
Molecular Mechanisms and Therapeutic Implications of Endothelial Dysfunction in Patients with Heart Failure
 ,
,  , , , , ,
, , , , ,
Abstract
:1. Introduction
2. Etiopathogenesis and Molecular Mechanisms of Heart Failure
2.1. Neurohormonal System Activation
2.2. Oxidative Stress
2.3. Calcium Regulation
2.4. Impaired Metabolism and Mitochondrial Energetics
2.5. Inflammation
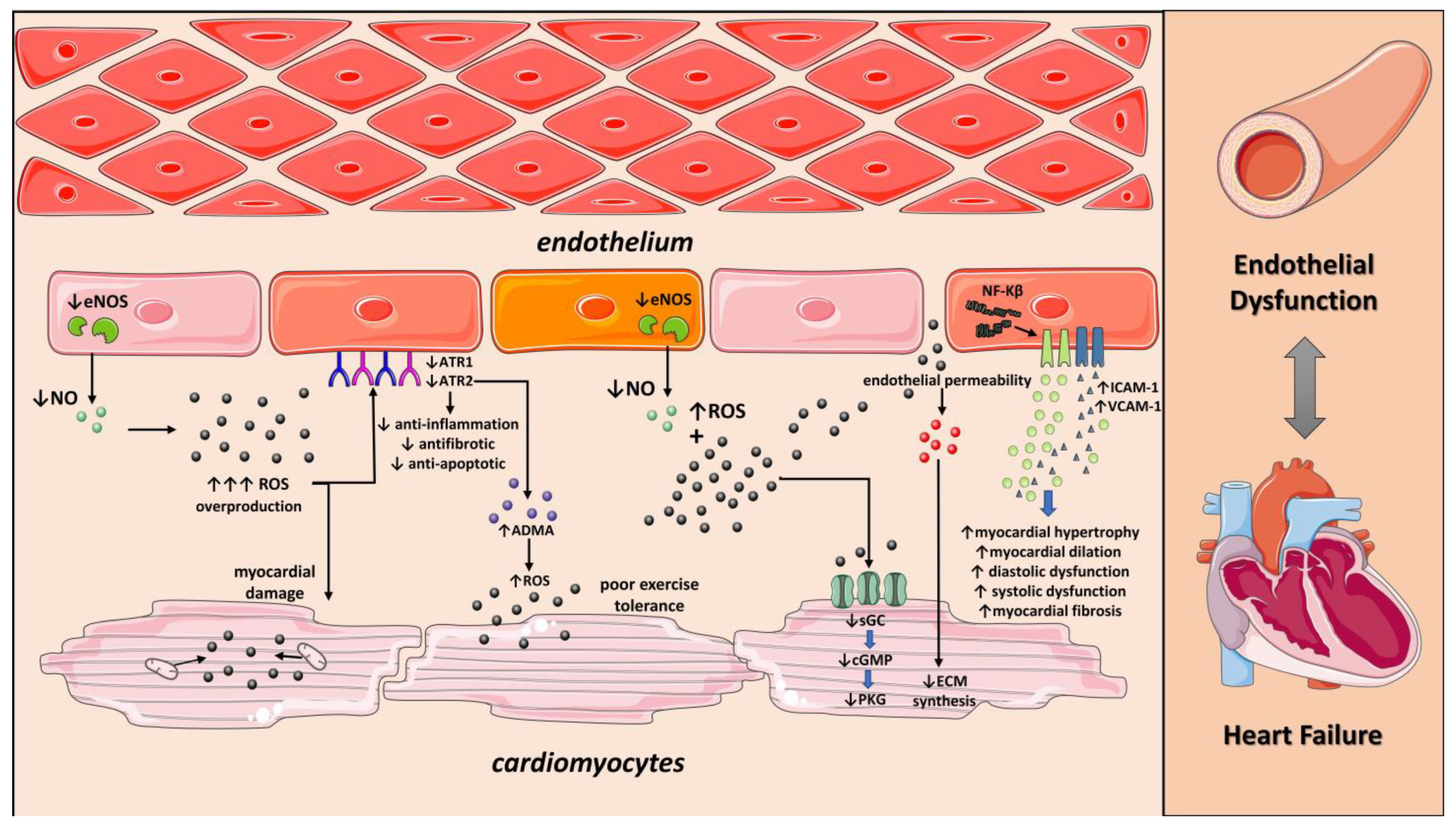
3. The Endothelium in Heart Failure
3.1. Endothelial Function in Patients with Heart Failure
3.2. Molecular Mechanisms of Endothelial Dysfunction in Patients with Heart Failure
3.2.1. Heart Failure with Reduced Ejection Fraction
3.2.2. Heart Failure with Preserved Ejection Fraction
3.2.3. Heart Failure of Ischemic Etiology
3.2.4. Non-Ischemic Heart Failure
3.2.5. Right Heart Failure Complicating Pulmonary Arterial Hypertension
3.3. Endothelial Function in the Course of Heart Failure
4. Modification of Endothelial Dysfunction in Patients with Heart Failure
4.1. Effects of Exercise Training
4.2. Drugs for the Treatment of Endothelial Dysfunction in Patients with Heart Failure
4.2.1. Statins
4.2.2. Beta Blockers
4.2.3. ACE Inhibitors/ARBs
4.2.4. Mineralocorticoid Receptor Antagonists (MRA)
4.2.5. SGLT2 Inhibitors
4.2.6. Sacubitril-Valsartan (ARNIs)
4.2.7. Endothelin Receptor Antagonists (ERAs)
5. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Bozkurt, B.; Coats, A.J.; Tsutsui, H.; Abdelhamid, M.; Adamopoulos, S.; Albert, N.; Anker, S.D.; Atherton, J.; Bohm, M.; Butler, J.; et al. Universal Definition and Classification of Heart Failure: A Report of the Heart Failure Society of America, Heart Failure Association of the European Society of Cardiology, Japanese Heart Failure Society and Writing Committee of the Universal Definition of Heart Failure. J. Card. Fail. 2021, 27, 387–413. [Google Scholar] [CrossRef]
- Becher, P.M.; Lund, L.H.; Coats, A.J.S.; Savarese, G. An update on global epidemiology in heart failure. Eur. Heart J. 2022, 43, 3005–3007. [Google Scholar] [CrossRef] [PubMed]
- Ho, J.E.; Lyass, A.; Lee, D.S.; Vasan, R.S.; Kannel, W.B.; Larson, M.G.; Levy, D. Predictors of new-onset heart failure: Differences in preserved versus reduced ejection fraction. Circ. Heart Fail. 2013, 6, 279–286. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paulus, W.J.; Tschope, C. A novel paradigm for heart failure with preserved ejection fraction: Comorbidities drive myocardial dysfunction and remodeling through coronary microvascular endothelial inflammation. J. Am. Coll Cardiol. 2013, 62, 263–271. [Google Scholar] [CrossRef] [Green Version]
- Taqueti, V.R.; Solomon, S.D.; Shah, A.M.; Desai, A.S.; Groarke, J.D.; Osborne, M.T.; Hainer, J.; Bibbo, C.F.; Dorbala, S.; Blankstein, R.; et al. Coronary microvascular dysfunction and future risk of heart failure with preserved ejection fraction. Eur. Heart J. 2018, 39, 840–849. [Google Scholar] [CrossRef]
- Daiber, A.; Xia, N.; Steven, S.; Oelze, M.; Hanf, A.; Kroller-Schon, S.; Munzel, T.; Li, H. New Therapeutic Implications of Endothelial Nitric Oxide Synthase (eNOS) Function/Dysfunction in Cardiovascular Disease. Int. J. Mol. Sci. 2019, 20, 187. [Google Scholar] [CrossRef] [Green Version]
- Breitenstein, S.; Roessig, L.; Sandner, P.; Lewis, K.S. Novel sGC Stimulators and sGC Activators for the Treatment of Heart Failure. Handb. Exp. Pharmacol. 2017, 243, 225–247. [Google Scholar] [CrossRef]
- Cornuault, L.; Rouault, P.; Duplaa, C.; Couffinhal, T.; Renault, M.A. Endothelial Dysfunction in Heart Failure With Preserved Ejection Fraction: What are the Experimental Proofs? Front. Physiol. 2022, 13, 906272. [Google Scholar] [CrossRef]
- Lee, D.S.; Gona, P.; Vasan, R.S.; Larson, M.G.; Benjamin, E.J.; Wang, T.J.; Tu, J.V.; Levy, D. Relation of disease pathogenesis and risk factors to heart failure with preserved or reduced ejection fraction: Insights from the framingham heart study of the national heart, lung, and blood institute. Circulation 2009, 119, 3070–3077. [Google Scholar] [CrossRef] [Green Version]
- Tota, B.; Cerra, M.C.; Gattuso, A. Catecholamines, cardiac natriuretic peptides and chromogranin A: Evolution and physiopathology of a ‘whip-brake’ system of the endocrine heart. J. Exp. Biol. 2010, 213, 3081–3103. [Google Scholar] [CrossRef] [Green Version]
- Krum, H. New and emerging pharmacological strategies in the management of chronic heart failure. Curr. Opin. Pharmacol. 2001, 1, 126–133. [Google Scholar] [CrossRef] [PubMed]
- Bonnefont-Rousselot, D.; Mahmoudi, A.; Mougenot, N.; Varoquaux, O.; Le Nahour, G.; Fouret, P.; Lechat, P. Catecholamine effects on cardiac remodelling, oxidative stress and fibrosis in experimental heart failure. Redox. Rep. 2002, 7, 145–151. [Google Scholar] [CrossRef] [PubMed]
- von Lueder, T.G.; Kotecha, D.; Atar, D.; Hopper, I. Neurohormonal Blockade in Heart Failure. Card. Fail. Rev. 2017, 3, 19–24. [Google Scholar] [CrossRef] [PubMed]
- Andreozzi, F.; Laratta, E.; Sciacqua, A.; Perticone, F.; Sesti, G. Angiotensin II impairs the insulin signaling pathway promoting production of nitric oxide by inducing phosphorylation of insulin receptor substrate-1 on Ser312 and Ser616 in human umbilical vein endothelial cells. Circ. Res. 2004, 94, 1211–1218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reina-Couto, M.; Afonso, J.; Carvalho, J.; Morgado, L.; Ronchi, F.A.; de Oliveira Leite, A.P.; Dias, C.C.; Casarini, D.E.; Bettencourt, P.; Albino-Teixeira, A.; et al. Interrelationship between renin-angiotensin-aldosterone system and oxidative stress in chronic heart failure patients with or without renal impairment. Biomed. Pharmacother. 2021, 133, 110938. [Google Scholar] [CrossRef]
- Kaschina, E.; Namsolleck, P.; Unger, T. AT2 receptors in cardiovascular and renal diseases. Pharmacol. Res. 2017, 125, 39–47. [Google Scholar] [CrossRef] [PubMed]
- Ide, T.; Tsutsui, H.; Kinugawa, S.; Utsumi, H.; Kang, D.; Hattori, N.; Uchida, K.; Arimura, K.; Egashira, K.; Takeshita, A. Mitochondrial electron transport complex I is a potential source of oxygen free radicals in the failing myocardium. Circ. Res. 1999, 85, 357–363. [Google Scholar] [CrossRef] [Green Version]
- Dubois-Deruy, E.; Peugnet, V.; Turkieh, A.; Pinet, F. Oxidative Stress in Cardiovascular Diseases. Antioxidants 2020, 9, 864. [Google Scholar] [CrossRef]
- Bugger, H.; Pfeil, K. Mitochondrial ROS in myocardial ischemia reperfusion and remodeling. Biochim. Biophys. Acta Mol. Basis Dis. 2020, 1866, 165768. [Google Scholar] [CrossRef]
- Ho, E.; Karimi Galougahi, K.; Liu, C.C.; Bhindi, R.; Figtree, G.A. Biological markers of oxidative stress: Applications to cardiovascular research and practice. Redox. Biol. 2013, 1, 483–491. [Google Scholar] [CrossRef] [Green Version]
- Cesselli, D.; Jakoniuk, I.; Barlucchi, L.; Beltrami, A.P.; Hintze, T.H.; Nadal-Ginard, B.; Kajstura, J.; Leri, A.; Anversa, P. Oxidative stress-mediated cardiac cell death is a major determinant of ventricular dysfunction and failure in dog dilated cardiomyopathy. Circ. Res. 2001, 89, 279–286. [Google Scholar] [CrossRef] [Green Version]
- Ide, T.; Tsutsui, H.; Hayashidani, S.; Kang, D.; Suematsu, N.; Nakamura, K.; Utsumi, H.; Hamasaki, N.; Takeshita, A. Mitochondrial DNA damage and dysfunction associated with oxidative stress in failing hearts after myocardial infarction. Circ. Res. 2001, 88, 529–535. [Google Scholar] [CrossRef] [Green Version]
- Kiyuna, L.A.; Albuquerque, R.P.E.; Chen, C.H.; Mochly-Rosen, D.; Ferreira, J.C.B. Targeting mitochondrial dysfunction and oxidative stress in heart failure: Challenges and opportunities. Free Radic. Biol. Med. 2018, 129, 155–168. [Google Scholar] [CrossRef] [PubMed]
- Piquereau, J.; Ventura-Clapier, R. Maturation of Cardiac Energy Metabolism During Perinatal Development. Front. Physiol. 2018, 9, 959. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miranda-Silva, D.; Lima, T.; Rodrigues, P.; Leite-Moreira, A.; Falcao-Pires, I. Mechanisms underlying the pathophysiology of heart failure with preserved ejection fraction: The tip of the iceberg. Heart Fail. Rev. 2021, 26, 453–478. [Google Scholar] [CrossRef] [PubMed]
- Triposkiadis, F.; Xanthopoulos, A.; Butler, J. Cardiovascular Aging and Heart Failure: JACC Review Topic of the Week. J. Am. Coll Cardiol. 2019, 74, 804–813. [Google Scholar] [CrossRef]
- Schroder, F.; Handrock, R.; Beuckelmann, D.J.; Hirt, S.; Hullin, R.; Priebe, L.; Schwinger, R.H.; Weil, J.; Herzig, S. Increased availability and open probability of single L-type calcium channels from failing compared with nonfailing human ventricle. Circulation 1998, 98, 969–976. [Google Scholar] [CrossRef] [Green Version]
- Gomes, K.P.; Jadli, A.S.; de Almeida, L.G.N.; Ballasy, N.N.; Edalat, P.; Shandilya, R.; Young, D.; Belke, D.; Shearer, J.; Dufour, A.; et al. Proteomic Analysis Suggests Altered Mitochondrial Metabolic Profile Associated With Diabetic Cardiomyopathy. Front. Cardiovasc. Med. 2022, 9, 791700. [Google Scholar] [CrossRef]
