
Response and Resistance to Trametinib in MAP2K1-Mutant Triple-Negative Melanoma

 , , and
, , and
Abstract
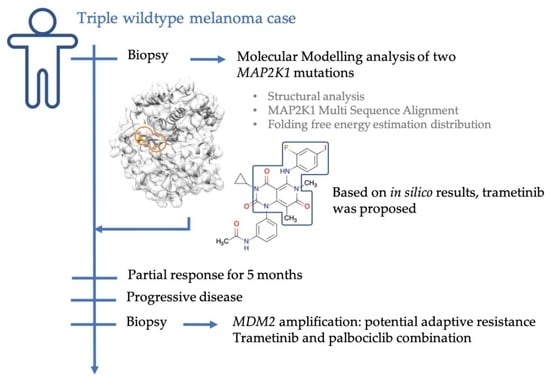
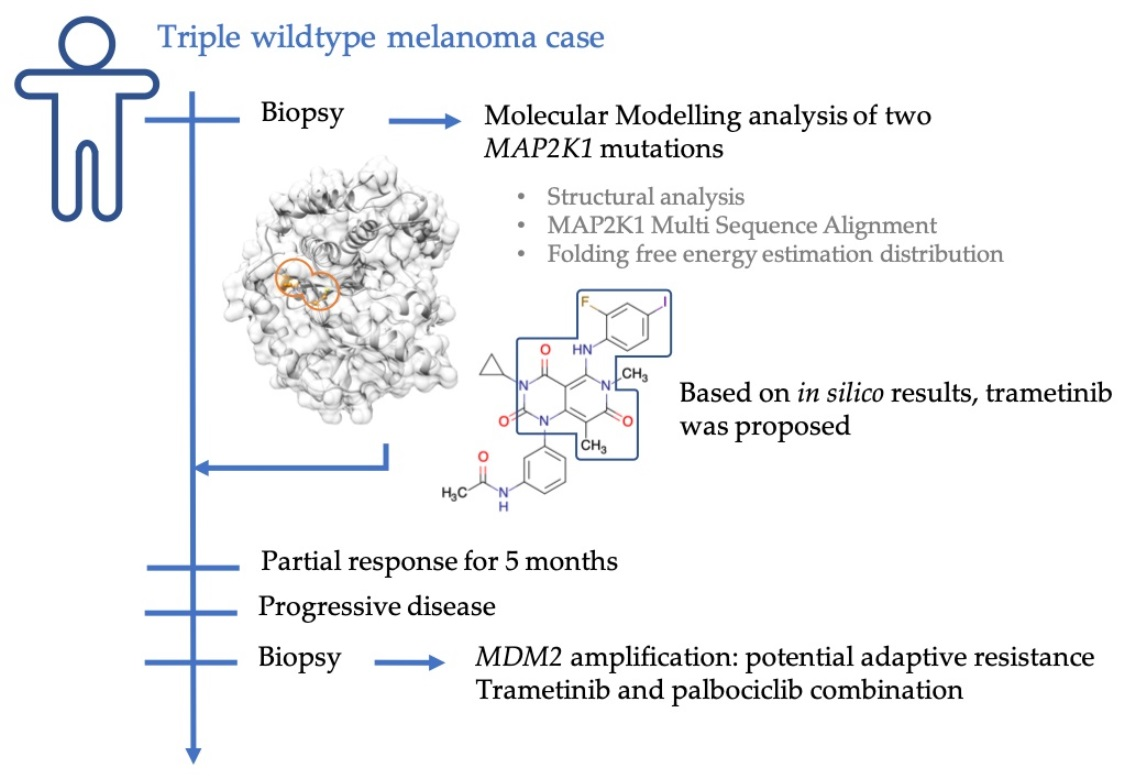
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
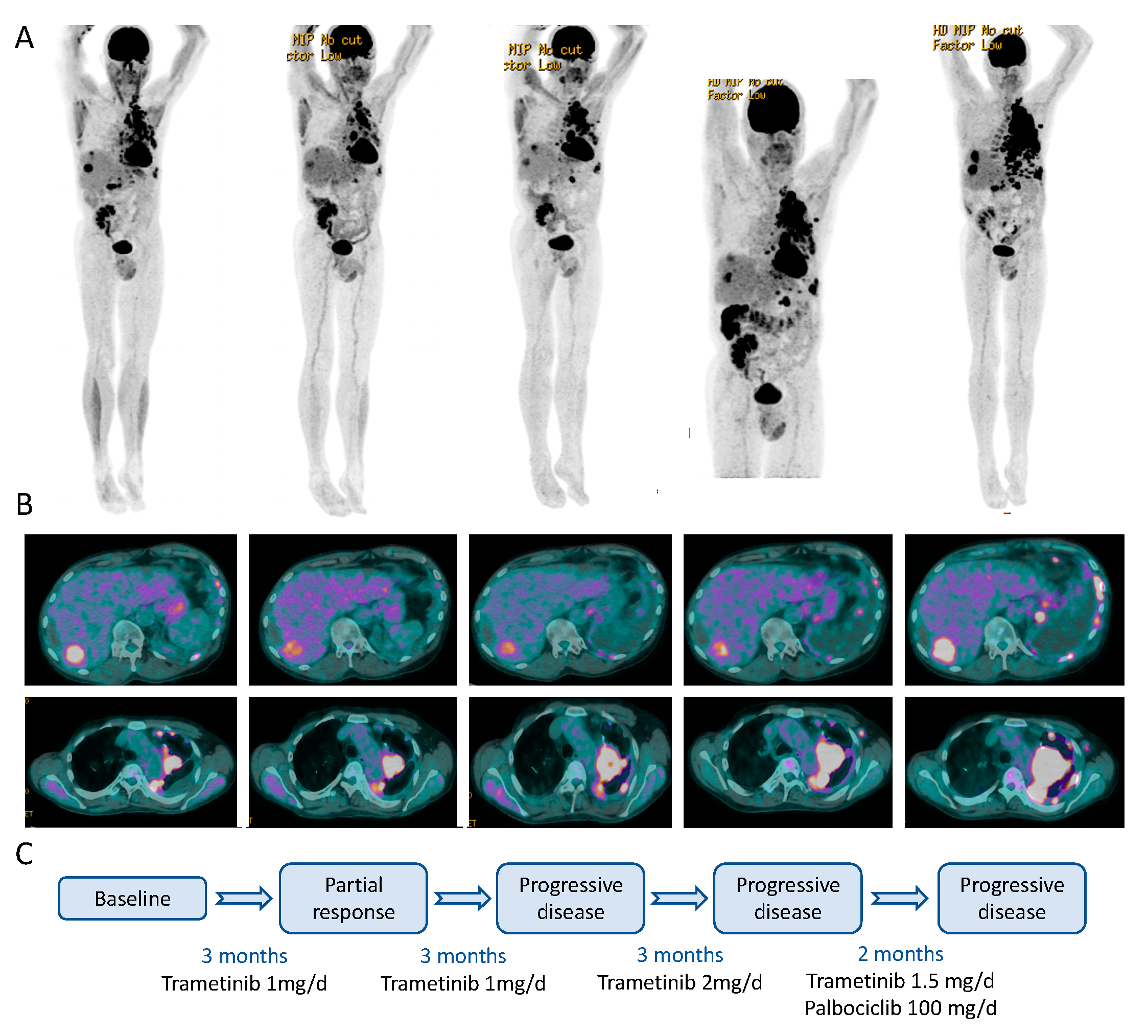
2.1. Case Description
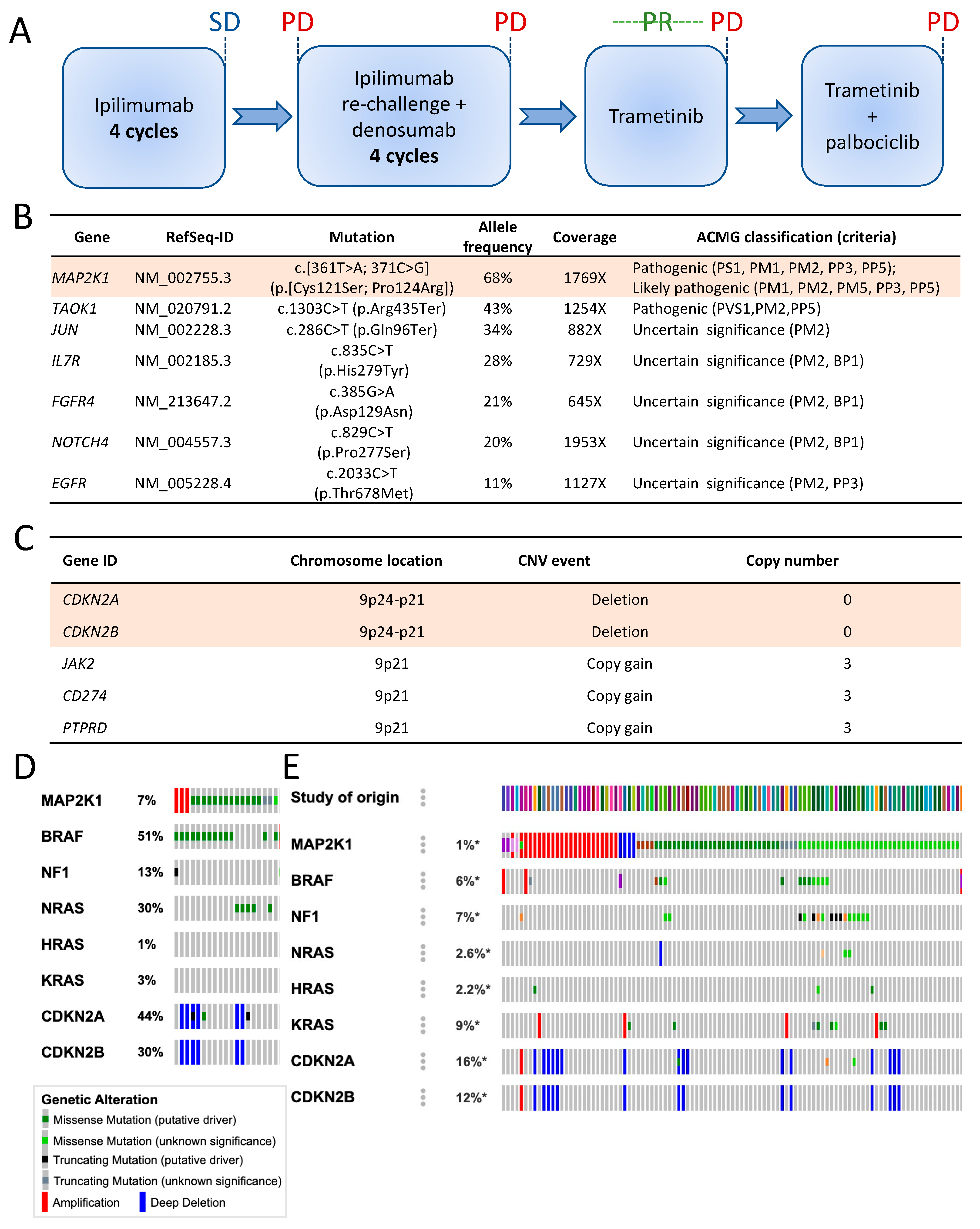
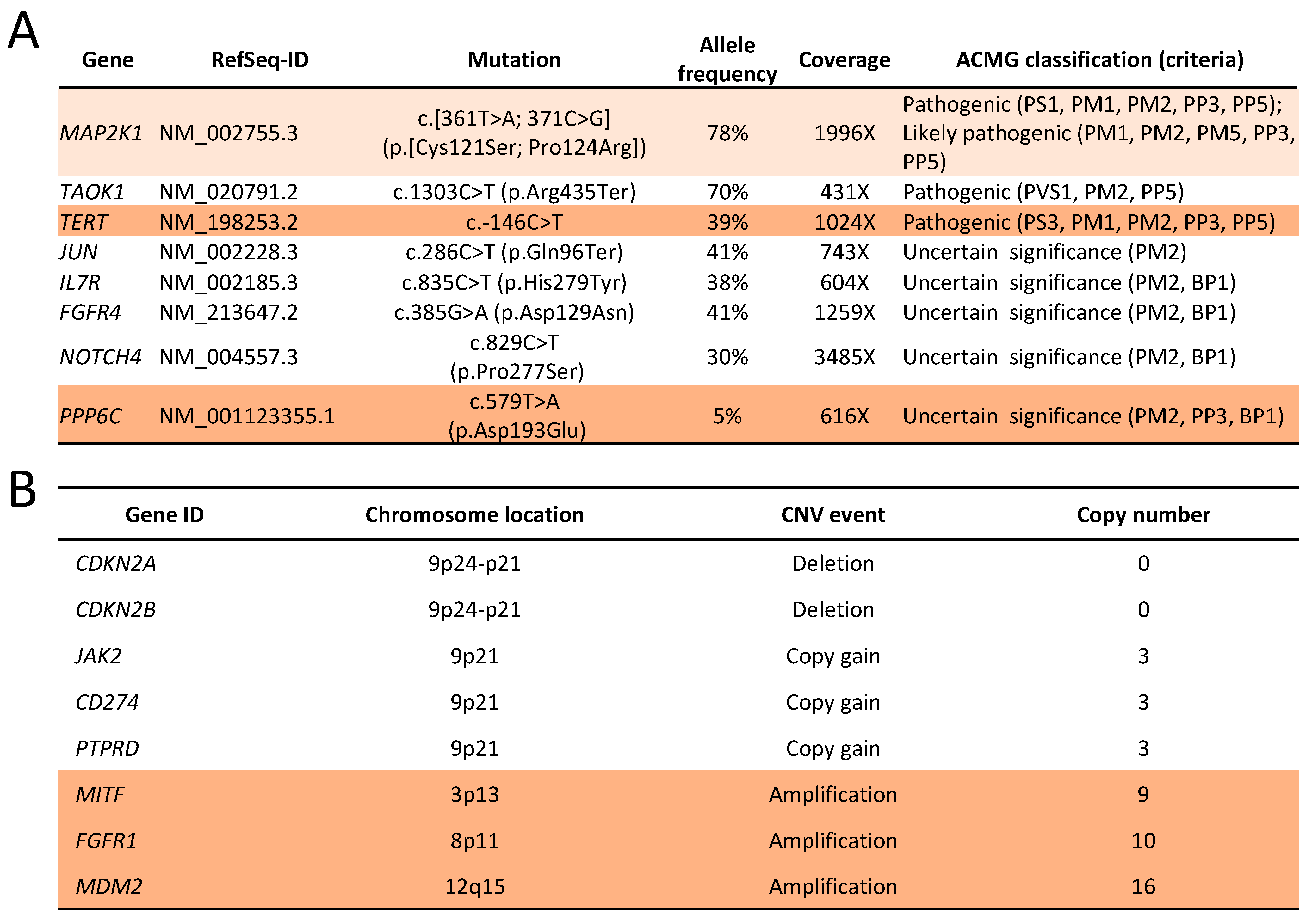
2.2. Genomic Analysis
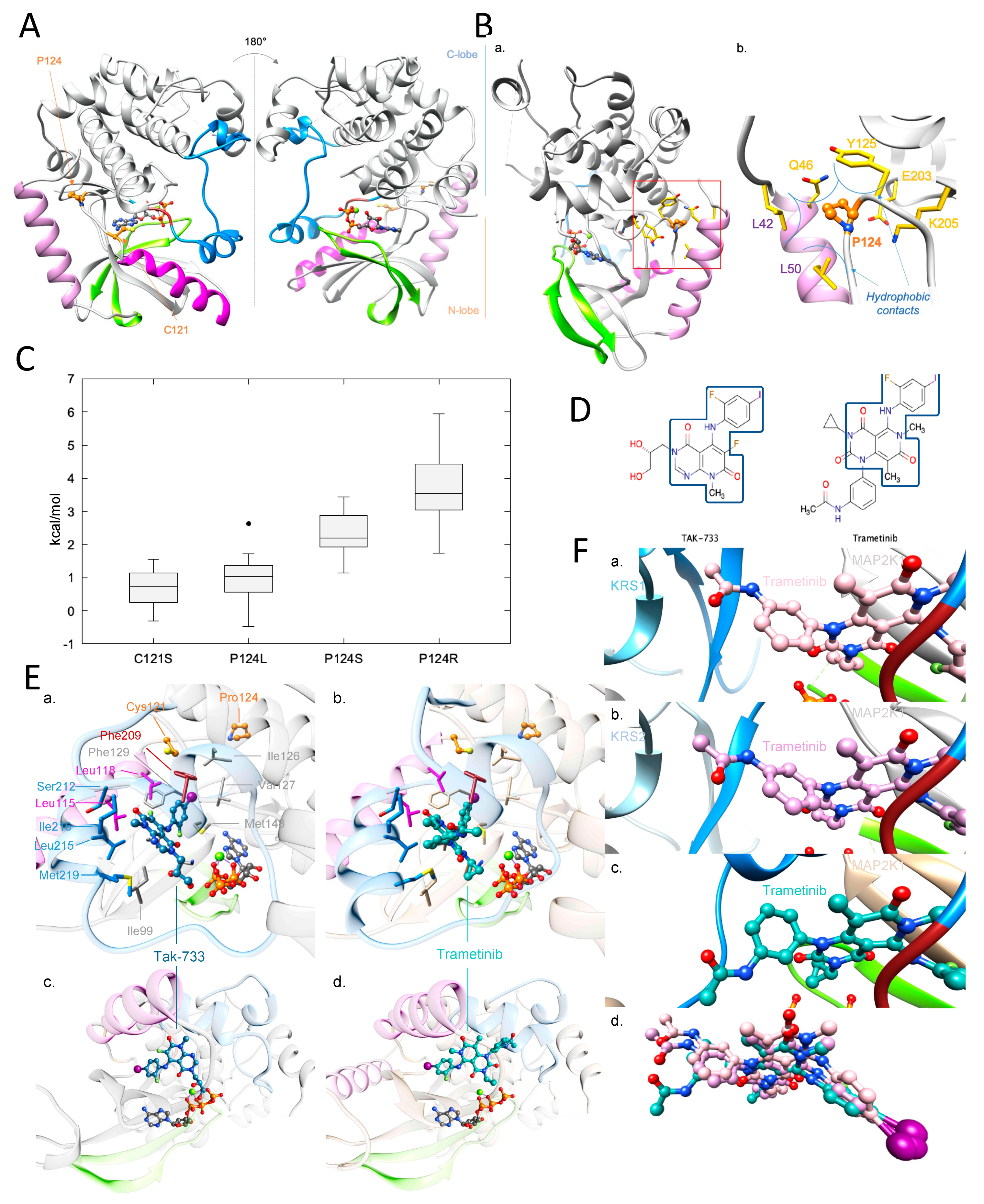
2.3. Molecular Modelling Analysis of MAP2K1 Mutations
2.4. Investigation of a Potential MAP2K1 Inhibitor and Mutations
2.5. Clinical Course
2.6. Genomic Changes in Response to MEK Inhibition
3. Discussion
4. Materials and Methods
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sullivan, R.J.; Flaherty, K. MAP kinase signaling and inhibition in melanoma. Oncogene 2013, 32, 2373–2379. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davies, H.; Bignell, G.R.; Cox, C.; Stephens, P.; Edkins, S.; Clegg, S.; Teague, J.; Woffendin, H.; Garnett, M.J.; Bottomley, W.; et al. Mutations of the BRAF gene in human cancer. Nature 2002, 417, 949–954. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robert, C.; Karaszewska, B.; Schachter, J.; Rutkowski, P.; Mackiewicz, A.; Stroiakovski, D.; Lichinitser, M.; Dummer, R.; Grange, F.; Mortier, L.; et al. Improved overall survival in melanoma with combined dabrafenib and trametinib. N. Engl. J. Med. 2015, 372, 30–39. [Google Scholar] [CrossRef] [Green Version]
- Akbani, R.; Akdemir, K.C.; Aksoy, B.A.; Albert, M.; Ally, A.; Amin, S.B.; Arachchi, H.; Arora, A.; Auman, J.T.; Ayala, B.; et al. Genomic Classification of Cutaneous Melanoma. Cell 2015, 161, 1681–1696. [Google Scholar] [CrossRef] [Green Version]
- Nissan, M.H.; Pratilas, C.A.; Jones, A.M.; Ramirez, R.; Won, H.; Liu, C.; Tiwari, S.; Kong, L.; Hanrahan, A.J.; Yao, Z.; et al. Loss of NF1 in Cutaneous Melanoma Is Associated with RAS Activation and MEK Dependence. Cancer Res. 2014, 74, 2340–2350. [Google Scholar] [CrossRef] [Green Version]
