117. [Google Scholar] [CrossRef]
- Swapna, N.; Vamsi, U.M.; Usha, G.; Padma, T. Risk conferred by FokI polymorphism of vitamin D receptor (VDR) gene for essential hypertension. Indian J. Hum. Genet. 2011, 17, 201–206. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Magiełda-Stola, J.; Kurzawińska, G.; Ożarowski, M.; Karpiński, T.M.; Drews, K.; Seremak-Mrozikiewicz, A. The Significance of VDR Genetic Polymorphisms in the Etiology of Preeclampsia in Pregnant Polish Women. Diagnostics 2021, 11, 1698. [Google Scholar] [CrossRef]
- Santos, B.R.; Casanova, G.; Silva, T.R.; Marchesan, L.B.; Oppermann, K.; Spritzer, P.M. Are vitamin D deficiency and VDR gene polymorphisms associated with high blood pressure as defined by the ACC/AHA 2017 criteria in postmenopausal women? Maturitas 2021, 149, 26–33. [Google Scholar] [CrossRef] [PubMed]
- Si, S.; Mo, M.; Cheng, H.; Peng, Z.; Alifu, X.; Zhou, H.; Chi, P.; Zhuang, Y.; Yu, Y. The Association of Vitamin D and Its Pathway Genes’ Polymorphisms with Hypertensive Disorders of Pregnancy: A Prospective Cohort Study. Nutrients 2022, 14, 2355. [Google Scholar]
- Beveridge, L.A.; Struthers, A.D.; Khan, F.; Jorde, R.; Scragg, R.; Macdonald, H.M.; Alvarez, J.A.; Boxer, R.S.; Dalbeni, A.; Gepner, A.D.; et al. Effect of Vitamin D Supplementation on Blood Pressure: A Systematic Review and Meta-analysis Incorporating Individual Patient Data. JAMA Intern. Med. 2015, 175, 745–754. [Google Scholar] [CrossRef] [Green Version]
- Lind, L.; Lithell, H.; Skarfors, E.; Wide, L.; Ljunghall, S. Reduction of blood pressure by treatment with alphacalcidol. A double-blind, placebo-controlled study in subjects with impaired glucose tolerance. Acta Med. Scand. 1988, 223, 211–217. [Google Scholar] [CrossRef]
- Lind, L.; Wengle, B.; Wide, L.; Sörensen, O.H.; Ljunghall, S. Hypertension in primary hyperparathyroidism--reduction of blood pressure by long-term treatment with vitamin D (alphacalcidol). A double-blind, placebo-controlled study. Am. J. Hypertens. 1988, 1, 397–402. [Google Scholar] [CrossRef]
- Panahi, Y.; Namazi, S.; Rostami-Yalmeh, J.; Sahebi, E.; Khalili, N.; Jamialahmadi, T.; Sahebkar, A. Effect of Vitamin D Supplementation on the Regulation of Blood Pressure in Iranian Patients with Essential Hypertension: A Clinical Trial. Adv. Exp. Med. Biol. 2021, 1328, 501–511. [Google Scholar] [CrossRef]
- Chen, W.R.; Liu, Z.Y.; Shi, Y.; Yin, D.W.; Wang, H.; Sha, Y.; Chen, Y.D. Vitamin D and nifedipine in the treatment of Chinese patients with grades I-II essential hypertension: A randomized placebo-controlled trial. Atherosclerosis 2014, 235, 102–109. [Google Scholar] [CrossRef] [PubMed]
- Sheikh, V.; Mozaianimonfared, A.; Gharakhani, M.; Poorolajal, J.; Ph, D. Effect of vitamin D supplementation versus placebo on essential hypertension in patients with vitamin D deficiency: A double-blind randomized clinical trial. J. Clin. Hypertens. 2020, 22, 1867–1873. [Google Scholar] [CrossRef] [PubMed]
- Bernini, G.; Carrara, D.; Bacca, A.; Carli, V.; Virdis, A.; Rugani, I.; Duranti, E.; Ghiadoni, L.; Bernini, M.; Taddei, S. Effect of acute and chronic vitamin D administration on systemic renin angiotensin system in essential hypertensives and controls. J. Endocrinol. Investig. 2013, 36, 216–220. [Google Scholar] [CrossRef] [PubMed]
- Vaidya, A.; Sun, B.; Larson, C.; Forman, J.P.; Williams, J.S. Vitamin D3 therapy corrects the tissue sensitivity to angiotensin ii akin to the action of a converting enzyme inhibitor in obese hypertensives: An interventional study. J. Clin. Endocrinol. Metab. 2012, 97, 2456–2465. [Google Scholar] [CrossRef]
- Carrara, D.; Bernini, M.; Bacca, A.; Rugani, I.; Duranti, E.; Virdis, A.; Ghiadoni, L.; Taddei, S.; Bernini, G. Cholecalciferol administration blunts the systemic renin-angiotensin system in essential hypertensives with hypovitaminosis D. J. Renin. Angiotensin Aldosterone Syst. 2014, 15, 82–87. [Google Scholar] [CrossRef]
- Carrara, D.; Bruno, R.M.; Bacca, A.; Taddei, S.; Duranti, E.; Ghiadoni, L.; Bernini, G. Cholecalciferol treatment downregulates renin-angiotensin system and improves endothelial function in essential hypertensive patients with hypovitaminosid D. J. Hypertens. 2016, 34, 2199–2205. [Google Scholar] [CrossRef]
