
Transcytosable Peptide-Paclitaxel Prodrug Nanoparticle for Targeted Treatment of Triple-Negative Breast Cancer
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. The Targeting and Drug Delivery Properties of TAR Peptide
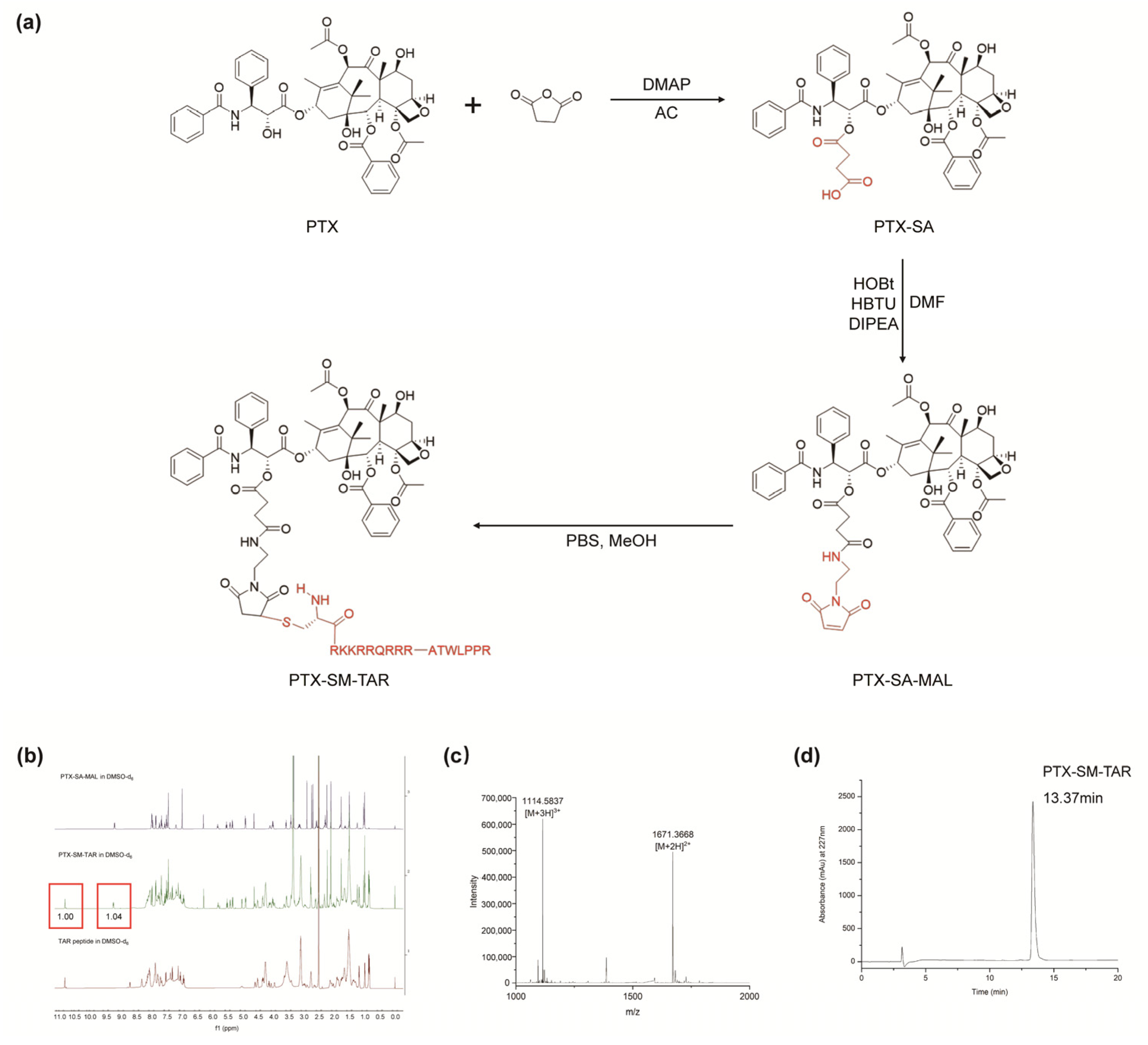
2.2. Synthesis and Characterization of PTX-SM-TAR
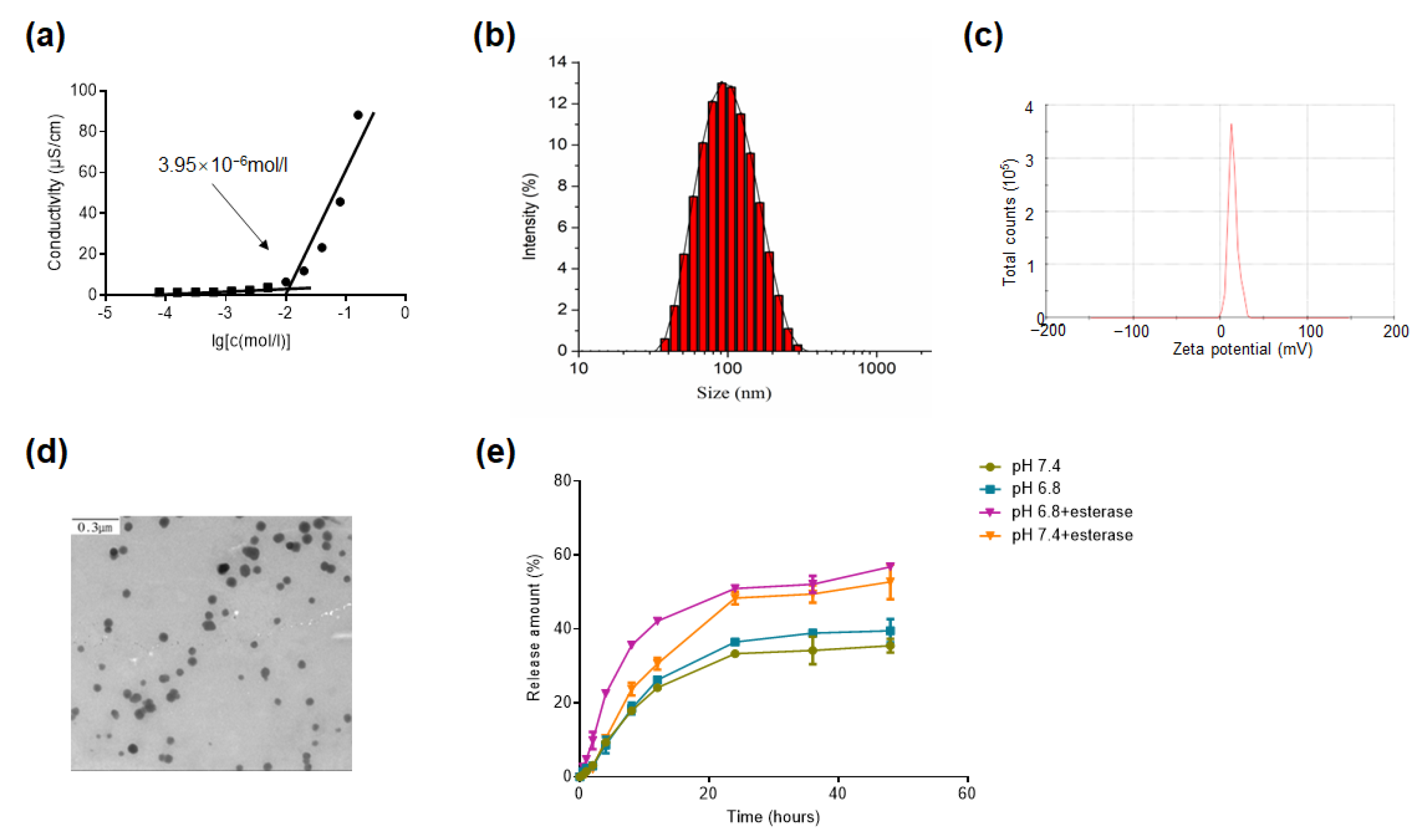
2.3. CAC, Morphology, Size, and Zeta Potential of PTX-SM-TAR NPs
2.4. In Vitro PTX Release from PTX-SM-TAR NPs
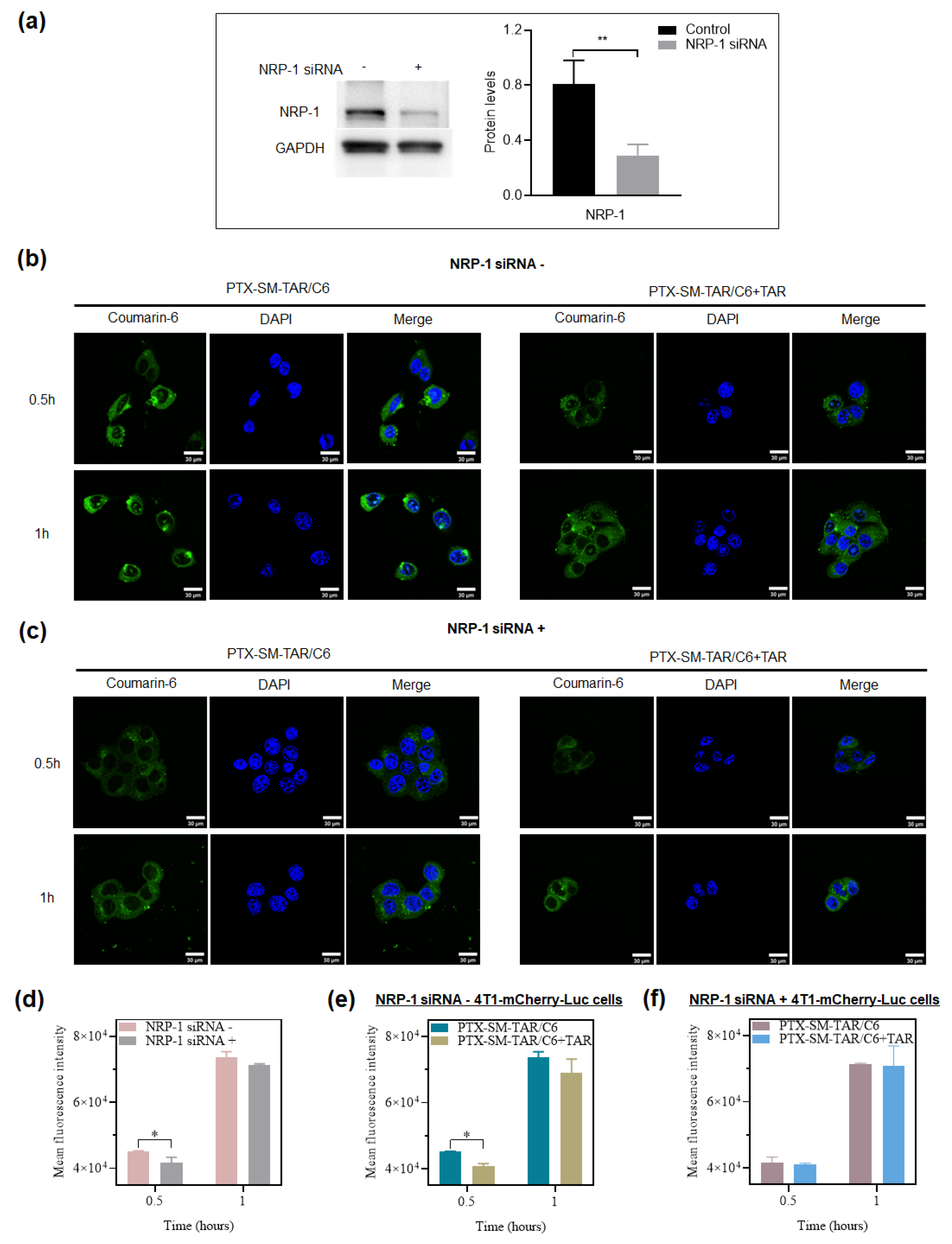
2.5. Targeting Properties of PTX-SM-TAR NPs to NRP-1 In Vitro
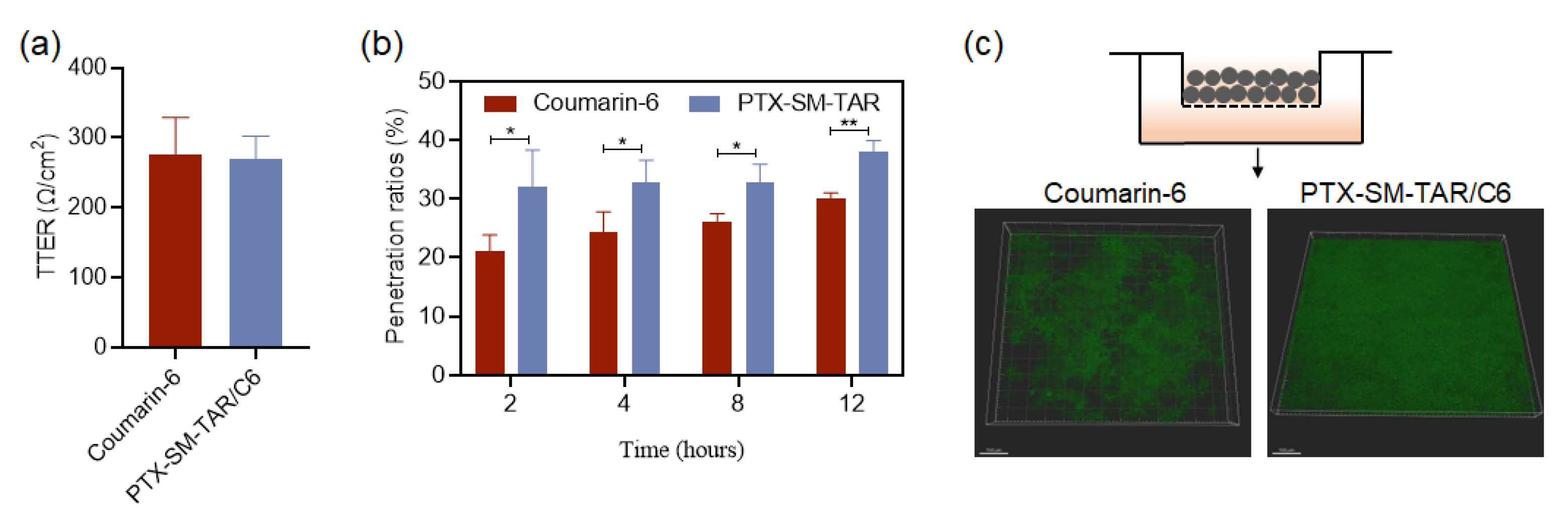
2.6. Penetration of PTX-SM-TAR/C6 NPs in the Vascular Barrier In Vitro
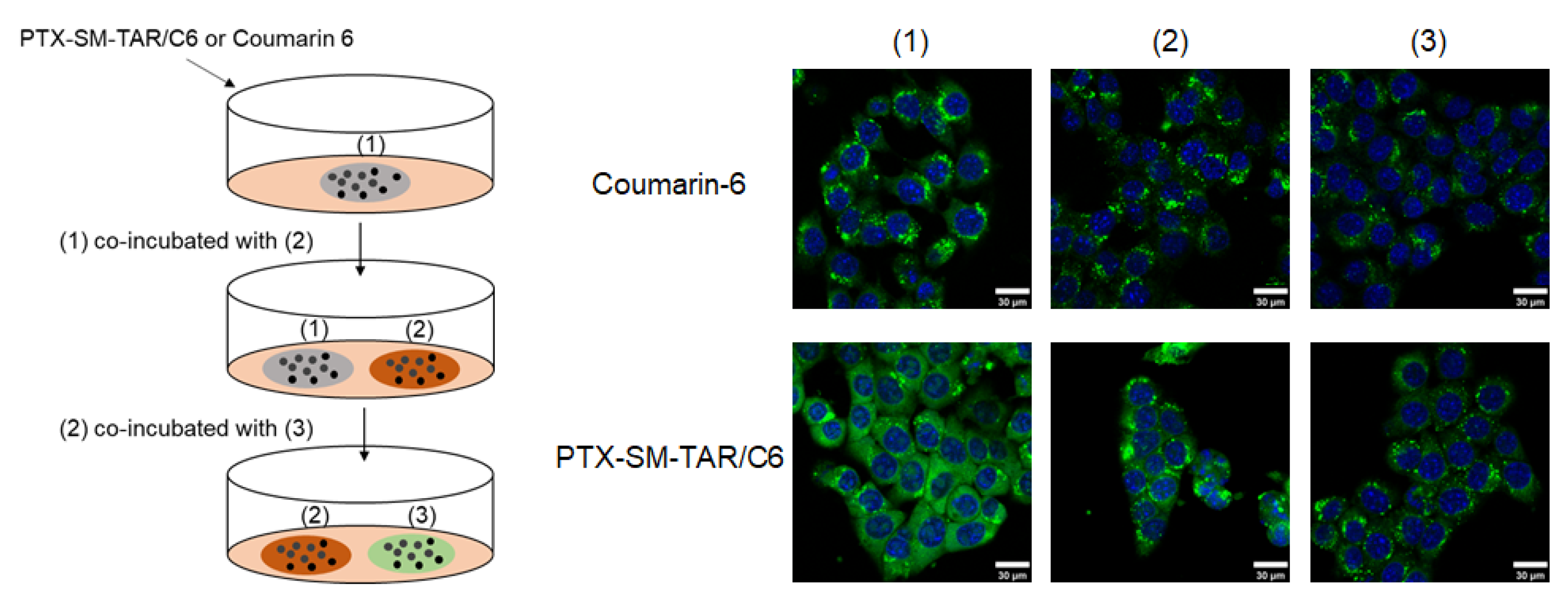
2.7. Transcellular Migration of PTX-SM-TAR/C6 NPs in 4T1-mCherry-Luc Cells
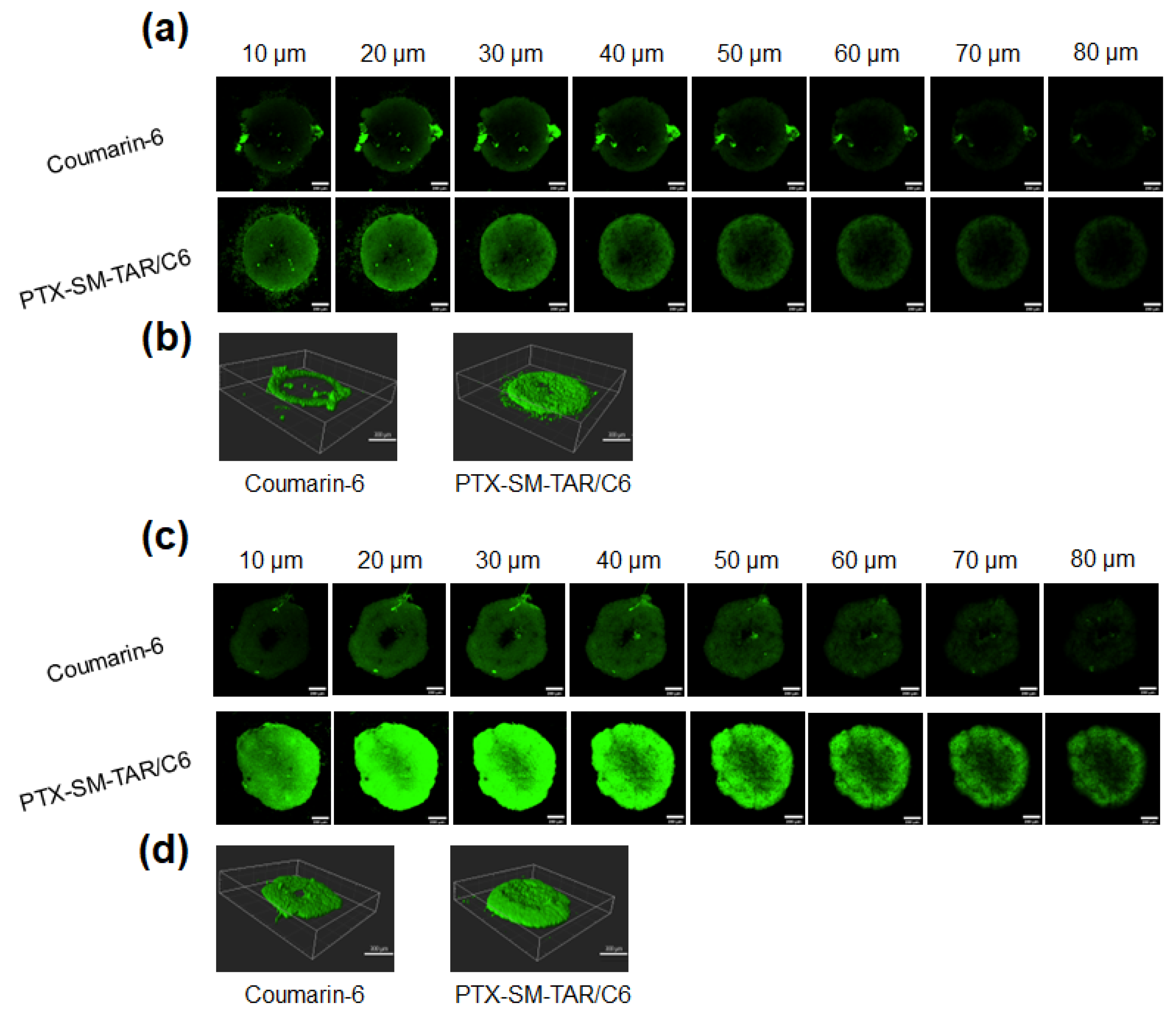
2.8. The Penetrating Efficiency of PTX-SM-TAR NPs in 3D Tumor Spheroids
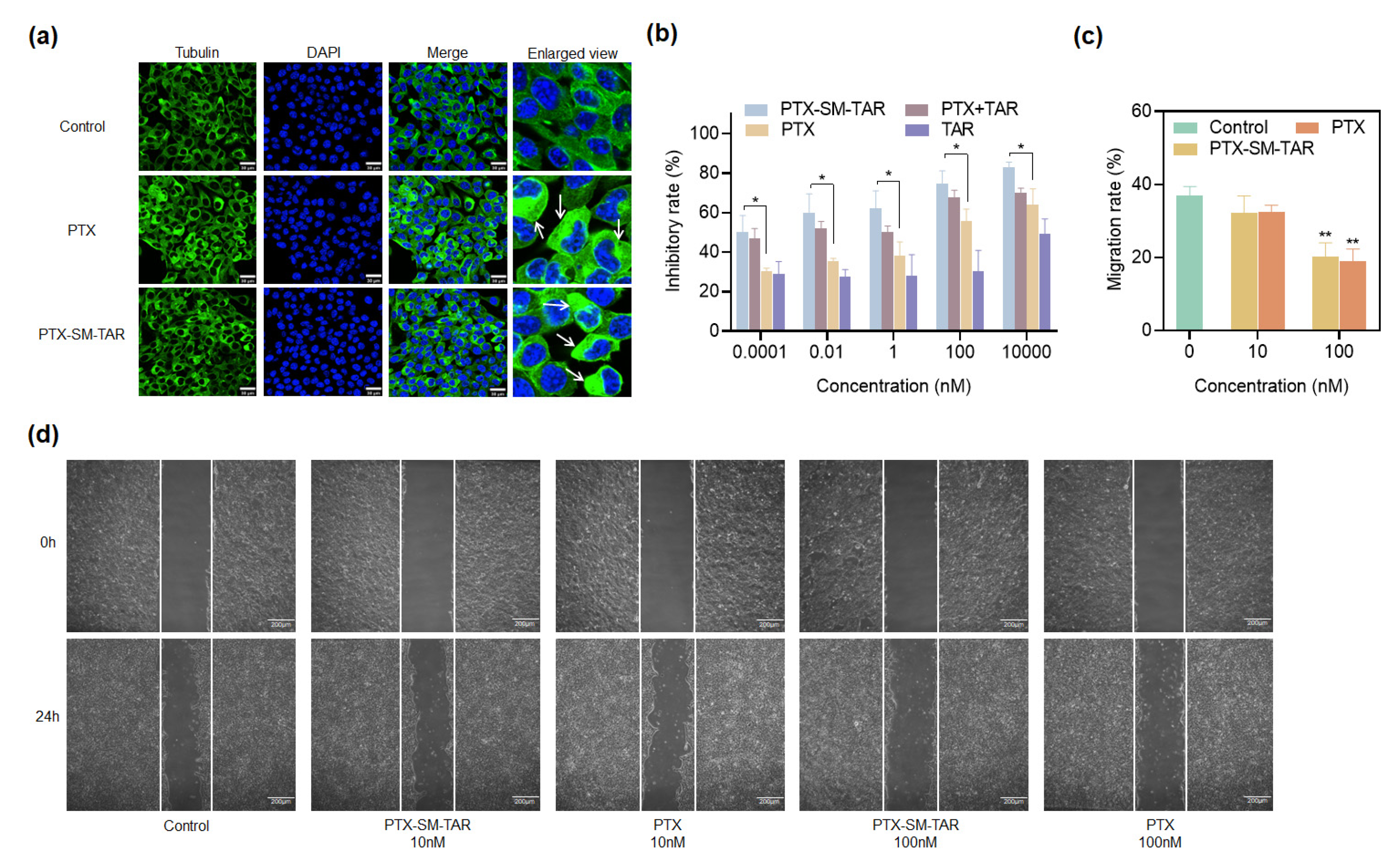
2.9. Anti-Tubulin and Anti-Proliferation Effects on 4T1-mCherry-Luc Cells
2.10. PTX-SM-TAR NPs Inhibit Migration on 4T1-mCherry-Luc Cells
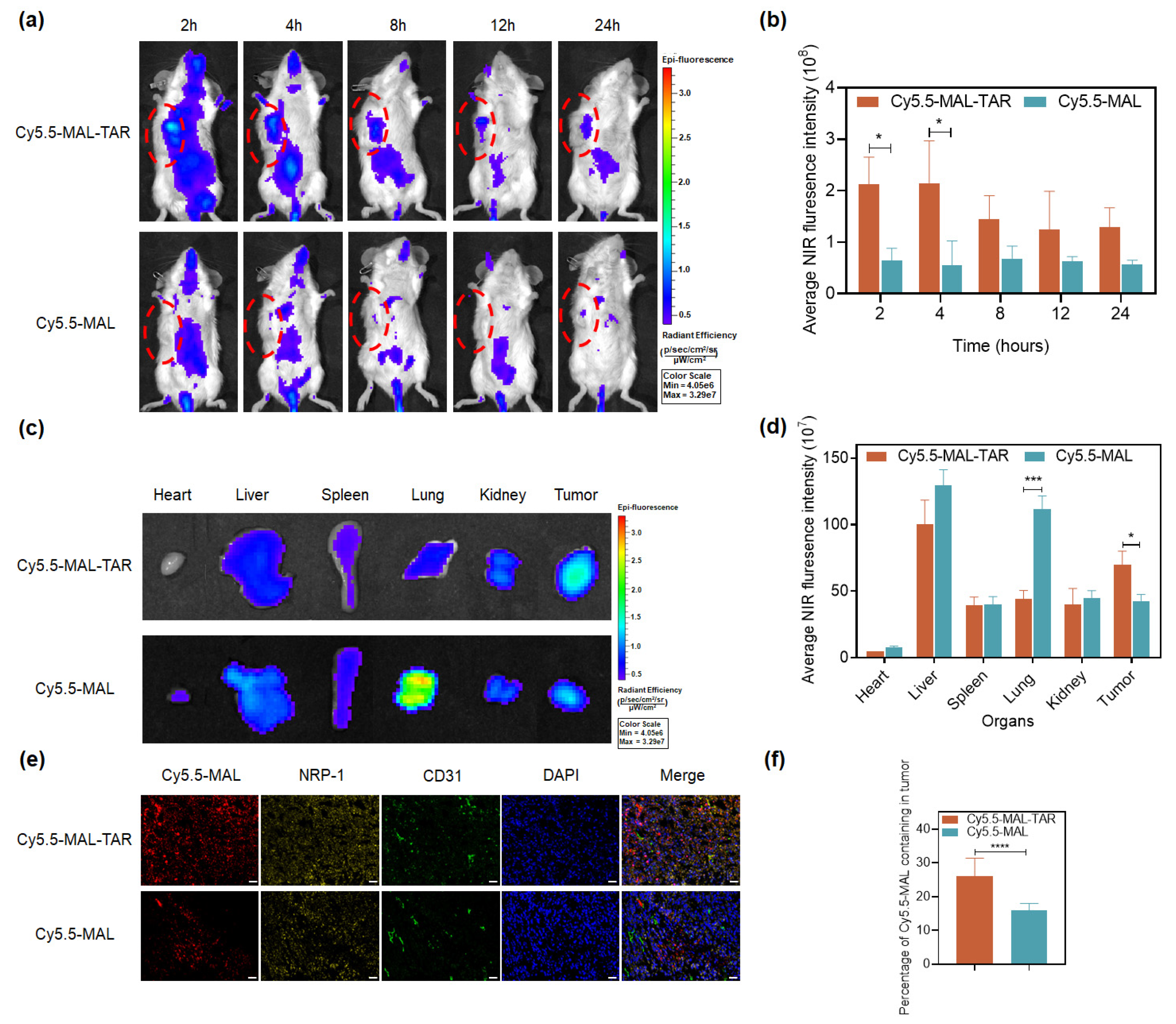
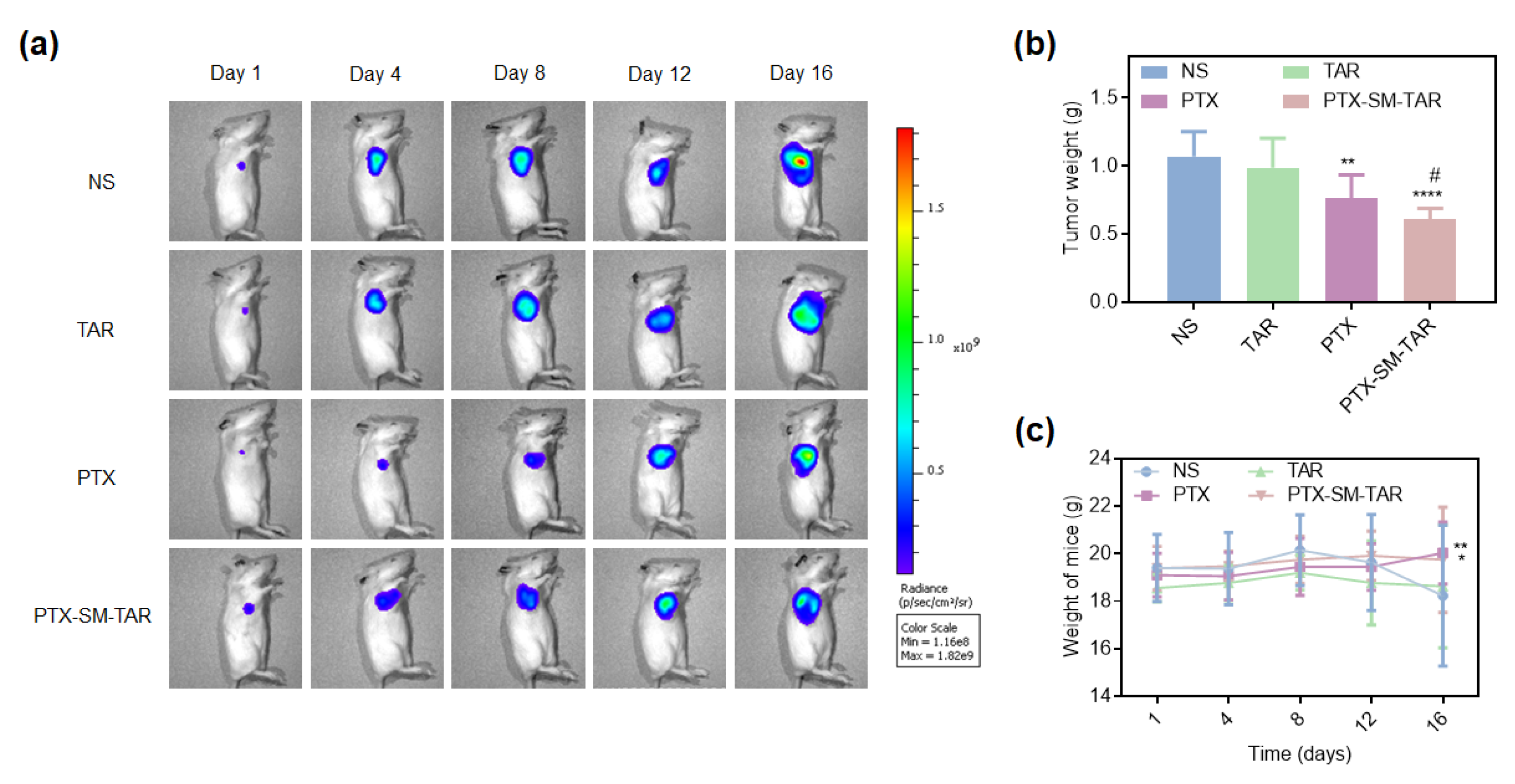
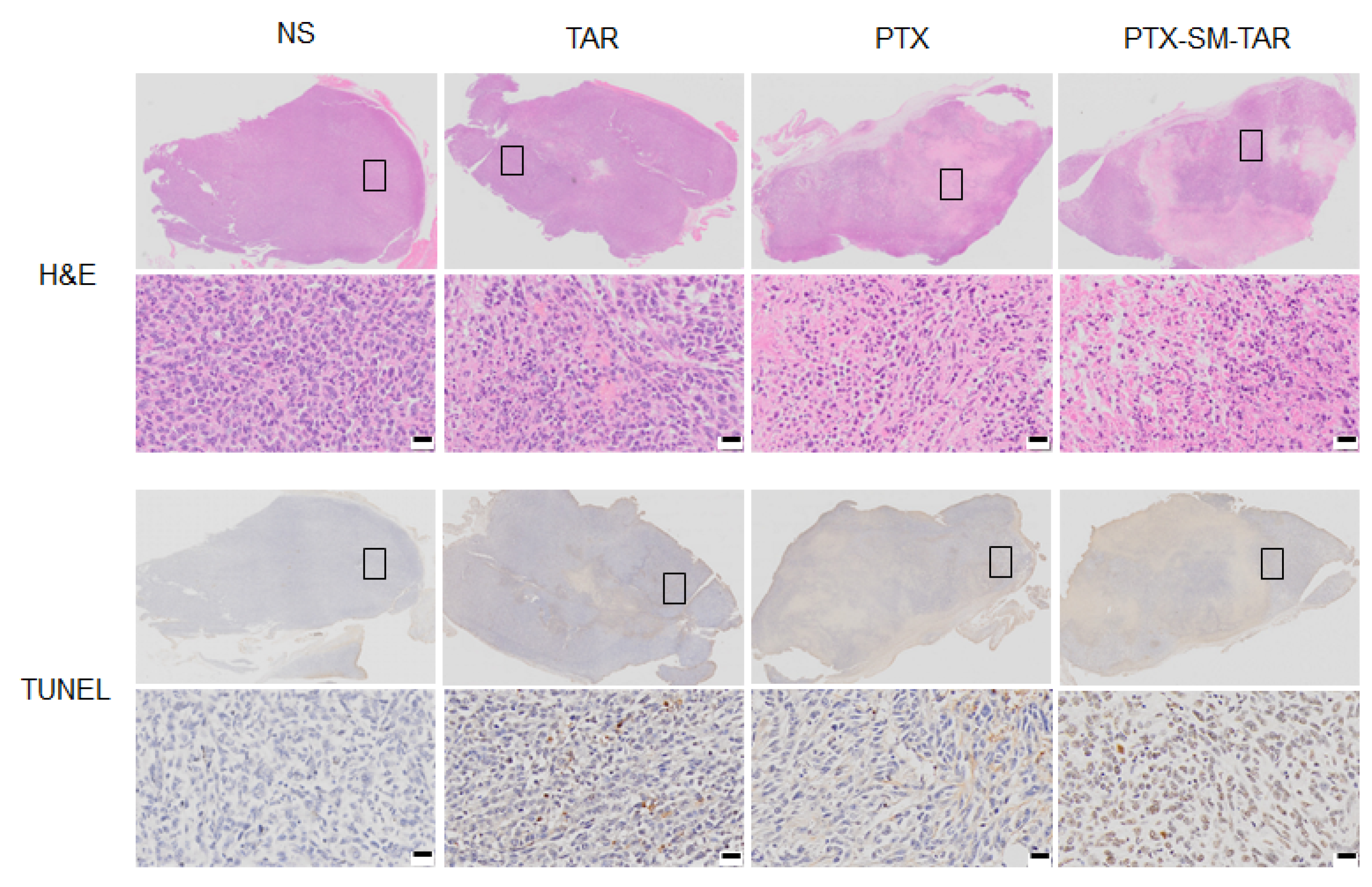
2.11. Evaluation of Antitumor Efficacy In Vivo
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Cell lines and Cell Culture
4.3. Animal Model
4.4. Validation of the Targeting and Drug Delivery Properties of TAR Peptide
4.5. Synthesis of PTX-SA
4.6. Synthesis of PTX-SA-MAL
4.7. Synthesis of PTX-SM-TAR
4.8. Determination of Critical Aggregation Concentration (CAC)
4.9. Characterization of PTX-SM-TAR Nanoparticles (NPs)
4.10. In Vitro Release Study for PTX-SM-TAR NPs
4.11. Transfection of siRNA into 4T1-mCherry-Luc Cells
4.12. Western Blotting Analysis
4.13. Cellular Uptake of PTX-SM-TAR/C6 NPs
4.14. Penetration of PTX-SM-TAR/C6 NPs in the Vascular Barrier In Vitro
4.15. Transcellular Migration of PTX-SM-TAR/C6 NPs in 4T1-mCherry-Luc Cells
4.16. Penetration of PTX-SM-TAR/C6 NPs in 3D Tumor Spheroids