- Dummer, R.; Schadendorf, D.; Ascierto, P.A.; Arance, A.; Dutriaux, C.; Di Giacomo, A.M.; Rutkowski, P.; Del Vecchio, M.; Gutzmer, R.; Mandala, M.; et al. Binimetinib versus dacarbazine in patients with advanced NRAS-mutant melanoma (NEMO): A multicentre, open-label, randomised, phase 3 trial. Lancet Oncol. 2017, 18, 435–445. [Google Scholar] [CrossRef]
- Wagle, N.; Emery, C.; Berger, M.F.; Davis, M.J.; Sawyer, A.; Pochanard, P.; Kehoe, S.M.; Johannessen, C.M.; MacConaill, L.E.; Hahn, W.C.; et al. Dissecting Therapeutic Resistance to RAF Inhibition in Melanoma by Tumor Genomic Profiling. J. Clin. Oncol. 2011, 29, 3085–3096. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bard, J.-P.; Caruba, C. High prevalence of somatic MAP2K1 mutations in BRAF V600E–negative Langerhans cell histiocytosis. Comptes. Rendus l’Académie Des. Sci. 1982, 294, 103–106. [Google Scholar] [CrossRef]
- Kinoshita-Kikuta, E.; Kinoshita, E.; Ueda, S.; Ino, Y.; Kimura, Y.; Hirano, H.; Koike, T. Increase in constitutively active MEK1 species by introduction of MEK1 mutations identified in cancers. Biochim. Biophys. Acta-Proteins Proteom. 2019, 1867, 62–70. [Google Scholar] [CrossRef] [PubMed]
- Nelson, D.S.; van Halteren, A.; Quispel, W.T.; van den Bos, C.; Bovée, J.V. MAP2K1 and MAP3K1 Mutations in Langerhans Cell Histiocytosis. Genes Chromosomes Cancer 2015, 54, 361–368. [Google Scholar] [CrossRef]
- Wu, P.-K.; Park, J.-I. MEK1/2 Inhibitors: Molecular Activity and Resistance Mechanisms. Semin. Oncol. 2015, 42, 849–862. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Emery, C.M.; Vijayendran, K.G.; Zipser, M.C.; Sawyer, A.M.; Niu, L.; Kim, J.J.; Hatton, C.; Chopra, R.; Oberholzer, P.A.; Karpova, M.B.; et al. MEK1 mutations confer resistance to MEK and B-RAF inhibition. Proc. Natl. Acad. Sci. USA 2009, 106, 20411–20416. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Narita, Y.; Okamoto, K.; Kawada, M.I.; Takase, K.; Minoshima, Y.; Kodama, K.; Iwata, M.; Miyamoto, N.; Sawada, K. Novel ATP-Competitive MEK Inhibitor E6201 Is Effective against Vemurafenib-Resistant Melanoma Harboring the MEK1-C121S Mutation in a Preclinical Model. Mol. Cancer Ther. 2014, 13, 823–832. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mansour, S.J.; Candia, J.M.; Matsuura, J.E.; Manning, M.C.; Ahn, N.G. Interdependent Domains Controlling the Enzymatic Activity of Mitogen-Activated Protein Kinase Kinase 1. Biochemistry 1996, 35, 15529–15536. [Google Scholar] [CrossRef] [PubMed]
- Modi, V.; Dunbrack, R.L. Defining a new nomenclature for the structures of active and inactive kinases. Proc. Natl. Acad. Sci. USA 2019, 116, 6818–6827. [Google Scholar] [CrossRef] [Green Version]
- Möbitz, H. The ABC of protein kinase conformations. Biochim. Biophys. Acta-Proteins Proteom. 2015, 1854, 1555–1566. [Google Scholar] [CrossRef]
- Fischmann, T.O.; Smith, C.K.; Mayhood, T.W.; Myers, J.J.E.; Reichert, P.; Mannarino, A.; Carr, D.; Zhu, H.; Wong, J.; Yang, R.-S.; et al. Crystal Structures of MEK1 Binary and Ternary Complexes with Nucleotides and Inhibitors. Biochemistry 2009, 48, 2661–2674. [Google Scholar] [CrossRef]
- Dong, Q.; Dougan, D.R.; Gong, X.; Halkowycz, P.; Jin, B.; Kanouni, T.; O’Connell, S.M.; Scorah, N.; Shi, L.; Wallace, M.B.; et al. Discovery of TAK-733, a potent and selective MEK allosteric site inhibitor for the treatment of cancer. Bioorg. Med. Chem. Lett. 2011, 21, 1315–1319. [Google Scholar] [CrossRef]
- Khan, Z.M.; Real, A.M.; Marsiglia, W.M.; Chow, A.; Duffy, M.E.; Yerabolu, J.R.; Scopton, A.P.; Dar, A.C. Structural basis for the action of the drug trametinib at KSR-bound MEK. Nature 2020, 588, 509–514. [Google Scholar] [CrossRef]