- Qasemi, R.; Ghavamzadeh, S.; Faghfouri, A.H.; Valizadeh, N.; Mohammadi, A.; Sayyadi, H. The effect of vitamin D supplementation on flow-mediated dilatation, oxidized LDL and intracellular adhesion molecule 1 on type 2 diabetic patients with hypertension: A randomized, placebo-controlled, double-blind trial. Diabetes Metab. Syndr. 2021, 15, 102200. [Google Scholar] [CrossRef]
- Mozaffari-Khosravi, H.; Loloei, S.; Mirjalili, M.R.; Barzegar, K. The effect of vitamin D supplementation on blood pressure in patients with elevated blood pressure and vitamin D deficiency: A randomized, double-blind, placebo-controlled trial. Blood Press. Monit. 2015, 20, 83–91. [Google Scholar] [CrossRef] [Green Version]
- Goel, R.K.; Lal, H. Role of vitamin d supplementation in hypertension. Indian J. Clin. Biochem. 2011, 26, 88–90. [Google Scholar] [CrossRef] [Green Version]
- Ullah, M.I.; Uwaifo, G.I.; Nicholas, W.C.; Koch, C.A. Does vitamin D deficiency cause hypertension? Current evidence from clinical studies and potential mechanisms. Int. J. Endocrinol. 2010, 2010, 579640. [Google Scholar] [CrossRef] [PubMed] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Category | Systolic (mmHg) | Diastolic (mmHg) | |
|---|---|---|---|
| Optimal | <120 | And | <80 |
| Normal | 120–129 | And/or | 80–84 |
| High normal | 130–139 | And/or | 85–89 |
| Grade 1 hypertension | 140–159 | And/or | 90–99 |
| Grade 2 hypertension | 160–179 | And/or | 100–109 |
| Grade 3 hypertension | 180 | And/or | 110 |
| Isolated systolic hypertension | 140 | And | <90 |
| Antihypertensive Agent | Mechanism of Action |
|---|---|
| Diuretics | Diuretics produce a negative salt and water balance via similar pathways and prevent sodium retention in the long-term [26]. Several diuretics exist with different potencies and renal sites of action [26]. Loop diuretics (furosemide, bumetanide) exert their mechanism on NKCC transporters (NKCC1 and NKCC2), inhibiting the intracellular Cl- concentration, and Na+ and Cl- reabsorption in the renal tubule [27]. Thiazides act in the cortical diluting segment of the distal renal tubule, inducing diuresis by excreting mainly of sodium and chloride [28], inhibiting the thiazide-sensitive sodium-chloride cotransporter, which reabsorbs 5–10% of the filtered sodium load in the kidney [29]. |
| ACE-inhibitors | RAAS is a key element in BP regulation and homeostasis of the body’s fluid volume. Renin breaks down angiotensinogen into angiotensin. In pulmonary blood vessels, angiotensin I interacts with ACE, which converts angiotensin I (Ang I) into the octapeptide hormone angiotensin II (Ang II) [30]. ACE-inhibitors prevent the conversion of Ang I to Ang II, thus inhibiting aldosterone release, promoting vasodilation [30]. |
| Angiotensin receptor blockers | Ang II acts on the angiotensin II type 1 receptor (AT1R), which triggers vasoconstriction [30]. The AT1R is the principal regulator of BP and fluid volume homeostasis, hereby playing a vital role in cardiovascular and renal pathophysiology. Over-stimulation of the AT1R is implicated in hypertension. AT1R is the primary target receptor for the antihypertensive drug ARBs, therefore over-stimulation can be greatly reduced by treating with ARBs, thus reducing BP [31]. |
| Beta-adrenergic receptor blockers | Stimulation of the -adrenergic receptor (-AR) by sympathetic neuronal activation (SNS), circulating catecholamines, or adrenergic agonists is proven to increase the heart rate, contraction force, and rate of cardiac relaxation [32]. -AR can be divided into three subtypes, -AR, -AR, and -AR, with the expression of mainly -AR (75–80%) and -AR in human cardiac tissue [32]. BARBs bind selectively to -ARs, implementing competitive antagonism to the effects of the -adrenergic stimuli [33]. Proposed antihypertensive functions of BARBs are [34]:
|
| Calcium channel blockers | Inhibit the flow of extracellular calcium by blocking ion-specific channels extending over the cell wall. Several types of ion-specific calcium channels have been identified; the mechanism of CCB action in humans is located in the L-type channels. Inhibiting calcium influx causes vascular smooth muscles to relax, evoking vasodilation and subsequently lowering the BP [35]. |