- Dashwood, A.; Cheesman, E.; Beard, N.; Haqqani, H.; Wong, Y.W.; Molenaar, P. Understanding How Phosphorylation and Redox Modifications Regulate Cardiac Ryanodine Receptor Type 2 Activity to Produce an Arrhythmogenic Phenotype in Advanced Heart Failure. ACS Pharmacol. Transl. Sci. 2020, 3, 563–582. [Google Scholar] [CrossRef]
- Rupee, S.; Rupee, K.; Singh, R.B.; Hanoman, C.; Ismail, A.M.A.; Smail, M.; Singh, J. Diabetes-induced chronic heart failure is due to defects in calcium transporting and regulatory contractile proteins: Cellular and molecular evidence. Heart Fail. Rev. 2022. [Google Scholar] [CrossRef]
- Sipido, K.R.; Volders, P.G.; Vos, M.A.; Verdonck, F. Altered Na/Ca exchange activity in cardiac hypertrophy and heart failure: A new target for therapy? Cardiovasc. Res. 2002, 53, 782–805. [Google Scholar] [CrossRef] [PubMed]
- Gwathmey, J.K.; Liao, R.; Ingwall, J.S. Comparison of twitch force and calcium handling in papillary muscles from right ventricular pressure overload hypertrophy in weanling and juvenile ferrets. Cardiovasc. Res. 1995, 29, 475–481. [Google Scholar] [CrossRef] [PubMed]
- Werner, C.; Baumhakel, M.; Teo, K.K.; Schmieder, R.; Mann, J.; Unger, T.; Yusuf, S.; Bohm, M. RAS blockade with ARB and ACE inhibitors: Current perspective on rationale and patient selection. Clin. Res. Cardiol. 2008, 97, 418–431. [Google Scholar] [CrossRef] [PubMed]
- Studer, R.; Reinecke, H.; Bilger, J.; Eschenhagen, T.; Bohm, M.; Hasenfuss, G.; Just, H.; Holtz, J.; Drexler, H. Gene expression of the cardiac Na(+)-Ca2+ exchanger in end-stage human heart failure. Circ. Res. 1994, 75, 443–453. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schwinger, R.H.; Munch, G.; Bolck, B.; Karczewski, P.; Krause, E.G.; Erdmann, E. Reduced Ca(2+)-sensitivity of SERCA 2a in failing human myocardium due to reduced serin-16 phospholamban phosphorylation. J. Mol. Cell Cardiol. 1999, 31, 479–491. [Google Scholar] [CrossRef]
- Liu, G.; Li, S.Q.; Hu, P.P.; Tong, X.Y. Altered sarco(endo)plasmic reticulum calcium adenosine triphosphatase 2a content: Targets for heart failure therapy. Diab. Vasc. Dis. Res. 2018, 15, 322–335. [Google Scholar] [CrossRef] [Green Version]
- Bertero, E.; Maack, C. Calcium Signaling and Reactive Oxygen Species in Mitochondria. Circ. Res. 2018, 122, 1460–1478. [Google Scholar] [CrossRef]
- Jiang, M.; Xie, X.; Cao, F.; Wang, Y. Mitochondrial Metabolism in Myocardial Remodeling and Mechanical Unloading: Implications for Ischemic Heart Disease. Front. Cardiovasc. Med. 2021, 8, 789267. [Google Scholar] [CrossRef]
- Sabbah, H.N. Targeting the Mitochondria in Heart Failure: A Translational Perspective. JACC Basic Transl. Sci. 2020, 5, 88–106. [Google Scholar] [CrossRef]
- Adaniya, S.M.; J, O.U.; Cypress, M.W.; Kusakari, Y.; Jhun, B.S. Posttranslational modifications of mitochondrial fission and fusion proteins in cardiac physiology and pathophysiology. Am. J. Physiol. Cell Physiol. 2019, 316, C583–C604. [Google Scholar] [CrossRef]
- Karamanlidis, G.; Nascimben, L.; Couper, G.S.; Shekar, P.S.; del Monte, F.; Tian, R. Defective DNA replication impairs mitochondrial biogenesis in human failing hearts. Circ Res 2010, 106, 1541–1548. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, H.; Yu, M.; Li, Z.; Zhang, E.; Ma, H. Identification and analysis of mitochondria-related key genes of heart failure. J. Transl. Med. 2022, 20, 410. [Google Scholar] [CrossRef]
- Van Linthout, S.; Tschope, C. Inflammation—Cause or Consequence of Heart Failure or Both? Curr. Heart Fail. Rep. 2017, 14, 251–265. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murphy, S.P.; Kakkar, R.; McCarthy, C.P.; Januzzi, J.L., Jr. Inflammation in Heart Failure: JACC State-of-the-Art Review. J. Am. Coll. Cardiol. 2020, 75, 1324–1340. [Google Scholar] [CrossRef] [PubMed]
- Sansonetti, M.; De Windt, L.J. Non-coding RNAs in cardiac inflammation: Key drivers in the pathophysiology of heart failure. Cardiovasc. Res. 2022, 118, 2058–2073. [Google Scholar] [CrossRef]
- Chen, G.Y.; Nunez, G. Sterile inflammation: Sensing and reacting to damage. Nat. Rev. Immunol. 2010, 10, 826–837. [Google Scholar] [CrossRef] [Green Version]
- Sekiguchi, K.; Li, X.; Coker, M.; Flesch, M.; Barger, P.M.; Sivasubramanian, N.; Mann, D.L. Cross-regulation between the renin-angiotensin system and inflammatory mediators in cardiac hypertrophy and failure. Cardiovasc. Res. 2004, 63, 433–442. [Google Scholar] [CrossRef] [Green Version]
- Mentz, R.J.; O’Connor, C.M. Pathophysiology and clinical evaluation of acute heart failure. Nat. Rev. Cardiol. 2016, 13, 28–35. [Google Scholar] [CrossRef]
- Harjola, V.P.; Mullens, W.; Banaszewski, M.; Bauersachs, J.; Brunner-La Rocca, H.P.; Chioncel, O.; Collins, S.P.; Doehner, W.; Filippatos, G.S.; Flammer, A.J.; et al. Organ dysfunction, injury and failure in acute heart failure: From pathophysiology to diagnosis and management. A review on behalf of the Acute Heart Failure Committee of the Heart Failure Association (HFA) of the European Society of Cardiology (ESC). Eur. J. Heart. Fail. 2017, 19, 821–836. [Google Scholar] [CrossRef] [Green Version]
- Levine, B.; Kalman, J.; Mayer, L.; Fillit, H.M.; Packer, M. Elevated circulating levels of tumor necrosis factor in severe chronic heart failure. N. Engl. J. Med. 1990, 323, 236–241. [Google Scholar] [CrossRef]
- Tromp, J.; Khan, M.A.; Klip, I.T.; Meyer, S.; de Boer, R.A.; Jaarsma, T.; Hillege, H.; van Veldhuisen, D.J.; van der Meer, P.; Voors, A.A. Biomarker Profiles in Heart Failure Patients With Preserved and Reduced Ejection Fraction. J. Am. Heart Assoc. 2017, 6, e003989. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, W.; Li, Y.; Jiang, F.; Liu, H. Correlation between serum levels of microRNA-21 and inflammatory factors in patients with chronic heart failure. Medicine (Baltimore) 2022, 101, e30596. [Google Scholar] [CrossRef] [PubMed]
- Kablak-Ziembicka, A.; Badacz, R.; Przewlocki, T. Clinical Application of Serum microRNAs in Atherosclerotic Coronary Artery Disease. J. Clin. Med. 2022, 11, 6849. [Google Scholar] [CrossRef] [PubMed]
- Nahrendorf, M.; Swirski, F.K. Innate immune cells in ischaemic heart disease: Does myocardial infarction beget myocardial infarction? Eur. Heart J. 2016, 37, 868–872. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Puhl, S.L.; Steffens, S. Neutrophils in Post-myocardial Infarction Inflammation: Damage vs. Resolution? Front. Cardiovasc. Med. 2019, 6, 25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oikonomou, E.; Tsaplaris, P.; Anastasiou, A.; Xenou, M.; Lampsas, S.; Siasos, G.; Pantelidis, P.; Theofilis, P.; Tsatsaragkou, A.; Katsarou, O.; et al. Interleukin-1 in Coronary Artery Disease. Curr. Top. Med. Chem. 2022, 22, 2368–2389. [Google Scholar] [CrossRef] [PubMed]
- Takimoto, E.; Kass, D.A. Role of oxidative stress in cardiac hypertrophy and remodeling. Hypertension 2007, 49, 241–248. [Google Scholar] [CrossRef] [PubMed]
- Tang, T.T.; Yuan, J.; Zhu, Z.F.; Zhang, W.C.; Xiao, H.; Xia, N.; Yan, X.X.; Nie, S.F.; Liu, J.; Zhou, S.F.; et al. Regulatory T cells ameliorate cardiac remodeling after myocardial infarction. Basic Res. Cardiol. 2012, 107, 232. [Google Scholar] [CrossRef]
- Zouggari, Y.; Ait-Oufella, H.; Bonnin, P.; Simon, T.; Sage, A.P.; Guerin, C.; Vilar, J.; Caligiuri, G.; Tsiantoulas, D.; Laurans, L.; et al. B lymphocytes trigger monocyte mobilization and impair heart function after acute myocardial infarction. Nat. Med. 2013, 19, 1273–1280. [Google Scholar] [CrossRef]
- Kourek, C.; Karatzanos, E.; Psarra, K.; Georgiopoulos, G.; Delis, D.; Linardatou, V.; Gavrielatos, G.; Papadopoulos, C.; Nanas, S.; Dimopoulos, S. Endothelial progenitor cells mobilization after maximal exercise according to heart failure severity. World J. Cardiol. 2020, 12, 526–539. [Google Scholar] [CrossRef]
- Kawai, C. From myocarditis to cardiomyopathy: Mechanisms of inflammation and cell death: Learning from the past for the future. Circulation 1999, 99, 1091–1100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuhl, U.; Pauschinger, M.; Seeberg, B.; Lassner, D.; Noutsias, M.; Poller, W.; Schultheiss, H.P. Viral persistence in the myocardium is associated with progressive cardiac dysfunction. Circulation 2005, 112, 1965–1970. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beydoun, N.; Feinstein, M.J. Heart Failure in Chronic Infectious and Inflammatory Conditions: Mechanistic Insights from Clinical Heterogeneity. Curr. Heart Fail. Rep. 2022, 19, 267–278. [Google Scholar] [CrossRef] [PubMed]
- Oikonomou, E.; Lampsas, S.; Theofilis, P.; Souvaliotis, N.; Papamikroulis, G.A.; Katsarou, O.; Kalogeras, K.; Pantelidis, P.; Papaioannou, T.G.; Tsatsaragkou, A.; et al. Impaired left ventricular deformation and ventricular-arterial coupling in post-COVID-19: Association with autonomic dysregulation. Heart Vessels 2023, 38, 381–393. [Google Scholar] [CrossRef]