- Gao, Y.; Chang, M.T.; McKay, D.; Na, N.; Zhou, B.; Yaeger, R.D.; Torres, N.M.; Muniz, K.; Drosten, M.; Barbacid, M.; et al. Allele-Specific Mechanisms of Activation of MEK1 Mutants Determine Their Properties. Cancer Discov. 2018, 8, 648–661. [Google Scholar] [CrossRef] [Green Version]
- Nikolaev, S.I.; Rimoldi, D.; Iseli, C.; Valsesia, A.; Robyr, D.; Gehrig, C.; Harshman, K.; Guipponi, M.; Bukach, O.; Zoete, V.; et al. Exome sequencing identifies recurrent somatic MAP2K1 and MAP2K2 mutations in melanoma. Nat. Genet. 2012, 44, 133–139. [Google Scholar] [CrossRef] [PubMed]
- Shi, H.; Moriceau, G.; Kong, X.; Koya, R.C.; Nazarian, R.; Pupo, G.M.; Bacchiocchi, A.; Dahlman, K.B.; Chmielowski, B.; Sosman, J.A.; et al. Preexisting MEK1 Exon 3 Mutations in V600E/K BRAF Melanomas Do Not Confer Resistance to BRAF Inhibitors. Cancer Discov. 2012, 2, 414–424. [Google Scholar] [CrossRef] [Green Version]
- Delgado, J.; Radusky, L.G.; Cianferoni, D.; Serrano, L. FoldX 5.0: Working with RNA, small molecules and a new graphical interface. Bioinformatics 2019, 35, 4168–4169. [Google Scholar] [CrossRef] [Green Version]
- Hanrahan, A.J.; Sylvester, B.E.; Chang, M.T.; Elzein, A.; Gao, J.; Han, W.; Liu, Y.; Xu, D.; Gao, S.P.; Gorelick, A.N.; et al. Leveraging Systematic Functional Analysis to Benchmark an In Silico Framework Distinguishes Driver from Passenger MEK Mutants in Cancer. Cancer Res. 2020, 80, 4233–4243. [Google Scholar] [CrossRef] [PubMed]
- Koukos, P.I.; Réau, M.; Bonvin, A.M.J.J. Shape-Restrained Modeling of Protein–Small-Molecule Complexes with High Ambiguity Driven DOCKing. J. Chem. Inf. Model. 2021, 61, 4807–4818. [Google Scholar] [CrossRef]
- Alayed, K.; Medeiros, L.J.; Patel, K.P.; Zuo, Z.; Li, S.; Verma, S.; Galbincea, J.; Cason, R.C.; Luthra, R.; Yin, C.C. BRAF and MAP2K1 mutations in Langerhans cell histiocytosis: A study of 50 cases. Hum. Pathol. 2016, 52, 61–67. [Google Scholar] [CrossRef] [PubMed]