| Title (Year of Completion) | Design | Intervention/Treatment | Endpoints/Outcomes | Results | Conclusions |
|---|---|---|---|---|---|
| Short-term vascular effects and oxidative status of calcium and vitamin D supplementation of postmenopausal hypertensive black women NCT04255992 [104] (2020) | Double-arm, double-blind, randomized, and parallel clinical trial 22 participants | Patients with stable antihypertensive therapy for at least 3 months Calcium arm: 1000 mg calcium tablet per day for 8 weeks Vitamin D/calcium arm: 1000 mg/800UI of vitamin D/calcium tablet per day for 8 weeks | Primary: Change in 24 h BP profile from baseline to week 8 Secondary: Change in serum malondialdehyde concentration from baseline to week 8 | Calcium vs. vitamin D/calcium arm: no significant differences in diurnal SBP reductions (4.7 [−2.5–10] vs. 4.7 [0.6–8.10] mmHg, p = 0.630) and HsCRP reductions (1.68 [0.43–5.58] vs. 1.46 [0.36–4.63] mg/L, p = 0.540). However, the vitamin D/calcium combination reduced uric acid levels significantly better than calcium alone (by 16 [9.63–24] vs. 11 [8–18] mg/L, p = 0.020). All values are given as median [interquartile range]. | Supplementation with both calcium alone and calcium and vitamin D in postmenopausal hypertensive women is associated with a significant reduction of diurnal BP and inflammatory biomarkers. |
| Study to investigate the effects of vitamin D administration on plasma renin activity in patients with stable chronic heart failure (VitD-CHF) NCT01092130 [105] (2013) | Open-label, blinded-endpoint, randomized, prospective trial 101 participants | Patients treated with ACE-inhibitors or ARBs and BARBs Control arm: no additional medication for 6 weeks Intervention arm: 2000 IU vitamin D daily, for 6 weeks | Primary: plasma renin activity (PRA) after 6 weeks Secondary: evaluation of the effect of vitamin D on plasma values of additional markers of RAAS activity, on different markers of the vitamin D cascade, on plasma levels of NT-proBNP, on urinary levels of markers of glomerular and tubular damage, on extracellular matrix markers, and on NYHA-class. Safety endpoints were biochemical indices of kidney function and bone homeostasis | Significant increase in both 25(OH)D and 1,25(OH)2D levels in the intervention group compared to control (80 [75–87] vs. 44 [39–49] nmol/L, p < 0.001 and 194 (179–211] vs.132 [121–143] nmol/L, p < 0.001, respectively) after 6 weeks. Significant decrease in both PRA and PRC in the intervention group compared to control (5.2 [2.9–9.5] vs. 7.3 [4.5–11.8] ng/mL, p = 0.002 and 55 [32–93] vs. 72 [47–111] ng/mL, p = 0.02, respectively) after 6 weeks. All values given as geometric means [95%CI]. | Supplementation with dietary vitamin D3 (2.000 IU/d) in CHF patients increased vitamin D levels and lowered both PRA and PRC effectively compared to control. |
| Physiologic interactions between the adrenal- and the parathyroid glands NCT02572960 [106] (2018) | Double-blind, placebo-controlled trial 81 participants | Patients with secondary hyperparathyroidism due to vitamin D deficiency Arm 1: Vitamin D3 70 µg/day for 12 weeks valsartan 80 mg/day for 2 weeks Arm 2: Vitamin D3 70 µg/day for 12 weeks placebo valsartan daily for 2 weeks Arm 3: Placebo vitamin D3/day for 12 weeks valsartan 80 mg/day for 2 weeks Arm 4: Placebo vitamin D3/day for 12 weeks placebo Valsartan daily for 2 weeks | Primary: Aldosterone at baseline and after 12 weeks of vitamin D3 treatment Secondary: PTH at baseline and after 2 weeks of ARB treatment, arterial stiffness, 24 h BP, physiological parameters | Valsartan (ARB) reduced DBP by −5.0 [−8.8; −1.0) mmHg vs. −2.6 [−7.5; 3.7] mmHg in the placebo group (p < 0.05). SBP was not significantly different in both groups. Renin increased strongly compared to placebo (81.2 [44.3; 180.0]) vs. 1.35 [−23.2; 46.3] pg/mL, p < 0.001), while the aldosterone ratio decreased significantly from valsartan treatment (−126 [−253; −52] vs. −8 [−59; 15], p < 0.0001). An addition of vitamin D3 had no further effect Vitamin D3 supplementation reduced PTH by −3.1 [−9.4; 9.1]% vs. a 5.7 [−5.2; 23.8]% increase in the placebo group (p = 0.1). All values are given as median [interquartile range]. | No effect of ARB treatment on PTH plasma concentration. No correlation between calcium homeostasis and RAAS. Vitamin D3 supplementation reduced PTH, but did not affect BP, cardiac conduction, or the RAAS. |