4.17. Anti-Tubulin Effects of PTX-SM-TAR NPs
4.18. Anti-Proliferative Activity of PTX-SM-TAR NPs
4.19. Migration Assay of PTX-SM-TAR NPs
4.20. In Vivo Tumor Evaluation Studies
4.21. Histology Analysis
4.22. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Borri, F.; Granaglia, A. Pathology of triple negative breast cancer. Semin. Cancer Biol. 2021, 72, 136–145. [Google Scholar] [CrossRef]
- Yin, L.; Duan, J.J.; Bian, X.W.; Shi, C.Y. Triple-negative breast cancer molecular subtyping and treatment progress. Breast Cancer Res. 2020, 22, 61. [Google Scholar] [CrossRef] [PubMed]
- Mo, H.N.; Xu, B.H. Progress in systemic therapy for triple-negative breast cancer. Front. Med. 2021, 15, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Gupta, G.K.; Collier, A.L.; Lee, D.; Hoefer, R.A.; Zheleva, V.; Reesema, L.L.S.; Tang-Tan, A.M.; Guye, M.L.; Chang, D.Z.; Winston, J.S. Perspectives on Triple-Negative Breast Cancer: Current Treatment Strategies, Unmet Needs, and Potential Targets for Future Therapies. Cancers 2020, 12, 2392. [Google Scholar] [CrossRef] [PubMed]
- Hwang, S.Y.; Park, S.; Kwon, Y. Recent therapeutic trends and promising targets in triple negative breast cancer. Pharmacol. Ther. 2019, 199, 30–57. [Google Scholar] [CrossRef]
- Wu, S.Y.; Wang, H.; Shao, Z.M.; Jiang, Y.Z. Triple-negative breast cancer: New treatment strategies in the era of precision medicine. Sci. China-Life Sci. 2021, 64, 372–388. [Google Scholar] [CrossRef]
- Huynh, M.M.; Pambid, M.R.; Jayanthan, A.; Dorr, A.; Los, G.; Dunn, S.E. The dawn of targeted therapies for triple negative breast cancer (TNBC): A snapshot of investigational drugs in phase I and II trials. Expert Opin. Investig. Drugs 2020, 29, 1199–1208. [Google Scholar] [CrossRef]
- Nagayama, A.; Vidula, N.; Ellisen, L.; Bardia, A. Novel antibody-drug conjugates for triple negative breast cancer. Ther. Adv. Med. Oncol. 2020, 12, 1758835920915980. [Google Scholar] [CrossRef]
- Lyons, T.G. Targeted Therapies for Triple-Negative Breast Cancer. Curr. Treat. Options Oncol. 2019, 20, 82. [Google Scholar] [CrossRef]