- Rajendran, P.; Rengarajan, T.; Thangavel, J.; Nishigaki, Y.; Sakthisekaran, D.; Sethi, G.; Nishigaki, I. The vascular endothelium and human diseases. Int. J. Biol. Sci. 2013, 9, 1057–1069. [Google Scholar] [CrossRef] [Green Version]
- Forstermann, U.; Munzel, T. Endothelial nitric oxide synthase in vascular disease: From marvel to menace. Circulation 2006, 113, 1708–1714. [Google Scholar] [CrossRef] [Green Version]
- Premer, C.; Kanelidis, A.J.; Hare, J.M.; Schulman, I.H. Rethinking Endothelial Dysfunction as a Crucial Target in Fighting Heart Failure. Mayo Clin. Proc. Innov. Qual. Outcomes 2019, 3, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Marti, C.N.; Gheorghiade, M.; Kalogeropoulos, A.P.; Georgiopoulou, V.V.; Quyyumi, A.A.; Butler, J. Endothelial dysfunction, arterial stiffness, and heart failure. J. Am. Coll. Cardiol. 2012, 60, 1455–1469. [Google Scholar] [CrossRef] [Green Version]
- Giannitsi, S.; Bougiakli, M.; Bechlioulis, A.; Naka, K. Endothelial dysfunction and heart failure: A review of the existing bibliography with emphasis on flow mediated dilation. JRSM Cardiovasc. Dis. 2019, 8, 2048004019843047. [Google Scholar] [CrossRef]
- Colombo, P.C.; Doran, A.C.; Onat, D.; Wong, K.Y.; Ahmad, M.; Sabbah, H.N.; Demmer, R.T. Venous congestion, endothelial and neurohormonal activation in acute decompensated heart failure: Cause or effect? Curr. Heart Fail. Rep. 2015, 12, 215–222. [Google Scholar] [CrossRef] [Green Version]
- Al Houri, H.N.; Jomaa, S.; Jabra, M.; Alhouri, A.N.; Latifeh, Y. Pathophysiology of stress cardiomyopathy: A comprehensive literature review. Ann. Med. Surg. 2022, 82, 104671. [Google Scholar] [CrossRef]
- Chignalia, A.Z.; Isbatan, A.; Patel, M.; Ripper, R.; Sharlin, J.; Shosfy, J.; Borlaug, B.A.; Dull, R.O. Pressure-dependent NOS activation contributes to endothelial hyperpermeability in a model of acute heart failure. Biosci. Rep. 2018, 38, BSR20181239. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alem, M.M. Endothelial Dysfunction in Chronic Heart Failure: Assessment, Findings, Significance, and Potential Therapeutic Targets. Int. J. Mol. Sci. 2019, 20, 3198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oikonomou, E.; Souvaliotis, N.; Lampsas, S.; Siasos, G.; Poulakou, G.; Theofilis, P.; Papaioannou, T.G.; Haidich, A.B.; Tsaousi, G.; Ntousopoulos, V.; et al. Endothelial dysfunction in acute and long standing COVID-19: A prospective cohort study. Vascul. Pharmacol. 2022, 144, 106975. [Google Scholar] [CrossRef] [PubMed]
- Kaneko, M.; Matsumoto, Y.; Hayashi, H.; Kobayashi, A.; Yamazaki, N. Oxygen free radicals and calcium homeostasis in the heart. Mol. Cell Biochem. 1994, 139, 91–100. [Google Scholar] [CrossRef] [Green Version]
- Meerson, F.Z.; Kagan, V.E.; Kozlov Yu, P.; Belkina, L.M.; Arkhipenko Yu, V. The role of lipid peroxidation in pathogenesis of ischemic damage and the antioxidant protection of the heart. Basic Res. Cardiol. 1982, 77, 465–485. [Google Scholar] [CrossRef]
- Dai, M.; Wang, Y.; Peng, L.; Liu, X.; Lu, J.; He, L.; Li, K.; Qian, D.; Wang, R. Relationship between Quantitative Parameters of Echocardiography and Vascular Endothelial Function in Patients with Chronic Heart Failure and Its Predictive Value for Short-Term MACE Risk. J. Healthc. Eng. 2021, 2021, 1985962. [Google Scholar] [CrossRef]
- Shao, Z.; Schuster, A.; Borowski, A.G.; Thakur, A.; Li, L.; Wilson Tang, W.H. Soluble angiotensin converting enzyme 2 levels in chronic heart failure is associated with decreased exercise capacity and increased oxidative stress-mediated endothelial dysfunction. Transl. Res. 2019, 212, 80–88. [Google Scholar] [CrossRef]
- Hulshoff, M.S.; Xu, X.; Krenning, G.; Zeisberg, E.M. Epigenetic Regulation of Endothelial-to-Mesenchymal Transition in Chronic Heart Disease. Arterioscler. Thromb. Vasc. Biol. 2018, 38, 1986–1996. [Google Scholar] [CrossRef] [Green Version]
- An, L.; An, S.; Jia, Z.; Wang, H.; Yang, Z.; Xu, C.; Teng, X.; Wang, J.; Liu, X.; Cao, Q.; et al. Atorvastatin improves left ventricular remodeling and cardiac function in rats with congestive heart failure by inhibiting RhoA/Rho kinase-mediated endothelial nitric oxide synthase. Exp. Ther. Med. 2019, 17, 960–966. [Google Scholar] [CrossRef] [Green Version]
- Berezin, A.E.; Kremzer, A.A.; Samura, T.A.; Berezina, T.A. Altered signature of apoptotic endothelial cell-derived microvesicles predicts chronic heart failure phenotypes. Biomark. Med. 2019, 13, 737–750. [Google Scholar] [CrossRef] [PubMed]
- Huang, W.P.; Yin, W.H.; Chen, J.S.; Huang, P.H.; Chen, J.W.; Lin, S.J. Fenofibrate Reverses Dysfunction of EPCs Caused by Chronic Heart Failure. J. Cardiovasc. Transl. Res. 2020, 13, 158–170. [Google Scholar] [CrossRef] [PubMed]
- Murohara, T.; Asahara, T.; Silver, M.; Bauters, C.; Masuda, H.; Kalka, C.; Kearney, M.; Chen, D.; Symes, J.F.; Fishman, M.C.; et al. Nitric oxide synthase modulates angiogenesis in response to tissue ischemia. J. Clin. Invest. 1998, 101, 2567–2578. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giordano, F.J.; Gerber, H.P.; Williams, S.P.; VanBruggen, N.; Bunting, S.; Ruiz-Lozano, P.; Gu, Y.; Nath, A.K.; Huang, Y.; Hickey, R.; et al. A cardiac myocyte vascular endothelial growth factor paracrine pathway is required to maintain cardiac function. Proc. Natl. Acad. Sci. USA 2001, 98, 5780–5785. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chong, A.Y.; Blann, A.D.; Patel, J.; Freestone, B.; Hughes, E.; Lip, G.Y. Endothelial dysfunction and damage in congestive heart failure: Relation of flow-mediated dilation to circulating endothelial cells, plasma indexes of endothelial damage, and brain natriuretic peptide. Circulation 2004, 110, 1794–1798. [Google Scholar] [CrossRef] [Green Version]
- Lampsas, S.; Tsaplaris, P.; Pantelidis, P.; Oikonomou, E.; Marinos, G.; Charalambous, G.; Souvaliotis, N.; Mystakidi, V.C.; Goliopoulou, A.; Katsianos, E.; et al. The Role of Endothelial Related Circulating Biomarkers in COVID-19. A Systematic Review and Meta-analysis. Curr. Med. Chem. 2022, 29, 3790–3805. [Google Scholar] [CrossRef]
- Prasad, A.; Higano, S.T.; Al Suwaidi, J.; Holmes, D.R., Jr.; Mathew, V.; Pumper, G.; Lennon, R.J.; Lerman, A. Abnormal coronary microvascular endothelial function in humans with asymptomatic left ventricular dysfunction. Am. Heart J. 2003, 146, 549–554. [Google Scholar] [CrossRef]
- Taher, R.; Sara, J.D.; Toya, T.; Borlaug, B.A.; Lerman, L.O.; Lerman, A. Peripheral endothelial dysfunction is a novel risk factor for systolic dysfunction and heart failure progression. Int. J. Cardiol. Heart Vasc. 2020, 30, 100584. [Google Scholar] [CrossRef]
- Triposkiadis, F.; Butler, J.; Abboud, F.M.; Armstrong, P.W.; Adamopoulos, S.; Atherton, J.J.; Backs, J.; Bauersachs, J.; Burkhoff, D.; Bonow, R.O.; et al. The continuous heart failure spectrum: Moving beyond an ejection fraction classification. Eur. Heart J. 2019, 40, 2155–2163. [Google Scholar] [CrossRef]
- Kim, Y.H.; Kitai, T.; Morales, R.; Kiefer, K.; Chaikijurajai, T.; Tang, W.H.W. Usefulness of Serum Biomarkers of Endothelial Glycocalyx Damage in Prognosis of Decompensated Patients with Heart Failure with Reduced Ejection Fraction. Am. J. Cardiol. 2022, 176, 73–78. [Google Scholar] [CrossRef]
- Nijst, P.; Cops, J.; Martens, P.; Swennen, Q.; Dupont, M.; Tang, W.H.W.; Mullens, W. Endovascular shedding markers in patients with heart failure with reduced ejection fraction: Results from a single-center exploratory study. Microcirculation 2018, 25, e12432. [Google Scholar] [CrossRef]
- Arnlov, J.; Sang, Y.; Ballew, S.H.; Vaidya, D.; Michos, E.D.; Jacobs, D.R., Jr.; Lima, J.; Shlipak, M.G.; Bertoni, A.G.; Coresh, J.; et al. Endothelial dysfunction and the risk of heart failure in a community-based study: The Multi-Ethnic Study of Atherosclerosis. ESC Heart Fail. 2020, 7, 4231–4240. [Google Scholar] [CrossRef] [PubMed]
- Martinelli, N.C.; Santos, K.G.; Biolo, A.; La Porta, V.L.; Cohen, C.R.; Silvello, D.; Andrades, M.E.; Clausell, N.; Rohde, L.E. Polymorphisms of endothelial nitric oxide synthase gene in systolic heart failure: An haplotype analysis. Nitric. Oxide 2012, 26, 141–147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shantsila, E.; Wrigley, B.; Shantsila, A.; Tapp, L.D.; Blann, A.D.; Gill, P.S.; Lip, G.Y. Ethnic differences in macrovascular and microvascular function in systolic heart failure. Circ. Heart Fail. 2011, 4, 754–762. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lampsas, S.; Oikonomou, E.; Pantelidis, P.; Theofilis, P.; Grammatopoulos, K.; Marathonitis, A.; Vavuranakis, M.A.; Siasos, G.; Tousoulis, D.; Vavuranakis, M. Lipoprotein (a) Levels and Abdominal Aortic Aneurysm. A Systematic Review and Meta-analysis. Curr. Pharm. Des. 2022, 28, 3492–3499. [Google Scholar] [CrossRef]