| Effects of vitamin D on blood pressure and cardiovascular risk factors NCT02136771 [66,107] (2014) | Single-center, double-blind, placebo-controlled, parallel-group study | Hypertensive patients with 25-hydroxyvitamin D levels below 30 ng/mL Treatment arm: 2800 IU vitamin D3 per day as oily drops for 8 weeks Placebo arm: Oily drops only as placebo for 8 weeks | Primary: 24-h systolic ambulatory blood pressure after 8 weeks Secondary: 24-h diastolic ambulatory blood pressure, plasma renin concentration, plasma aldosterone concentration, NT-pro-BNP, 24-h urinary albumin excretion, triglycerides, HDL-cholesterol | No significant reduction of 24 h SBP was observed (−0.4 [−2.8 to 1.9] mmHg, p = 0.712). Triglycerides increased significantly in the vitamin D group with a mean treatment of 17 [1–33] mg/dL (p = 0.013). A significant increase in 25(OH)D (mean treatment effect 11.5 [9.4–13.7] ng/mL: p < 0.001) and a significant decrease in PTH (−5.7 [−9.3 to −2.1] pg/mL; p = 0.003). All values given as mean [95%CI]. | No significant effects of vitamin D supplementation on BP and several cardiovascular risk factors were shown; however, a significant increase in triglycerides was observed. |
| DAYLIGHT: Vitamin D therapy in individuals at high risk of hypertension NCT01240512 [108] (2017) | Double-blind, randomized, controlled trial 534 participants | Patients with 25-hydroxyvitamin D <25 ng/mL, SBP 120–159 mmHg, DBP ≤ 99 mmHg, no antihypertensive medication High dose arm: 4000 IU/d vitamin D3 for 6 months Low dose arm: 400 IU/d vitamin D3 for 6 months | Primary: change in 24 h SBP after 6 months Secondary: change in 24 h DBP, change in mean daytime and nighttime ambulatory systolic and diastolic blood pressure, change in mean clinic systolic and diastolic blood pressure, change in mean clinic pulse pressure, all after 6 months | There was no significant difference in the primary end point or in any of the secondary end points. No evidence of association between change in 25-hydroxyvitamin D and change in 24-h systolic blood pressure after 6 months. | Supplementation with vitamin D did not reduce BP in vitamin D deficient individuals with either prehypertension or stage 1 hypertension. |
| The VALIDATE-D study NCT01635062 [109] (2017) | Randomized, double-blinded, and placebo-controlled study 18 participants | Patients with treated type-two diabetes, normal blood pressure or stage 1 hypertension (treated or untreated), and normal kidney function Intervention arm: calcitriol (titrated up to 0.75 µg/d) for 3 weeks Placebo arm: placebo for 3 weeks | Primary: change in circulating RAS activity after 2 weeks Secondary: change in renal plasma flow and urinary protein excretion after 2 weeks | Increases in 1,25(OH)2D (45.4 ± 18.2 to 61.8 ± 11.2 pg/mL, p = 0.03) with calcitriol administration vs. no change in the placebo group. No significant differences in PRA, serum or urinary aldosterone, baseline ang II-stimulated MAP, or basal and ang II-stimulated RPF between interventions was found. Values are given as mean ± SD. | Calcitriol raises 1,25(OH)2D levels compared to placebo, but this has no significant effect on the change in circulating RAS activity or vascular hemodynamics. |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jensen, N.S.; Wehland, M.; Wise, P.M.; Grimm, D. Latest Knowledge on the Role of Vitamin D in Hypertension. Int. J. Mol. Sci. 2023, 24, 4679. https://doi.org/10.3390/ijms24054679
Jensen NS, Wehland M, Wise PM, Grimm D. Latest Knowledge on the Role of Vitamin D in Hypertension. International Journal of Molecular Sciences. 2023; 24(5):4679. https://doi.org/10.3390/ijms24054679
Chicago/Turabian StyleJensen, Niklas S., Markus Wehland, Petra M. Wise, and Daniela Grimm. 2023. "Latest Knowledge on the Role of Vitamin D in Hypertension" International Journal of Molecular Sciences 24, no. 5: 4679. https://doi.org/10.3390/ijms24054679