- Diamond, E.L.; Durham, B.H.; Ulaner, G.A.; Drill, E.; Buthorn, J.; Ki, M.; Bitner, L.; Cho, H.; Young, R.J.; Francis, J.H.; et al. Efficacy of MEK inhibition in patients with histiocytic neoplasms. Nature 2019, 567, 521–524. [Google Scholar] [CrossRef]
- Van Allen, E.M.; Wagle, N.; Sucker, A.; Treacy, D.J.; Johannessen, C.M.; Goetz, E.M.; Place, C.S.; Taylor-Weiner, A.; Whittaker, S.; Kryukov, G.V.; et al. The Genetic Landscape of Clinical Resistance to RAF Inhibition in Metastatic Melanoma. Cancer Discov. 2014, 4, 94–109. [Google Scholar] [CrossRef] [Green Version]
- Finn, R.S.; Martin, M.; Rugo, H.S.; Jones, S.; Im, S.-A.; Gelmon, K.; Harbeck, N.; Lipatov, O.N.; Walshe, J.M.; Moulder, S.; et al. Palbociclib and Letrozole in Advanced Breast Cancer. N. Engl. J. Med. 2016, 375, 1925–1936. [Google Scholar] [CrossRef]
- Asghar, U.S.; Kanani, R.; Roylance, R.; Mittnacht, S. Systematic Review of Molecular Biomarkers Predictive of Resistance to CDK4/6 Inhibition in Metastatic Breast Cancer. JCO Precis. Oncol. 2022, in press. [Google Scholar] [CrossRef]
- Clark, A.S.; Hong, F.; Finn, R.S.; DeMichele, A.M.; Mitchell, E.P.; Zweibel, J.; Arnaldez, F.I.; McShane, L.M.; Li, S.; Gray, R.J.; et al. Abstract LB-010: Molecular analysis for therapy choice (NCI-MATCH, EAY131) arm Z1B: Phase II trial of palbociclib for CCND1, 2 or 3 amplified tumors. Cancer Res. 2019, 79, LB-010. [Google Scholar] [CrossRef]
- Nassar, K.W.; Hintzsche, J.D.; Bagby, S.M.; Espinoza, V.; Langouët-Astrié, C.; Amato, C.M.; Chimed, T.-S.; Fujita, M.; Robinson, W.; Tan, A.C.; et al. Targeting CDK4/6 Represents a Therapeutic Vulnerability in Acquired BRAF/MEK Inhibitor–Resistant Melanoma. Mol. Cancer Ther. 2021, 20, 2049–2060. [Google Scholar] [CrossRef] [PubMed]