- Bai, X.; Ni, J.; Beretov, J.; Graham, P.; Li, Y. Triple-negative breast cancer therapeutic resistance: Where is the Achilles’ heel? Cancer Lett. 2021, 497, 100–111. [Google Scholar] [CrossRef] [PubMed]
- Khadela, A.; Soni, S.; Shah, A.C.; Pandya, A.J.; Megha, K.; Kothari, N.; Cb, A. Unveiling the antibody-drug conjugates portfolio in battling Triple-negative breast cancer: Therapeutic trends and Future horizon. Med. Oncol. 2022, 40, 25. [Google Scholar] [CrossRef]
- Yang, C.P.H.; Horwitz, S.B. Taxol((R)): The First Microtubule Stabilizing Agent. Int. J. Mol. Sci. 2017, 18, 1733. [Google Scholar] [CrossRef] [Green Version]
- Scribano, C.M.; Wan, J.; Esbona, K.; Tucker, J.B.; Lasek, A.; Zhou, A.S.; Zasadil, L.M.; Molini, R.; Fitzgerald, J.; Lager, A.M. Chromosomal instability sensitizes patient breast tumors to multipolar divisions induced by paclitaxel. Sci. Transl. Med. 2021, 13, eabd4811. [Google Scholar] [CrossRef] [PubMed]
- Mahtani, R.; Kittaneh, M.; Kalinsky, K.; Mamounas, E.; Badve, S.; Vogel, C.; Lower, E.; Schwartzberg, L.; Pegram, M. Advances in Therapeutic Approaches for Triple-Negative Breast Cancer. Clin. Breast Cancer 2021, 21, 383–390. [Google Scholar] [CrossRef] [PubMed]
- Bernabeu, E.; Cagel, M.; Lagomarsino, E.; Moretton, M.; Chiappetta, D.A. Paclitaxel: What has been done and the challenges remain ahead. Int. J. Pharm. 2017, 526, 474–495. [Google Scholar] [CrossRef] [PubMed]
- Haddad, R.; Alrabadi, N.; Altaani, B.; Li, T.L. Paclitaxel Drug Delivery Systems: Focus on Nanocrystals’ Surface Modifications. Polymers 2022, 14, 658. [Google Scholar] [CrossRef]
- Wang, Y.; Iqbal, H.; Ur-Rehman, U.; Zhai, L.N.; Yuan, Z.Y.; Razzaq, A.; Lv, M.; Wei, H.Y.; Ning, X.; Xin, J.; et al. Albumin-based nanodevices for breast cancer diagnosis and therapy. J. Drug Eelivery Sci. Technol. 2023, 79, 104072. [Google Scholar] [CrossRef]
- Gallego-Jara, J.; Lozano-Terol, G.; Sola-Martinez, R.; Cánovas-Díaz, M.; Puente, T.D. A Compressive Review about Taxol(R): History and Future Challenges. Molecules 2020, 25, 5986. [Google Scholar] [CrossRef]