- Zhang, P.; Xu, X.; Hu, X.; van Deel, E.D.; Zhu, G.; Chen, Y. Inducible nitric oxide synthase deficiency protects the heart from systolic overload-induced ventricular hypertrophy and congestive heart failure. Circ. Res. 2007, 100, 1089–1098. [Google Scholar] [CrossRef]
- Munzel, T.; Gori, T.; Keaney, J.F., Jr.; Maack, C.; Daiber, A. Pathophysiological role of oxidative stress in systolic and diastolic heart failure and its therapeutic implications. Eur. Heart J. 2015, 36, 2555–2564. [Google Scholar] [CrossRef] [Green Version]
- Ansari-Ramandi, M.M.; Maleki, M.; Alizadehasl, A.; Amin, A.; Taghavi, S.; Alemzadeh-Ansari, M.J.; Kazem Moussavi, A.; Naderi, N. Safety and effect of high dose allopurinol in patients with severe left ventricular systolic dysfunction. J. Cardiovasc. Thorac. Res. 2017, 9, 102–107. [Google Scholar] [CrossRef] [Green Version]
- Cordwin, D.J.; Berei, T.J.; Pogue, K.T. The Role of sGC Stimulators and Activators in Heart Failure With Reduced Ejection Fraction. J. Cardiovasc. Pharmacol. Ther. 2021, 26, 593–600. [Google Scholar] [CrossRef]
- Aimo, A.; Castiglione, V.; Vergaro, G.; Panichella, G.; Senni, M.; Lombardi, C.M.; Emdin, M. The place of vericiguat in the landscape of treatment for heart failure with reduced ejection fraction. Heart Fail. Rev. 2022, 27, 1165–1171. [Google Scholar] [CrossRef]
- Yin, W.H.; Chen, J.W.; Chen, Y.H.; Lin, S.J. Fenofibrate Modulates HO-1 and Ameliorates Endothelial Expression of Cell Adhesion Molecules in Systolic Heart Failure. Acta Cardiol. Sin. 2013, 29, 251–260. [Google Scholar] [PubMed]
- Theofilis, P.; Lampsas, S.; Oikonomou, E.; Siasos, G.; Vavuranakis, M.A.; Marinos, G.; Tsioufis, K.; Vavuranakis, M.; Tousoulis, D. Endothelial dysfunction in convalescent COVID-19 patients: Systematic review and meta-analysis. Arch. Cardiovasc. Dis. 2022, 115, 675–677. [Google Scholar] [CrossRef] [PubMed]
- Tang, W.H.; Shrestha, K.; Tong, W.; Wang, Z.; Troughton, R.W.; Borowski, A.G.; Klein, A.L.; Hazen, S.L. Nitric oxide bioavailability and adiponectin production in chronic systolic heart failure: Relation to severity of cardiac dysfunction. Transl. Res. 2013, 162, 26–33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bonadei, I.; Sciatti, E.; Vizzardi, E.; Fabbricatore, D.; Pagnoni, M.; Rossi, L.; Carubelli, V.; Lombardi, C.M.; Metra, M. Effects of ivabradine on endothelial function, aortic properties and ventricular-arterial coupling in chronic systolic heart failure patients. Cardiovasc. Ther. 2018, 36, e12323. [Google Scholar] [CrossRef] [PubMed]
- Andrei, S.; Nguyen, M.; Longrois, D.; Popescu, B.A.; Bouhemad, B.; Guinot, P.G. Ventriculo-Arterial Coupling Is Associated With Oxygen Consumption and Tissue Perfusion in Acute Circulatory Failure. Front. Cardiovasc. Med. 2022, 9, 842554. [Google Scholar] [CrossRef]
- Salvador, A.M.; Moss, M.E.; Aronovitz, M.; Mueller, K.B.; Blanton, R.M.; Jaffe, I.Z.; Alcaide, P. Endothelial mineralocorticoid receptor contributes to systolic dysfunction induced by pressure overload without modulating cardiac hypertrophy or inflammation. Physiol. Rep. 2017, 5, e13313. [Google Scholar] [CrossRef]
- Zhang, H.; Feng, L.; Wan, Q.; Hong, Y.; Li, Y.; Cheng, G.; Han, X. Osteoprotegerin is associated with depletion of circulating endothelial progenitor cells and elevation in pulmonary arterial pressure in patients with systolic heart failure. Acta Cardiol. 2015, 70, 435–441. [Google Scholar] [CrossRef]
- Zhang, H.; Feng, L.; Wan, Q.L.; Hong, Y.; Li, Y.M.; Cheng, G.C.; Han, X.Q. Sleep-disordered breathing is associated with depletion of circulating endothelial progenitor cells and elevation in pulmonary arterial pressure in patients with decompensated systolic heart failure. J. Geriatr. Cardiol. 2015, 12, 424–430. [Google Scholar] [CrossRef]
- Susic, L.; Maricic, L.; Vincelj, J.; Vadoci, M.; Susic, T. Understanding the association between endothelial dysfunction and left ventricle diastolic dysfunction in development of coronary artery disease and heart failure. Acta Biomed. 2021, 92, e2021204. [Google Scholar] [CrossRef]
- Gamrat, A.; Surdacki, M.A.; Chyrchel, B.; Surdacki, A. Endothelial Dysfunction: A Contributor to Adverse Cardiovascular Remodeling and Heart Failure Development in Type 2 Diabetes beyond Accelerated Atherogenesis. J. Clin. Med. 2020, 9, 2090. [Google Scholar] [CrossRef]
- Cauwenberghs, N.; Godderis, S.; Sabovcik, F.; Cornelissen, V.; Kuznetsova, T. Subclinical heart remodeling and dysfunction in relation to peripheral endothelial dysfunction: A general population study. Microcirculation 2021, 28, e12731. [Google Scholar] [CrossRef] [PubMed]
- Sorop, O.; Heinonen, I.; van Kranenburg, M.; van de Wouw, J.; de Beer, V.J.; Nguyen, I.T.N.; Octavia, Y.; van Duin, R.W.B.; Stam, K.; van Geuns, R.J.; et al. Multiple common comorbidities produce left ventricular diastolic dysfunction associated with coronary microvascular dysfunction, oxidative stress, and myocardial stiffening. Cardiovasc. Res. 2018, 114, 954–964. [Google Scholar] [CrossRef] [PubMed]
- Abelanet, A.; Camoin, M.; Rubin, S.; Bougaran, P.; Delobel, V.; Pernot, M.; Forfar, I.; Guilbeau-Frugier, C.; Gales, C.; Bats, M.L.; et al. Increased Capillary Permeability in Heart Induces Diastolic Dysfunction Independently of Inflammation, Fibrosis, or Cardiomyocyte Dysfunction. Arterioscler. Thromb. Vasc. Biol. 2022, 42, 745–763. [Google Scholar] [CrossRef] [PubMed]
- Dhot, J.; Ferron, M.; Prat, V.; Persello, A.; Roul, D.; Stevant, D.; Guijarro, D.; Piriou, N.; Aillerie, V.; Erraud, A.; et al. Overexpression of endothelial beta(3) -adrenergic receptor induces diastolic dysfunction in rats. ESC Heart Fail. 2020, 7, 4159–4171. [Google Scholar] [CrossRef] [PubMed]
- Erkens, R.; Kramer, C.M.; Luckstadt, W.; Panknin, C.; Krause, L.; Weidenbach, M.; Dirzka, J.; Krenz, T.; Mergia, E.; Suvorava, T.; et al. Left ventricular diastolic dysfunction in Nrf2 knock out mice is associated with cardiac hypertrophy, decreased expression of SERCA2a, and preserved endothelial function. Free Radic. Biol. Med. 2015, 89, 906–917. [Google Scholar] [CrossRef] [Green Version]
- Heinonen, I.; Sorop, O.; van Dalen, B.M.; Wust, R.C.I.; van de Wouw, J.; de Beer, V.J.; Octavia, Y.; van Duin, R.W.B.; Hoogstrate, Y.; Blonden, L.; et al. Cellular, mitochondrial and molecular alterations associate with early left ventricular diastolic dysfunction in a porcine model of diabetic metabolic derangement. Sci. Rep. 2020, 10, 13173. [Google Scholar] [CrossRef]
- Waddingham, M.T.; Sonobe, T.; Tsuchimochi, H.; Edgley, A.J.; Sukumaran, V.; Chen, Y.C.; Hansra, S.S.; Schwenke, D.O.; Umetani, K.; Aoyama, K.; et al. Diastolic dysfunction is initiated by cardiomyocyte impairment ahead of endothelial dysfunction due to increased oxidative stress and inflammation in an experimental prediabetes model. J. Mol. Cell Cardiol. 2019, 137, 119–131. [Google Scholar] [CrossRef] [Green Version]
- Gevaert, A.B.; Lemmens, K.; Vrints, C.J.; Van Craenenbroeck, E.M. Targeting Endothelial Function to Treat Heart Failure with Preserved Ejection Fraction: The Promise of Exercise Training. Oxid. Med. Cell Longev. 2017, 2017, 4865756. [Google Scholar] [CrossRef] [Green Version]
- Hirai, D.M.; Zelt, J.T.; Jones, J.H.; Castanhas, L.G.; Bentley, R.F.; Earle, W.; Staples, P.; Tschakovsky, M.E.; McCans, J.; O’Donnell, D.E.; et al. Dietary nitrate supplementation and exercise tolerance in patients with heart failure with reduced ejection fraction. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2017, 312, R13–R22. [Google Scholar] [CrossRef] [Green Version]
- Mocan, M.; Mocan Hognogi, L.D.; Anton, F.P.; Chiorescu, R.M.; Goidescu, C.M.; Stoia, M.A.; Farcas, A.D. Biomarkers of Inflammation in Left Ventricular Diastolic Dysfunction. Dis. Markers 2019, 2019, 7583690. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kloch, M.; Stolarz-Skrzypek, K.; Olszanecka, A.; Wojciechowska, W.; Bednarski, A.; Stefaniak, J.; Czarnecka, D. Inflammatory markers and left ventricular diastolic dysfunction in a family-based population study. Kardiol. Pol. 2019, 77, 33–39. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zeng, H.; He, X.; Chen, J.X. A Sex-Specific Role of Endothelial Sirtuin 3 on Blood Pressure and Diastolic Dysfunction in Female Mice. Int. J. Mol. Sci. 2020, 21, 9744. [Google Scholar] [CrossRef] [PubMed]
- Teuber, J.P.; Essandoh, K.; Hummel, S.L.; Madamanchi, N.R.; Brody, M.J. NADPH Oxidases in Diastolic Dysfunction and Heart Failure with Preserved Ejection Fraction. Antioxidants 2022, 11, 1822. [Google Scholar] [CrossRef] [PubMed]