- Razak, A.R.A.; Bauer, S.; Suarez, C.; Lin, C.-C.; Quek, R.; Hütter-Krönke, M.L.; Cubedo, R.; Ferretti, S.; Guerreiro, N.; Jullion, A.; et al. Co-Targeting of MDM2 and CDK4/6 with Siremadlin and Ribociclib for the Treatment of Patients with Well-Differentiated or Dedifferentiated Liposarcoma: Results from a Proof-of-Concept, Phase Ib Study. Clin. Cancer Res. 2022, 28, 1087–1097. [Google Scholar] [CrossRef] [PubMed]
- Czyz, M. Fibroblast Growth Factor Receptor Signaling in Skin Cancers. Cells 2019, 8, 540. [Google Scholar] [CrossRef] [Green Version]
- Chen, G.J.; Weylie, B.; Hu, C.; Zhu, J.; Forough, R. FGFR1/PI3K/AKT signaling pathway is a novel target for antiangiogenic effects of the cancer drug Fumagillin (TNP-470). J. Cell. Biochem. 2007, 101, 1492–1504. [Google Scholar] [CrossRef]
- Tran, K.B.; Kolekar, S.; Jabed, A.; Jaynes, P.; Shih, J.-H.; Wang, Q.; Flanagan, J.U.; Rewcastle, G.W.; Baguley, B.C.; Shepherd, P.R. Diverse mechanisms activate the PI 3-kinase/mTOR pathway in melanomas: Implications for the use of PI 3-kinase inhibitors to overcome resistance to inhibitors of BRAF and MEK. BMC Cancer 2021, 21, 136. [Google Scholar] [CrossRef]
- UniProt Consortium. UniProt: The universal protein knowledgebase in 2021. Nucleic Acids Res. 2021, 49, D480–D489. [Google Scholar] [CrossRef]
- Edgar, R.C. MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef] [Green Version]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [Green Version]
- Moorhouse, M.; Barry, P. The Protein Databank. In Bioinformatics Biocomputing and Perl; John Wiley & Sons: Hoboken, NJ, USA, 2005. [Google Scholar] [CrossRef]
- Amaning, K.; Lowinski, M.; Vallee, F.; Steier, V.; Marcireau, C.; Ugolini, A.; Delorme, C.; Foucalt, F.; McCort, G.; Derimay, N.; et al. The use of virtual screening and differential scanning fluorimetry for the rapid identification of fragments active against MEK1. Bioorg. Med. Chem. Lett. 2013, 23, 3620–3626. [Google Scholar] [CrossRef]