- Raza, F.; Zafar, H.; Khan, M.W.; Ullah, A.; Khan, A.U.; Baseer, A.; Fareed, R.; Sohail, M. Recent advances in the targeted delivery of paclitaxel nanomedicine for cancer therapy. Mater. Adv. 2022, 3, 2268–2290. [Google Scholar] [CrossRef]
- Chen, Q.; Xu, S.; Liu, S.; Wang, Y.; Liu, G.X. Emerging nanomedicines of paclitaxel for cancer treatment. J. Control. Release 2022, 342, 280–294. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.K.; Chen, H.Y.; Wang, F.S.; Zhang, X.K. The development of peptide-drug conjugates (PDCs) strategies for paclitaxel. Expert Opin. Drug Deliv. 2022, 19, 147–161. [Google Scholar] [CrossRef] [PubMed]
- Alas, M.; Saghaeidehkordi, A.; Kaur, K. Peptide-Drug Conjugates with Different Linkers for Cancer Therapy. J. Med. Chem. 2021, 64, 216–232. [Google Scholar] [CrossRef] [PubMed]
- Wu, M.; Huang, W.; Yang, N.; Liu, Y.Y. Learn from antibody-drug conjugates: Consideration in the future construction of peptide-drug conjugates for cancer therapy. Exp. Hematol. Oncol. 2022, 11, 93. [Google Scholar] [CrossRef]
- Wang, Y.; Cheetham, A.G.; Angacian, G.; Su, H.; Xie, L.S.; Cui, H.G. Peptide-drug conjugates as effective prodrug strategies for targeted delivery. Adv. Drug Deliv. Rev. 2017, 110, 112–126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heh, E.; Allen, J.; Ramirez, F.; Lovasz, D.; Fernandez, L.; Hogg, T.; Riva, H.; Holland, N.; Chacon, J. Peptide Drug Conjugates and Their Role in Cancer Therapy. Int. J. Mol. Sci. 2023, 24, 829. [Google Scholar] [CrossRef]
- Cooper, B.M.; Iegre, J.; O’ Donovan, D.H.; Halvarsson, M.Ö.; Spring, D.R. Peptides as a platform for targeted therapeutics for cancer: Peptide-drug conjugates (PDCs). Chem. Soc. Rev. 2021, 50, 1480–1494. [Google Scholar] [CrossRef] [PubMed]
- Hoppenz, P.; Els-Heindl, S.; Beck-Sickinger, A.G. Peptide-Drug Conjugates and Their Targets in Advanced Cancer Therapies. Front. Chem. 2020, 8, 571. [Google Scholar] [CrossRef]