- Yin, J.; Kukucka, M.; Hoffmann, J.; Sterner-Kock, A.; Burhenne, J.; Haefeli, W.E.; Kuppe, H.; Kuebler, W.M. Sildenafil preserves lung endothelial function and prevents pulmonary vascular remodeling in a rat model of diastolic heart failure. Circ. Heart Fail. 2011, 4, 198–206. [Google Scholar] [CrossRef] [Green Version]
- Serpi, R.; Tolonen, A.M.; Huusko, J.; Rysa, J.; Tenhunen, O.; Yla-Herttuala, S.; Ruskoaho, H. Vascular endothelial growth factor-B gene transfer prevents angiotensin II-induced diastolic dysfunction via proliferation and capillary dilatation in rats. Cardiovasc. Res. 2011, 89, 204–213. [Google Scholar] [CrossRef] [Green Version]
- Sama, I.E.; Woolley, R.J.; Nauta, J.F.; Romaine, S.P.R.; Tromp, J.; Ter Maaten, J.M.; van der Meer, P.; Lam, C.S.P.; Samani, N.J.; Ng, L.L.; et al. A network analysis to identify pathophysiological pathways distinguishing ischaemic from non-ischaemic heart failure. Eur. J. Heart Fail. 2020, 22, 821–833. [Google Scholar] [CrossRef] [Green Version]
- Rubattu, S.; Bigatti, G.; Evangelista, A.; Lanzani, C.; Stanzione, R.; Zagato, L.; Manunta, P.; Marchitti, S.; Venturelli, V.; Bianchi, G.; et al. Association of atrial natriuretic peptide and type a natriuretic peptide receptor gene polymorphisms with left ventricular mass in human essential hypertension. J. Am. Coll Cardiol. 2006, 48, 499–505. [Google Scholar] [CrossRef] [Green Version]
- Shantsila, E.; Wrigley, B.J.; Blann, A.D.; Gill, P.S.; Lip, G.Y. A contemporary view on endothelial function in heart failure. Eur. J. Heart Fail. 2012, 14, 873–881. [Google Scholar] [CrossRef]
- Klosinska, M.; Rudzinski, T.; Grzelak, P.; Stefanczyk, L.; Drozdz, J.; Krzeminska-Pakula, M. Endothelium-dependent and -independent vasodilation is more attenuated in ischaemic than in non-ischaemic heart failure. Eur. J. Heart Fail. 2009, 11, 765–770. [Google Scholar] [CrossRef]
- Tsigkou, V.; Siasos, G.; Oikonomou, E.; Zaromitidou, M.; Mourouzis, K.; Dimitropoulos, S.; Bletsa, E.; Gouliopoulos, N.; Stampouloglou, P.K.; Panoilia, M.E.; et al. The prognostic role of galectin-3 and endothelial function in patients with heart failure. Cardiol. J. 2022. [Google Scholar] [CrossRef]
- Molitor, M.; Rudi, W.S.; Garlapati, V.; Finger, S.; Schuler, R.; Kossmann, S.; Lagrange, J.; Nguyen, T.S.; Wild, J.; Knopp, T.; et al. Nox2+ myeloid cells drive vascular inflammation and endothelial dysfunction in heart failure after myocardial infarction via angiotensin II receptor type 1. Cardiovasc. Res. 2021, 117, 162–177. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Isaksen, K.; Halvorsen, B.; Munk, P.S.; Aukrust, P.; Larsen, A.I. Effects of interval training on inflammatory biomarkers in patients with ischemic heart failure. Scand. Cardiovasc. J. 2019, 53, 213–219. [Google Scholar] [CrossRef]
- Dimitropoulos, S.; Mystakidi, V.C.; Oikonomou, E.; Siasos, G.; Tsigkou, V.; Athanasiou, D.; Gouliopoulos, N.; Bletsa, E.; Kalampogias, A.; Charalambous, G.; et al. Association of Soluble Suppression of Tumorigenesis-2 (ST2) with Endothelial Function in Patients with Ischemic Heart Failure. Int. J. Mol. Sci. 2020, 21, 9385. [Google Scholar] [CrossRef] [PubMed]
- Tentolouris, C.; Tousoulis, D.; Antoniades, C.; Bosinakou, E.; Kotsopoulou, M.; Trikas, A.; Toutouzas, P.; Stefanadis, C. Endothelial function and proinflammatory cytokines in patients with ischemic heart disease and dilated cardiomyopathy. Int. J. Cardiol. 2004, 94, 301–305. [Google Scholar] [CrossRef] [PubMed]
- Hsu, C.P.; Lin, S.J.; Chung, M.Y.; Lu, T.M. Asymmetric dimethylarginine predicts clinical outcomes in ischemic chronic heart failure. Atherosclerosis 2012, 225, 504–510. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.; Wang, B.; Yang, Y.; Jia, Q.; Qi, Z.; Zhang, A.; Lv, S.; Zhang, J. Exosomes in ischemic heart disease: Novel carriers for bioinformation. Biomed. Pharmacother. 2019, 120, 109451. [Google Scholar] [CrossRef]
- Oikonomou, E.; Theofilis, P.; Lampsas, S.; Katsarou, O.; Kalogeras, K.; Marinos, G.; Tsatsaragkou, A.; Anastasiou, A.; Lysandrou, A.; Gounaridi, M.I.; et al. Current Concepts and Future Applications of Non-Invasive Functional and Anatomical Evaluation of Coronary Artery Disease. Life 2022, 12, 1803. [Google Scholar] [CrossRef]
- Oie, E.; Vinge, L.E.; Yndestad, A.; Sandberg, C.; Grogaard, H.K.; Attramadal, H. Induction of a myocardial adrenomedullin signaling system during ischemic heart failure in rats. Circulation 2000, 101, 415–422. [Google Scholar] [CrossRef] [Green Version]
- Bansal, S.S.; Ismahil, M.A.; Goel, M.; Zhou, G.; Rokosh, G.; Hamid, T.; Prabhu, S.D. Dysfunctional and Proinflammatory Regulatory T-Lymphocytes Are Essential for Adverse Cardiac Remodeling in Ischemic Cardiomyopathy. Circulation 2019, 139, 206–221. [Google Scholar] [CrossRef]
- Gueret, A.; Harouki, N.; Favre, J.; Galmiche, G.; Nicol, L.; Henry, J.P.; Besnier, M.; Thuillez, C.; Richard, V.; Kolkhof, P.; et al. Vascular Smooth Muscle Mineralocorticoid Receptor Contributes to Coronary and Left Ventricular Dysfunction After Myocardial Infarction. Hypertension 2016, 67, 717–723. [Google Scholar] [CrossRef] [Green Version]
- Abdel Hamid, M.; Bakhoum, S.W.; Sharaf, Y.; Sabry, D.; El-Gengehe, A.T.; Abdel-Latif, A. Circulating Endothelial Cells and Endothelial Function Predict Major Adverse Cardiac Events and Early Adverse Left Ventricular Remodeling in Patients With ST-Segment Elevation Myocardial Infarction. J. Interv. Cardiol. 2016, 29, 89–98. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berezin, A.E.; Kremzer, A.A. Circulating endothelial progenitor cells as markers for severity of ischemic chronic heart failure. J. Card. Fail. 2014, 20, 438–447. [Google Scholar] [CrossRef] [PubMed]
- Berezin, A.E.; Kremzer, A.A.; Samura, T.A.; Martovitskaya, Y.V. Circulating endothelial-derived apoptotic microparticles in the patients with ischemic symptomatic chronic heart failure: Relevance of pro-inflammatory activation and outcomes. Int. Cardiovasc. Res. J. 2014, 8, 116–123. [Google Scholar]
- Ponikowski, P.; Voors, A.A.; Anker, S.D.; Bueno, H.; Cleland, J.G.F.; Coats, A.J.S.; Falk, V.; Gonzalez-Juanatey, J.R.; Harjola, V.P.; Jankowska, E.A.; et al. 2016 ESC Guidelines for the diagnosis and treatment of acute and chronic heart failure: The Task Force for the diagnosis and treatment of acute and chronic heart failure of the European Society of Cardiology (ESC)Developed with the special contribution of the Heart Failure Association (HFA) of the ESC. Eur. Heart J. 2016, 37, 2129–2200. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shah, A.; Gkaliagkousi, E.; Ritter, J.M.; Ferro, A. Endothelial function and arterial compliance are not impaired in subjects with heart failure of non-ischemic origin. J. Card. Fail. 2010, 16, 114–120. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.W.; Ting, C.T.; Chen, Y.H.; Wu, T.C.; Hsu, N.W.; Lin, S.J.; Chang, M.S. Differential coronary microvascular function in patients with left ventricular dysfunction of unknown cause--implication for possible mechanism of myocardial ischemia in early stage of cardiomyopathy. Int. J. Cardiol. 1999, 69, 251–261. [Google Scholar] [CrossRef] [PubMed]
- Oikonomou, E.; Souvaliotis, N.; Lampsas, S.; Siasos, G.; Theofilis, P.; Korakas, E.; Lambadiari, V.; Ikonomidis, I.; Pesiridis, T.; Zakynthinos, G.; et al. The role of cardiometabolic risk factors and endothelial dysfunction in serum albumin levels of patients with COVID-19. Cardiol. J. 2022, 29, 1037–1039. [Google Scholar] [CrossRef] [PubMed]
- Hirakawa, K.; Yamamuro, M.; Uemura, T.; Takashio, S.; Kaikita, K.; Utsunomiya, D.; Nakayama, M.; Yamamoto, E.; Yamashita, Y.; Hokimoto, S.; et al. Correlation between microvascular dysfunction and B-type natriuretic peptide levels in non-ischemic heart failure patients with cardiac fibrosis. Int. J. Cardiol. 2017, 228, 881–885. [Google Scholar] [CrossRef]
- Knapp, M.; Tu, X.; Wu, R. Vascular endothelial dysfunction, a major mediator in diabetic cardiomyopathy. Acta Pharmacol. Sin. 2019, 40, 1–8. [Google Scholar] [CrossRef]
- Reichman-Warmusz, E.; Brzozowa-Zasada, M.; Wojciechowska, C.; Dudek, D.; Warmusz, O.; Wojnicz, R. Decreased immunoreactivity of von Willebrand factor may reflect persistent nature of the endothelial dysfunction in non-ischemic heart failure. Folia Histochem. Cytobiol. 2021, 59, 108–113. [Google Scholar] [CrossRef]
- Thorin, E. Different contribution of endothelial nitric oxide in the relaxation of human coronary arteries of ischemic and dilated cardiomyopathic hearts. J. Cardiovasc. Pharmacol. 2001, 37, 227–232. [Google Scholar] [CrossRef] [PubMed]
- Vittorio, T.J.; Zolty, R.; Garg, P.K.; Sarswat, N.; Tseng, C.H.; Jorde, U.P.; Colombo, P.C. Interdependence of cardiac and endothelial function in patients with symptomatic chronic heart failure of nonischemic etiology. Echocardiography 2009, 26, 916–921. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, H.; Nakamura, M.; Akatsu, T.; Arakawa, N.; Hiramori, K. Effects of nitric oxide inhibition on basal forearm blood flow in patients with nonischemic chronic heart failure. Heart Vessels 1998, 13, 142–146. [Google Scholar] [CrossRef] [PubMed]