- Linke, P.; Amaning, K.; Maschberger, M.; Vallee, F.; Steier, V.; Baaske, P.; Duhr, S.; Breitsprecher, D.; Rak, A. An Automated Microscale Thermophoresis Screening Approach for Fragment-Based Lead Discovery. J. Biomol. Screen. 2016, 21, 414–421. [Google Scholar] [CrossRef] [Green Version]
- Sini, P.; Gürtler, U.; Zahn, S.K.; Baumann, C.; Rudolph, D.; Baumgartinger, R.; Strauss, E.; Haslinger, C.; Tontsch-Grunt, U.; Waizenegger, I.C.; et al. Pharmacological Profile of BI 847325, an Orally Bioavailable, ATP-Competitive Inhibitor of MEK and Aurora Kinases. Mol. Cancer Ther. 2016, 15, 2388–2398. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meier, C.; Brookings, D.C.; Ceska, T.A.; Doyle, C.; Gong, H.; McMillan, D.; Saville, G.P.; Mushtaq, A.; Knight, D.; Reich, S.; et al. Engineering human MEK-1 for structural studies: A case study of combinatorial domain hunting. J. Struct. Biol. 2012, 177, 329–334. [Google Scholar] [CrossRef] [PubMed]
- Liau, N.P.D.; Wendorff, T.J.; Quinn, J.G.; Steffek, M.; Phung, W.; Liu, P.; Tang, J.; Irudayanathan, F.J.; Izadi, S.; Shaw, A.S.; et al. Negative regulation of RAF kinase activity by ATP is overcome by 14-3-3-induced dimerization. Nat. Struct. Mol. Biol. 2020, 27, 134–141. [Google Scholar] [CrossRef] [PubMed]
- O’Boyle, N.M.; Banck, M.; James, C.A.; Morley, C.; Vandermeersch, T.; Hutchison, G.R. Open Babel: An open chemical toolbox. J. Cheminform. 2011, 3, 33. [Google Scholar] [CrossRef] [Green Version]
- Zoete, V.; Schuepbach, T.; Bovigny, C.; Chaskar, P.; Daina, A.; Röhrig, U.F.; Michielin, O. Attracting cavities for docking. Replacing the rough energy landscape of the protein by a smooth attracting landscape. J. Comput. Chem. 2016, 37, 437–447. [Google Scholar] [CrossRef] [Green Version]