- Chavda, V.P.; Solanki, H.K.; Davidson, M.; Apostolopoulos, V.; Bojarska, J. Peptide-Drug Conjugates: A New Hope for Cancer Management. Molecules 2022, 27, 7232. [Google Scholar] [CrossRef]
- Ma, L.; Wang, C.; He, Z.; Cheng, B.; Zheng, L.; Huang, K. Peptide-Drug Conjugate: A Novel Drug Design Approach. Curr. Med. Chem. 2017, 24, 3373–3396. [Google Scholar] [CrossRef] [PubMed]
- Vrettos, E.I.; Tzakos, A.G. Construction of Peptide-Drug Conjugates for Selective Targeting of Malignant Tumor Cells. Methods Mol. Biol. 2021, 2207, 327–338. [Google Scholar]
- Tang, S.Y.; Wei, H.; Yu, C.Y. Peptide-functionalized delivery vehicles for enhanced cancer therapy. Int. J. Pharm. 2021, 593, 120141. [Google Scholar] [CrossRef] [PubMed]
- Nam, S.H.; Park, J.; Koo, H. Recent advances in selective and targeted drug/gene delivery systems using cell-penetrating peptides. Arch. Pharmacal. Res. 2023, 46, 18–34. [Google Scholar] [CrossRef] [PubMed]
- Fu, H.; Shi, K.R.; Hu, G.L.; Yang, Y.T.; Kuang, Q.F.; Lu, L.B.; Zhang, L.; Chen, W.F.; Dong, M.L.; Chen, Y.T.; et al. Tumor-Targeted Paclitaxel Delivery and Enhanced Penetration Using TAT-Decorated Liposomes Comprising Redox-Responsive Poly(Ethylene Glycol). J. Pharm. Sci. 2015, 104, 1160–1173. [Google Scholar] [CrossRef] [PubMed]
- Duan, Z.Q.; Chen, C.T.; Qin, J.; Liu, Q.; Wang, Q.; Xu, X.C.; Wang, J.X. Cell-penetrating peptide conjugates to enhance the antitumor effect of paclitaxel on drug-resistant lung cancer. Drug Deliv. 2017, 24, 752–764. [Google Scholar] [CrossRef] [Green Version]
- Dissanayake, S.; Denny, W.A.; Gamage, S.; Sarojini, V. Recent developments in anticancer drug delivery using cell penetrating and tumor targeting peptides. J. Control. Release 2017, 250, 62–76. [Google Scholar] [CrossRef]
- Deng, X.; Mai, R.Y.; Zhang, C.Y.; Yu, D.B.; Ren, Y.C.; Li, G.; Cheng, B.B.; Li, L.; Yu, Z.Q.; Chen, J.J. Discovery of novel cell-penetrating and tumor-targeting peptide-drug conjugate (PDC) for programmable delivery of paclitaxel and cancer treatment. Eur. J. Med. Chem. 2021, 213, 113050. [Google Scholar] [CrossRef] [PubMed]