- Tuder, R.M.; Archer, S.L.; Dorfmuller, P.; Erzurum, S.C.; Guignabert, C.; Michelakis, E.; Rabinovitch, M.; Schermuly, R.; Stenmark, K.R.; Morrell, N.W. Relevant issues in the pathology and pathobiology of pulmonary hypertension. J. Am. Coll. Cardiol. 2013, 62, D4–D12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ranchoux, B.; Harvey, L.D.; Ayon, R.J.; Babicheva, A.; Bonnet, S.; Chan, S.Y.; Yuan, J.X.; Perez, V.J. Endothelial dysfunction in pulmonary arterial hypertension: An evolving landscape (2017 Grover Conference Series). Pulm. Circ. 2018, 8, 2045893217752912. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Southgate, L.; Machado, R.D.; Graf, S.; Morrell, N.W. Molecular genetic framework underlying pulmonary arterial hypertension. Nat. Rev. Cardiol. 2020, 17, 85–95. [Google Scholar] [CrossRef] [PubMed]
- Santos-Gomes, J.; Le Ribeuz, H.; Bras-Silva, C.; Antigny, F.; Adao, R. Role of Ion Channel Remodeling in Endothelial Dysfunction Induced by Pulmonary Arterial Hypertension. Biomolecules 2022, 12, 484. [Google Scholar] [CrossRef]
- Friedman, D.; Szmuszkovicz, J.; Rabai, M.; Detterich, J.A.; Menteer, J.; Wood, J.C. Systemic endothelial dysfunction in children with idiopathic pulmonary arterial hypertension correlates with disease severity. J. Heart Lung Transplant. 2012, 31, 642–647. [Google Scholar] [CrossRef] [Green Version]
- Loscalzo, J. Endothelial dysfunction in pulmonary hypertension. N. Engl. J. Med. 1992, 327, 117–119. [Google Scholar] [CrossRef]
- Huertas, A.; Perros, F.; Tu, L.; Cohen-Kaminsky, S.; Montani, D.; Dorfmuller, P.; Guignabert, C.; Humbert, M. Immune dysregulation and endothelial dysfunction in pulmonary arterial hypertension: A complex interplay. Circulation 2014, 129, 1332–1340. [Google Scholar] [CrossRef]
- Levy, M.; Maurey, C.; Celermajer, D.S.; Vouhe, P.R.; Danel, C.; Bonnet, D.; Israel-Biet, D. Impaired apoptosis of pulmonary endothelial cells is associated with intimal proliferation and irreversibility of pulmonary hypertension in congenital heart disease. J. Am. Coll. Cardiol. 2007, 49, 803–810. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, C.; Townsley, M.I.; Alexeyev, M.; Voelkel, N.F.; Stevens, T. Endothelial hyperpermeability in severe pulmonary arterial hypertension: Role of store-operated calcium entry. Am. J. Physiol. Lung Cell Mol. Physiol. 2016, 311, L560–L569. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dai, Z.; Zhu, M.M.; Peng, Y.; Machireddy, N.; Evans, C.E.; Machado, R.; Zhang, X.; Zhao, Y.Y. Therapeutic Targeting of Vascular Remodeling and Right Heart Failure in Pulmonary Arterial Hypertension with a HIF-2alpha Inhibitor. Am. J. Respir. Crit. Care Med. 2018, 198, 1423–1434. [Google Scholar] [CrossRef]
- Morrell, N.W.; Adnot, S.; Archer, S.L.; Dupuis, J.; Lloyd Jones, P.; MacLean, M.R.; McMurtry, I.F.; Stenmark, K.R.; Thistlethwaite, P.A.; Weissmann, N.; et al. Cellular and molecular basis of pulmonary arterial hypertension. J. Am. Coll. Cardiol. 2009, 54, S20–S31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rabinovitch, M.; Guignabert, C.; Humbert, M.; Nicolls, M.R. Inflammation and immunity in the pathogenesis of pulmonary arterial hypertension. Circ. Res. 2014, 115, 165–175. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stenmark, K.R.; Yeager, M.E.; El Kasmi, K.C.; Nozik-Grayck, E.; Gerasimovskaya, E.V.; Li, M.; Riddle, S.R.; Frid, M.G. The adventitia: Essential regulator of vascular wall structure and function. Annu. Rev. Physiol. 2013, 75, 23–47. [Google Scholar] [CrossRef] [Green Version]
- Frid, M.G.; Kale, V.A.; Stenmark, K.R. Mature vascular endothelium can give rise to smooth muscle cells via endothelial-mesenchymal transdifferentiation: In vitro analysis. Circ. Res. 2002, 90, 1189–1196. [Google Scholar] [CrossRef] [Green Version]
- Cober, N.D.; VandenBroek, M.M.; Ormiston, M.L.; Stewart, D.J. Evolving Concepts in Endothelial Pathobiology of Pulmonary Arterial Hypertension. Hypertension 2022, 79, 1580–1590. [Google Scholar] [CrossRef]
- Wermuth, P.J.; Li, Z.; Mendoza, F.A.; Jimenez, S.A. Stimulation of Transforming Growth Factor-beta1-Induced Endothelial-To-Mesenchymal Transition and Tissue Fibrosis by Endothelin-1 (ET-1): A Novel Profibrotic Effect of ET-1. PLoS ONE 2016, 11, e0161988. [Google Scholar] [CrossRef] [Green Version]
- Guo, Y.; Li, P.; Bledsoe, G.; Yang, Z.R.; Chao, L.; Chao, J. Kallistatin inhibits TGF-beta-induced endothelial-mesenchymal transition by differential regulation of microRNA-21 and eNOS expression. Exp. Cell Res. 2015, 337, 103–110. [Google Scholar] [CrossRef] [Green Version]
- Boucherat, O.; Potus, F.; Bonnet, S. microRNA and Pulmonary Hypertension. Adv. Exp. Med. Biol. 2015, 888, 237–252. [Google Scholar] [CrossRef] [PubMed]
- Aldred, M.A.; Vijayakrishnan, J.; James, V.; Soubrier, F.; Gomez-Sanchez, M.A.; Martensson, G.; Galie, N.; Manes, A.; Corris, P.; Simonneau, G.; et al. BMPR2 gene rearrangements account for a significant proportion of mutations in familial and idiopathic pulmonary arterial hypertension. Hum. Mutat. 2006, 27, 212–213. [Google Scholar] [CrossRef] [PubMed]
- Gomez-Puerto, M.C.; van Zuijen, I.; Huang, C.J.; Szulcek, R.; Pan, X.; van Dinther, M.A.; Kurakula, K.; Wiesmeijer, C.C.; Goumans, M.J.; Bogaard, H.J.; et al. Autophagy contributes to BMP type 2 receptor degradation and development of pulmonary arterial hypertension. J. Pathol. 2019, 249, 356–367. [Google Scholar] [CrossRef] [PubMed]
- Han, Y.; Ali, M.K.; Dua, K.; Spiekerkoetter, E.; Mao, Y. Role of Long Non-Coding RNAs in Pulmonary Arterial Hypertension. Cells 2021, 10, 1892. [Google Scholar] [CrossRef] [PubMed]
- Xu, D.; Hu, Y.H.; Gou, X.; Li, F.Y.; Yang, X.Y.; Li, Y.M.; Chen, F. Oxidative Stress and Antioxidative Therapy in Pulmonary Arterial Hypertension. Molecules 2022, 27, 3724. [Google Scholar] [CrossRef] [PubMed]
- Ryan, J.J.; Archer, S.L. Emerging concepts in the molecular basis of pulmonary arterial hypertension: Part I: Metabolic plasticity and mitochondrial dynamics in the pulmonary circulation and right ventricle in pulmonary arterial hypertension. Circulation 2015, 131, 1691–1702. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cuthbertson, I.; Morrell, N.W.; Caruso, P. BMPR2 Mutation and Metabolic Reprogramming in Pulmonary Arterial Hypertension. Circ. Res. 2023, 132, 109–126. [Google Scholar] [CrossRef] [PubMed]
- Mumby, S.; Perros, F.; Hui, C.; Xu, B.L.; Xu, W.; Elyasigomari, V.; Hautefort, A.; Manaud, G.; Humbert, M.; Chung, K.F.; et al. Extracellular matrix degradation pathways and fatty acid metabolism regulate distinct pulmonary vascular cell types in pulmonary arterial hypertension. Pulm. Circ. 2021, 11, 2045894021996190. [Google Scholar] [CrossRef]
- Kobayashi, T.; Kim, J.D.; Naito, A.; Yanagisawa, A.; Jujo-Sanada, T.; Kasuya, Y.; Nakagawa, Y.; Sakao, S.; Tatsumi, K.; Suzuki, T. Multi-omics analysis of right ventricles in rat models of pulmonary arterial hypertension: Consideration of mitochondrial biogenesis by chrysin. Int. J. Mol. Med. 2022, 49, 1–12. [Google Scholar] [CrossRef]
- Legchenko, E.; Chouvarine, P.; Borchert, P.; Fernandez-Gonzalez, A.; Snay, E.; Meier, M.; Maegel, L.; Mitsialis, S.A.; Rog-Zielinska, E.A.; Kourembanas, S.; et al. PPARgamma agonist pioglitazone reverses pulmonary hypertension and prevents right heart failure via fatty acid oxidation. Sci. Transl. Med. 2018, 10, eaao0303. [Google Scholar] [CrossRef] [Green Version]
- Omura, J.; Satoh, K.; Kikuchi, N.; Satoh, T.; Kurosawa, R.; Nogi, M.; Ohtsuki, T.; Al-Mamun, M.E.; Siddique, M.A.H.; Yaoita, N.; et al. ADAMTS8 Promotes the Development of Pulmonary Arterial Hypertension and Right Ventricular Failure: A Possible Novel Therapeutic Target. Circ. Res. 2019, 125, 884–906. [Google Scholar] [CrossRef]
- Zuchi, C.; Tritto, I.; Carluccio, E.; Mattei, C.; Cattadori, G.; Ambrosio, G. Role of endothelial dysfunction in heart failure. Heart Fail. Rev. 2020, 25, 21–30. [Google Scholar] [CrossRef]
- Bank, A.J.; Lee, P.C.; Kubo, S.H. Endothelial dysfunction in patients with heart failure: Relationship to disease severity. J. Card. Fail. 2000, 6, 29–36. [Google Scholar] [CrossRef] [PubMed]
- Popovic, B.; Zannad, F.; Louis, H.; Clerc-Urmes, I.; Lakomy, C.; Gibot, S.; Denis, C.V.; Lacolley, P.; Regnault, V. Endothelial-driven increase in plasma thrombin generation characterising a new hypercoagulable phenotype in acute heart failure. Int. J. Cardiol. 2019, 274, 195–201. [Google Scholar] [CrossRef] [PubMed]
- Heitzer, T.; Baldus, S.; von Kodolitsch, Y.; Rudolph, V.; Meinertz, T. Systemic endothelial dysfunction as an early predictor of adverse outcome in heart failure. Arterioscler. Thromb. Vasc. Biol. 2005, 25, 1174–1179. [Google Scholar] [CrossRef] [PubMed]