- Brooks, B.R.; Brooks, C.L., 3rd; Mackerell, A.D., Jr.; Nilsson, L.; Petrella, R.J.; Roux, B.; Won, Y.; Archontis, G.; Bartels, C.; Boresch, S.; et al. CHARMM: The biomolecular simulation program. J. Comput. Chem. 2009, 30, 1545–1614. [Google Scholar] [CrossRef] [Green Version]
- Huang, J.; Mackerell, A.D. CHARMM36 All-Atom Additive Protein Force Field: Validation Based on Comparison to NMR Data. J. Comput. Chem. 2013, 34, 2135–2145. [Google Scholar] [CrossRef] [Green Version]
- Zoete, V.; Cuendet, M.A.; Grosdidier, A.; Michielin, O. SwissParam: A fast force field generation tool for small organic molecules. J. Comput. Chem. 2011, 32, 2359–2368. [Google Scholar] [CrossRef]
- Lee, M.S.; Feig, M.; Salsbury, F.R., Jr.; Brooks, C.L., 3rd. New analytic approximation to the standard molecular volume definition and its application to generalized Born calculations. J. Comput. Chem. 2003, 24, 1348–1356, Erratum in J. Comput. Chem. 2003, 24, 1821. [Google Scholar] [CrossRef]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Krebs, F.S.; Moura, B.; Missiaglia, E.; Aedo-Lopez, V.; Michielin, O.; Tsantoulis, P.; Bisig, B.; Trimech, M.; Zoete, V.; Homicsko, K. Response and Resistance to Trametinib in MAP2K1-Mutant Triple-Negative Melanoma. Int. J. Mol. Sci. 2023, 24, 4520. https://doi.org/10.3390/ijms24054520
Krebs FS, Moura B, Missiaglia E, Aedo-Lopez V, Michielin O, Tsantoulis P, Bisig B, Trimech M, Zoete V, Homicsko K. Response and Resistance to Trametinib in MAP2K1-Mutant Triple-Negative Melanoma. International Journal of Molecular Sciences. 2023; 24(5):4520. https://doi.org/10.3390/ijms24054520
Chicago/Turabian StyleKrebs, Fanny Seraphine, Bianca Moura, Edoardo Missiaglia, Veronica Aedo-Lopez, Olivier Michielin, Petros Tsantoulis, Bettina Bisig, Mounir Trimech, Vincent Zoete, and Krisztian Homicsko. 2023. "Response and Resistance to Trametinib in MAP2K1-Mutant Triple-Negative Melanoma" International Journal of Molecular Sciences 24, no. 5: 4520. https://doi.org/10.3390/ijms24054520