- Hua, D.; Tang, L.D.; Wang, W.T.; Tang, S.G.; Yu, L.; Zhou, X.X.; Wang, Q.; Sun, C.Y.; Shi, C.J.; Luo, W.J.; et al. Improved Antiglioblastoma Activity and BBB Permeability by Conjugation of Paclitaxel to a Cell-Penetrative MMP-2-Cleavable Peptide. Adv. Sci. 2021, 8, 2001960. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; Chen, H.Y.; Lu, L.; Zhao, C.Q.; Malichewe, C.V.; Wang, L.; Guo, X.L.; Zhang, X.K. Design and screening of a novel neuropilin-1 targeted penetrating peptide for anti-angiogenic therapy in glioma. Life Sci. 2021, 270, 119113. [Google Scholar] [CrossRef]
- Wang, S.; Li, C.; Yuan, Y.C.; Xiong, Y.X.; Xu, H.J.; Pan, W.S.; Pan, H.; Zhu, Z.H. Microvesicles as drug delivery systems: A new frontier for bionic therapeutics in cancer. J. Drug Deliv. Dcience Dechnol. 2023, 79, 104088. [Google Scholar] [CrossRef]
- Feng, Y.; Liao, Z.; Zhang, H.X.; Xie, X.X.; You, F.M.; Liao, X.L.; Wu, C.H.; Zhang, W.; Yang, H.; Liu, Y.Y. Emerging nanomedicines strategies focused on tumor microenvironment against cancer recurrence and metastasis. Chem. Eng. J. 2023, 452, 139506. [Google Scholar] [CrossRef]
- Zhao, L.; Chen, H.; Lu, L.; Wang, L.; Zhang, X.K.; Guo, X.L. New insights into the role of co-receptor neuropilins in tumour angiogenesis and lymphangiogenesis and targeted therapy strategies. J. Drug Target. 2021, 29, 155–167. [Google Scholar] [CrossRef] [PubMed]
- Hu, C.; Jiang, X. Role of NRP-1 in VEGF-VEGFR2-Independent Tumorigenesis. Target. Oncol. 2016, 11, 501–505. [Google Scholar] [CrossRef]
- Naik, A.; Al-Zeheimi, N.; Bakheit, C.S.; Riyami, M.A.; Jarrah, A.A.; Moundhri, M.S.A.; Habsi, Z.A.; Basheer, M.; Adham, S.A. Neuropilin-1 Associated Molecules in the Blood Distinguish Poor Prognosis Breast Cancer: A Cross-Sectional Study. Sci. Rep. 2017, 7, 3301. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Zeng, H.Y.; Wang, P.; Soker, S.; Mukhopadhyay, D. Neuropilin-1-mediated vascular permeability factor/vascular endothelial growth factor-dependent endothelial cell migration. J. Biol. Chem. 2003, 278, 48848–48860. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, H.Y.; Wang, X.H.; Zhang, Y.; Cheng, R.; Yuan, J.D.; Zhong, Z.Y. Systemic Delivery of NAC-1 siRNA by Neuropilin-Targeted Polymersomes Sensitizes Antiangiogenic Therapy of Metastatic Triple-Negative Breast Cancer. Biomacromolecules 2020, 21, 5119–5127. [Google Scholar] [CrossRef]
- Cano-Cortes, M.V.; Navarro-Marchal, S.A.; Ruiz-Blas, M.P.; Diaz-Mochon, J.J.; Marchal, J.A.; Sanchez-Martin, R.M. A versatile theranostic nanodevice based on an orthogonal bioconjugation strategy for efficient targeted treatment and monitoring of triple negative breast cancer. Nanomed. -Nanotechnol. Biol. Med. 2020, 24, 102120. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.Y.; Zhang, X.S.; Li, Z.Y.; Wang, Q.S.; Shi, Y.; Jiang, X.; Sun, X.Y. The miR-124-3p/Neuropilin-1 Axis Contributes to the Proliferation and Metastasis of Triple-Negative Breast Cancer Cells and Co-Activates the TGF-beta Pathway. Front. Oncol. 2021, 11, 654672. [Google Scholar] [CrossRef]
- Lu, L.; Chen, H.Y.; Hao, D.K.; Zhang, X.K.; Wang, F.S. The functions and applications of A7R in anti-angiogenic therapy, imaging and drug delivery systems. Asian J. Pharm. Sci. 2019, 14, 595–608. [Google Scholar] [CrossRef] [PubMed]
- Lu, L.; Chen, H.Y.; Wang, L.K.; Zhao, L.; Cheng, Y.N.; Wang, A.J.; Wang, F.S.; Zhang, X.K. A Dual Receptor Targeting- and BBB Penetrating-Peptide Functionalized Polyethyleneimine Nanocomplex for Secretory Endostatin Gene Delivery to Malignant Glioma. Int. J. Nanomed. 2020, 15, 8875–8892. [Google Scholar] [CrossRef] [PubMed]
- Park, J.; Choi, Y.; Chang, H.; Um, W.; Ryu, J.H.; Kwon, I.C. Alliance with EPR Effect: Combined Strategies to Improve the EPR Effect in the Tumor Microenvironment. Theranostics 2019, 9, 8073–8090. [Google Scholar] [CrossRef]
- Li, J.J.; Kataoka, K. Chemo-physical Strategies to Advance the in Vivo Functionality of Targeted Nanomedicine: The Next Generation. J. Am. Chem. Soc. 2021, 143, 538–599. [Google Scholar] [CrossRef]