- Katz, S.D.; Hryniewicz, K.; Hriljac, I.; Balidemaj, K.; Dimayuga, C.; Hudaihed, A.; Yasskiy, A. Vascular endothelial dysfunction and mortality risk in patients with chronic heart failure. Circulation 2005, 111, 310–314. [Google Scholar] [CrossRef] [Green Version]
- Akar, J.G.; Al-Chekakie, M.O.; Fugate, T.; Moran, L.; Froloshki, B.; Varma, N.; Santucci, P.; Wilber, D.J.; Matsumura, M.E. Endothelial dysfunction in heart failure identifies responders to cardiac resynchronization therapy. Heart Rhythm. 2008, 5, 1229–1235. [Google Scholar] [CrossRef]
- Suhrs, H.E.; Schroder, J.; Bove, K.B.; Mygind, N.D.; Frestad, D.; Michelsen, M.M.; Lange, T.; Gustafsson, I.; Kastrup, J.; Prescott, E. Inflammation, non-endothelial dependent coronary microvascular function and diastolic function-Are they linked? PLoS ONE 2020, 15, e0236035. [Google Scholar] [CrossRef]
- Hage, C.; Michaelsson, E.; Kull, B.; Miliotis, T.; Svedlund, S.; Linde, C.; Donal, E.; Daubert, J.C.; Gan, L.M.; Lund, L.H. Myeloperoxidase and related biomarkers are suggestive footprints of endothelial microvascular inflammation in HFpEF patients. ESC Heart Fail. 2020, 7, 1534–1546. [Google Scholar] [CrossRef]
- Chong, A.Y.; Freestone, B.; Patel, J.; Lim, H.S.; Hughes, E.; Blann, A.D.; Lip, G.Y. Endothelial activation, dysfunction, and damage in congestive heart failure and the relation to brain natriuretic peptide and outcomes. Am. J. Cardiol. 2006, 97, 671–675. [Google Scholar] [CrossRef]
- Maio, R.; Perticone, M.; Suraci, E.; Sciacqua, A.; Sesti, G.; Perticone, F. Endothelial dysfunction and C-reactive protein predict the incidence of heart failure in hypertensive patients. ESC Heart Fail. 2021, 8, 399–407. [Google Scholar] [CrossRef] [PubMed]
- Anastasiou, M.; Oikonomou, E.; Zagouri, F.; Siasos, G.; Antonopoulos, A.S.; Psaltopoulou, T.; Bamias, A.; Dimopoulos, M.A.; Tousoulis, D. Flow-Mediated Dilation of Brachial Artery as a Screening Tool for Anthracycline-Induced Cardiotoxicity. J. Am. Coll. Cardiol. 2017, 70, 3072. [Google Scholar] [CrossRef] [PubMed]
- Keteyian, S.J.; Michaels, A. Heart Failure in Cardiac Rehabilitation: A REVIEW AND PRACTICAL CONSIDERATIONS. J. Cardiopulm. Rehabil. Prev. 2022, 42, 296–303. [Google Scholar] [CrossRef] [PubMed]
- Dickstein, K.; Cohen-Solal, A.; Filippatos, G.; McMurray, J.J.; Ponikowski, P.; Poole-Wilson, P.A.; Stromberg, A.; van Veldhuisen, D.J.; Atar, D.; Hoes, A.W.; et al. ESC guidelines for the diagnosis and treatment of acute and chronic heart failure 2008: The Task Force for the diagnosis and treatment of acute and chronic heart failure 2008 of the European Society of Cardiology. Developed in collaboration with the Heart Failure Association of the ESC (HFA) and endorsed by the European Society of Intensive Care Medicine (ESICM). Eur. J. Heart Fail. 2008, 10, 933–989. [Google Scholar] [CrossRef]
- Leggio, M.; Fusco, A.; Loreti, C.; Limongelli, G.; Bendini, M.G.; Mazza, A.; Coraci, D.; Padua, L. Effects of exercise training in heart failure with preserved ejection fraction: An updated systematic literature review. Heart Fail. Rev. 2020, 25, 703–711. [Google Scholar] [CrossRef]
- Piepoli, M.F.; Conraads, V.; Corra, U.; Dickstein, K.; Francis, D.P.; Jaarsma, T.; McMurray, J.; Pieske, B.; Piotrowicz, E.; Schmid, J.P.; et al. Exercise training in heart failure: From theory to practice. A consensus document of the Heart Failure Association and the European Association for Cardiovascular Prevention and Rehabilitation. Eur. J. Heart Fail. 2011, 13, 347–357. [Google Scholar] [CrossRef]
- Pearson, M.J.; Smart, N.A. Effect of exercise training on endothelial function in heart failure patients: A systematic review meta-analysis. Int. J. Cardiol. 2017, 231, 234–243. [Google Scholar] [CrossRef]
- Sandri, M.; Viehmann, M.; Adams, V.; Rabald, K.; Mangner, N.; Hollriegel, R.; Lurz, P.; Erbs, S.; Linke, A.; Kirsch, K.; et al. Chronic heart failure and aging—Effects of exercise training on endothelial function and mechanisms of endothelial regeneration: Results from the Leipzig Exercise Intervention in Chronic heart failure and Aging (LEICA) study. Eur. J. Prev. Cardiol. 2016, 23, 349–358. [Google Scholar] [CrossRef]
- Chen, J.; Gu, S.; Song, Y.; Ji, X.; Zeng, W.; Wang, X.; Wang, Y.; Feng, Q. The impact of cardiomotor rehabilitation on endothelial function in elderly patients with chronic heart failure. BMC Cardiovasc. Disord. 2021, 21, 524. [Google Scholar] [CrossRef]
- Sales, A.R.K.; Azevedo, L.F.; Silva, T.O.C.; Rodrigues, A.G.; Oliveira, P.A.; Jordao, C.P.; Andrade, A.C.M.; Urias, U.; Guimaraes, G.V.; Bocchi, E.A.; et al. High-Intensity Interval Training Decreases Muscle Sympathetic Nerve Activity and Improves Peripheral Vascular Function in Patients With Heart Failure With Reduced Ejection Fraction. Circ. Heart Fail. 2020, 13, e007121. [Google Scholar] [CrossRef]
- Jaconiano, E.; Moreira-Goncalves, D. Unveiling the role of exercise training in targeting the inflammatory paradigm of heart failure with preserved ejection fraction: A narrative review. Heart Fail. Rev. 2022, 27, 163–190. [Google Scholar] [CrossRef] [PubMed]
- Kitzman, D.W.; Brubaker, P.H.; Herrington, D.M.; Morgan, T.M.; Stewart, K.P.; Hundley, W.G.; Abdelhamed, A.; Haykowsky, M.J. Effect of endurance exercise training on endothelial function and arterial stiffness in older patients with heart failure and preserved ejection fraction: A randomized, controlled, single-blind trial. J. Am. Coll. Cardiol. 2013, 62, 584–592. [Google Scholar] [CrossRef] [PubMed]
- Hibbert, B.; Simard, T.; Ramirez, F.D.; Pourdjabbar, A.; Raizman, J.E.; Maze, R.; Wilson, K.R.; Hawken, S.; O’Brien, E.R. The effect of statins on circulating endothelial progenitor cells in humans: A systematic review. J. Cardiovasc. Pharmacol. 2013, 62, 491–496. [Google Scholar] [CrossRef]
- Zou, G.; Song, L.; Chang, L.; Wang, H. Comparison and analysis of statins drug use in the treatment of diastolic dysfunction in patients. Pak. J. Pharm. Sci. 2018, 31, 1725–1730. [Google Scholar] [PubMed]
- Oikonomou, E.; Siasos, G.; Zaromitidou, M.; Hatzis, G.; Mourouzis, K.; Chrysohoou, C.; Zisimos, K.; Mazaris, S.; Tourikis, P.; Athanasiou, D.; et al. Atorvastatin treatment improves endothelial function through endothelial progenitor cells mobilization in ischemic heart failure patients. Atherosclerosis 2015, 238, 159–164. [Google Scholar] [CrossRef] [PubMed]
- Tousoulis, D.; Andreou, I.; Tsiatas, M.; Miliou, A.; Tentolouris, C.; Siasos, G.; Papageorgiou, N.; Papadimitriou, C.A.; Dimopoulos, M.A.; Stefanadis, C. Effects of rosuvastatin and allopurinol on circulating endothelial progenitor cells in patients with congestive heart failure: The impact of inflammatory process and oxidative stress. Atherosclerosis 2011, 214, 151–157. [Google Scholar] [CrossRef] [PubMed]
- Erbs, S.; Beck, E.B.; Linke, A.; Adams, V.; Gielen, S.; Krankel, N.; Mobius-Winkler, S.; Hollriegel, R.; Thiele, H.; Hambrecht, R.; et al. High-dose rosuvastatin in chronic heart failure promotes vasculogenesis, corrects endothelial function, and improves cardiac remodeling--results from a randomized, double-blind, and placebo-controlled study. Int. J. Cardiol. 2011, 146, 56–63. [Google Scholar] [CrossRef]
- Tousoulis, D.; Antoniades, C.; Bosinakou, E.; Kotsopoulou, M.; Pitsavos, C.; Vlachopoulos, C.; Panagiotakos, D.; Stefanadis, C. Effects of atorvastatin on reactive hyperemia and inflammatory process in patients with congestive heart failure. Atherosclerosis 2005, 178, 359–363. [Google Scholar] [CrossRef]
- Tousoulis, D.; Oikonomou, E.; Siasos, G.; Chrysohoou, C.; Zaromitidou, M.; Kioufis, S.; Maniatis, K.; Dilaveris, P.; Miliou, A.; Michalea, S.; et al. Dose-dependent effects of short term atorvastatin treatment on arterial wall properties and on indices of left ventricular remodeling in ischemic heart failure. Atherosclerosis 2013, 227, 367–372. [Google Scholar] [CrossRef]
- Gounari, P.; Tousoulis, D.; Antoniades, C.; Kampoli, A.M.; Stougiannos, P.; Papageorgiou, N.; Roulia, G.; Stefanadi, E.; Siasos, G.; Tsioufis, C.; et al. Rosuvastatin but not ezetimibe improves endothelial function in patients with heart failure, by mechanisms independent of lipid lowering. Int. J. Cardiol. 2010, 142, 87–91. [Google Scholar] [CrossRef]
- do Vale, G.T.; Ceron, C.S.; Gonzaga, N.A.; Simplicio, J.A.; Padovan, J.C. Three Generations of beta-blockers: History, Class Differences and Clinical Applicability. Curr. Hypertens. Rev. 2019, 15, 22–31. [Google Scholar] [CrossRef] [PubMed]
- Peller, M.; Ozieranski, K.; Balsam, P.; Grabowski, M.; Filipiak, K.J.; Opolski, G. Influence of beta-blockers on endothelial function: A meta-analysis of randomized controlled trials. Cardiol. J. 2015, 22, 708–716. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ambrosioni, E.; Bacchelli, S.; Esposti, D.D.; Borghi, C. Beta-blockade in hypertension and congestive heart failure. J. Cardiovasc. Pharmacol. 2001, 38 (Suppl. 3), S25–S31. [Google Scholar] [CrossRef] [PubMed]