- Liu, Y.; Huo, Y.Y.; Yao, L.; Xu, Y.W.; Meng, F.Q.; Li, H.F.; Sun, K.; Zhou, G.D.; Kohane, D.S.; Tao, K. Transcytosis of Nanomedicine for Tumor Penetration. Nano Lett. 2019, 19, 8010–8020. [Google Scholar] [CrossRef]
- Zhou, Q.; Shao, S.Q.; Wang, J.Q.; Xu, C.H.; Xiang, J.J.; Piao, Y.; Zhou, Z.X.; Yu, Q.S.; Tang, J.B.; Liu, X.R. Enzyme-activatable polymer-drug conjugate augments tumour penetration and treatment efficacy. Nat. Nanotechnol. 2019, 14, 799–809. [Google Scholar] [CrossRef]
- Okuyama, N.C.M.; Ribeiro, D.L.; da Rocha, C.Q.; Pereira, É.R.; Cólus, I.M.S.; Serpeloni, J.M. Three-dimensional cell cultures as preclinical models to assess the biological activity of phytochemicals in breast cancer. Toxicol. Appl. Pharmacol. 2023, 460, 116376. [Google Scholar] [CrossRef]
- Zhou, Y.; Zhou, W.; Chen, X.; Wang, Q.B.; Li, C.; Chen, Q.J.; Zhang, Y.; Lu, Y.F.; Ding, X.Y.; Jiang, C. Bone marrow mesenchymal stem cells-derived exosomes for penetrating and targeted chemotherapy of pancreatic cancer. Acta Pharm. Sin. B 2020, 10, 1563–1575. [Google Scholar] [CrossRef]










Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, L.; Zhao, C.; Lu, L.; Jiang, H.; Wang, F.; Zhang, X. Transcytosable Peptide-Paclitaxel Prodrug Nanoparticle for Targeted Treatment of Triple-Negative Breast Cancer. Int. J. Mol. Sci. 2023, 24, 4646. https://doi.org/10.3390/ijms24054646
Wang L, Zhao C, Lu L, Jiang H, Wang F, Zhang X. Transcytosable Peptide-Paclitaxel Prodrug Nanoparticle for Targeted Treatment of Triple-Negative Breast Cancer. International Journal of Molecular Sciences. 2023; 24(5):4646. https://doi.org/10.3390/ijms24054646
Chicago/Turabian StyleWang, Longkun, Chunqian Zhao, Lu Lu, Honglei Jiang, Fengshan Wang, and Xinke Zhang. 2023. "Transcytosable Peptide-Paclitaxel Prodrug Nanoparticle for Targeted Treatment of Triple-Negative Breast Cancer" International Journal of Molecular Sciences 24, no. 5: 4646. https://doi.org/10.3390/ijms24054646