- Chin, B.S.; Langford, N.J.; Nuttall, S.L.; Gibbs, C.R.; Blann, A.D.; Lip, G.Y. Anti-oxidative properties of beta-blockers and angiotensin-converting enzyme inhibitors in congestive heart failure. Eur. J. Heart Fail. 2003, 5, 171–174. [Google Scholar] [CrossRef]
- Bernjak, A.; Clarkson, P.B.; McClintock, P.V.; Stefanovska, A. Low-frequency blood flow oscillations in congestive heart failure and after beta1-blockade treatment. Microvasc. Res. 2008, 76, 224–232. [Google Scholar] [CrossRef]
- Turchetti, V.; Bellini, M.A.; Boschi, L.; Postorino, G.; Pallassini, A.; Richichi, M.G.; Trabalzini, L.; Guerrini, M.; Forconi, S. Haemorheological and endothelial-dependent alterations in heart failure after ACE inhibitor, calcium antagonist and beta blocker. Clin. Hemorheol. Microcirc. 2002, 27, 209–218. [Google Scholar]
- Poelzl, G.; Frick, M.; Lackner, B.; Huegel, H.; Alber, H.F.; Mair, J.; Herold, M.; Schwarzacher, S.P.; Pachinger, O.; Weidinger, F. Short-term improvement in submaximal exercise capacity by optimized therapy with ACE inhibitors and beta blockers in heart failure patients is associated with restoration of peripheral endothelial function. Int. J. Cardiol. 2006, 108, 48–54. [Google Scholar] [CrossRef]
- Castro, P.; Vukasovic, J.L.; Chiong, M.; Diaz-Araya, G.; Alcaino, H.; Copaja, M.; Valenzuela, R.; Greig, D.; Perez, O.; Corbalan, R.; et al. Effects of carvedilol on oxidative stress and chronotropic response to exercise in patients with chronic heart failure. Eur. J. Heart Fail. 2005, 7, 1033–1039. [Google Scholar] [CrossRef]
- Alfieri, A.B.; Briceno, L.; Fragasso, G.; Spoladore, R.; Palloshi, A.; Bassanelli, G.; Montano, C.; Arioli, F.; Cuko, A.; Ruotolo, G.; et al. Differential long-term effects of carvedilol on proinflammatory and antiinflammatory cytokines, asymmetric dimethylarginine, and left ventricular function in patients with heart failure. J. Cardiovasc. Pharmacol. 2008, 52, 49–54. [Google Scholar] [CrossRef]
- Falskov, B.; Hermann, T.S.; Raunso, J.; Christiansen, B.; Rask-Madsen, C.; Major-Pedersen, A.; Kober, L.; Torp-Pedersen, C.; Dominguez, H. Endothelial function is unaffected by changing between carvedilol and metoprolol in patients with heart failure--a randomized study. Cardiovasc. Diabetol. 2011, 10, 91. [Google Scholar] [CrossRef] [Green Version]
- Ding, Y.; Wei, Z.; Li, J.; Zhu, L. Effects of Metoprolol Succinate Combined with Entresto on Cardiac Function Indexes and Coagulation Function in Patients with Congestive Heart Failure. Comput. Math Methods Med. 2022, 2022, 9765884. [Google Scholar] [CrossRef]
- Gibbs, C.R.; Blann, A.D.; Watson, R.D.; Lip, G.Y. Abnormalities of hemorheological, endothelial, and platelet function in patients with chronic heart failure in sinus rhythm: Effects of angiotensin-converting enzyme inhibitor and beta-blocker therapy. Circulation 2001, 103, 1746–1751. [Google Scholar] [CrossRef] [Green Version]
- Fortini, F.; Vieceli Dalla Sega, F.; Marracino, L.; Severi, P.; Rapezzi, C.; Rizzo, P.; Ferrari, R. Well-Known and Novel Players in Endothelial Dysfunction: Updates on a Notch(ed) Landscape. Biomedicines 2021, 9, 997. [Google Scholar] [CrossRef] [PubMed]
- Joannides, R.; Bizet-Nafeh, C.; Costentin, A.; Iacob, M.; Derumeaux, G.; Cribier, A.; Thuillez, C. Chronic ACE inhibition enhances the endothelial control of arterial mechanics and flow-dependent vasodilatation in heart failure. Hypertension 2001, 38, 1446–1450. [Google Scholar] [CrossRef] [Green Version]
- Hryniewicz, K.; Dimayuga, C.; Hudaihed, A.; Androne, A.S.; Zheng, H.; Jankowski, K.; Katz, S.D. Inhibition of angiotensin-converting enzyme and phosphodiesterase type 5 improves endothelial function in heart failure. Clin. Sci. 2005, 108, 331–338. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Safonova, J.I.; Kozhevnikova, M.V.; Danilogorskaya, Y.A.; Zheleznykh, E.A.; Ilgisonis, I.S.; Privalova, E.V.; Khabarova, N.V.; Belenkov, Y.N. Possible pathway for heart failure with preserved ejection fraction prevention and treatment: The angiotensin-converting enzyme inhibitor effect on endothelial function in comorbid patients. Kardiologiia 2022, 62, 65–71. [Google Scholar] [CrossRef] [PubMed]
- Besler, C.; Doerries, C.; Giannotti, G.; Luscher, T.F.; Landmesser, U. Pharmacological approaches to improve endothelial repair mechanisms. Expert. Rev. Cardiovasc. Ther. 2008, 6, 1071–1082. [Google Scholar] [CrossRef]
- Imanishi, T.; Hano, T.; Nishio, I. Angiotensin II accelerates endothelial progenitor cell senescence through induction of oxidative stress. J. Hypertens. 2005, 23, 97–104. [Google Scholar] [CrossRef]
- Chuang, S. Mathematic models for cancer chemotherapy: Pharmacokinetic and cell kinetic considerations. Cancer Chemother. Rep. 1975, 59, 827–842. [Google Scholar]
- Levi, A.; Leshem-Lev, D.; Weissler-Snir, A.; Hasin, T.; Mats, I.; Murninkas, D.; Kornowski, R.; Lev, E.I.; Ben-Gal, T. The Effect of Mineralocorticoid Receptor Antagonists on Recruitment and Function of Endothelial Progenitor Cells in Patients with Congestive Heart Failure. Isr. Med. Assoc. J. 2018, 20, 233–238. [Google Scholar]
- Marumo, T.; Uchimura, H.; Hayashi, M.; Hishikawa, K.; Fujita, T. Aldosterone impairs bone marrow-derived progenitor cell formation. Hypertension 2006, 48, 490–496. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Farquharson, C.A.; Struthers, A.D. Spironolactone increases nitric oxide bioactivity, improves endothelial vasodilator dysfunction, and suppresses vascular angiotensin I/angiotensin II conversion in patients with chronic heart failure. Circulation 2000, 101, 594–597. [Google Scholar] [CrossRef] [Green Version]
- Abiose, A.K.; Mansoor, G.A.; Barry, M.; Soucier, R.; Nair, C.K.; Hager, D. Effect of spironolactone on endothelial function in patients with congestive heart failure on conventional medical therapy. Am. J. Cardiol. 2004, 93, 1564–1566. [Google Scholar] [CrossRef] [PubMed]
- Gager, G.M.; von Lewinski, D.; Sourij, H.; Jilma, B.; Eyileten, C.; Filipiak, K.; Hulsmann, M.; Kubica, J.; Postula, M.; Siller-Matula, J.M. Effects of SGLT2 Inhibitors on Ion Homeostasis and Oxidative Stress associated Mechanisms in Heart Failure. Biomed. Pharmacother. 2021, 143, 112169. [Google Scholar] [CrossRef] [PubMed]
- Cappetta, D.; De Angelis, A.; Bellocchio, G.; Telesca, M.; Cianflone, E.; Torella, D.; Rossi, F.; Urbanek, K.; Berrino, L. Sodium-Glucose Cotransporter 2 Inhibitors and Heart Failure: A Bedside-to-Bench Journey. Front. Cardiovasc. Med. 2021, 8, 810791. [Google Scholar] [CrossRef] [PubMed]
- Packer, M. Autophagy stimulation and intracellular sodium reduction as mediators of the cardioprotective effect of sodium-glucose cotransporter 2 inhibitors. Eur. J. Heart Fail. 2020, 22, 618–628. [Google Scholar] [CrossRef] [Green Version]
- Packer, M. SGLT2 Inhibitors Produce Cardiorenal Benefits by Promoting Adaptive Cellular Reprogramming to Induce a State of Fasting Mimicry: A Paradigm Shift in Understanding Their Mechanism of Action. Diabetes Care 2020, 43, 508–511. [Google Scholar] [CrossRef] [Green Version]
- Dyck, J.R.B.; Sossalla, S.; Hamdani, N.; Coronel, R.; Weber, N.C.; Light, P.E.; Zuurbier, C.J. Cardiac mechanisms of the beneficial effects of SGLT2 inhibitors in heart failure: Evidence for potential off-target effects. J. Mol. Cell Cardiol. 2022, 167, 17–31. [Google Scholar] [CrossRef]
- Theofilis, P.; Sagris, M.; Oikonomou, E.; Antonopoulos, A.S.; Siasos, G.; Tsioufis, K.; Tousoulis, D. Pleiotropic effects of SGLT2 inhibitors and heart failure outcomes. Diabetes Res. Clin. Pract. 2022, 188, 109927. [Google Scholar] [CrossRef]
- Kolijn, D.; Pabel, S.; Tian, Y.; Lodi, M.; Herwig, M.; Carrizzo, A.; Zhazykbayeva, S.; Kovacs, A.; Fulop, G.A.; Falcao-Pires, I.; et al. Empagliflozin improves endothelial and cardiomyocyte function in human heart failure with preserved ejection fraction via reduced pro-inflammatory-oxidative pathways and protein kinase Galpha oxidation. Cardiovasc. Res. 2021, 117, 495–507. [Google Scholar] [CrossRef]
- Koyani, C.N.; Plastira, I.; Sourij, H.; Hallstrom, S.; Schmidt, A.; Rainer, P.P.; Bugger, H.; Frank, S.; Malle, E.; von Lewinski, D. Empagliflozin protects heart from inflammation and energy depletion via AMPK activation. Pharmacol. Res. 2020, 158, 104870. [Google Scholar] [CrossRef]
- Heidenreich, P.A.; Bozkurt, B.; Aguilar, D.; Allen, L.A.; Byun, J.J.; Colvin, M.M.; Deswal, A.; Drazner, M.H.; Dunlay, S.M.; Evers, L.R.; et al. 2022 AHA/ACC/HFSA Guideline for the Management of Heart Failure: A Report of the American College of Cardiology/American Heart Association Joint Committee on Clinical Practice Guidelines. Circulation 2022, 145, e895–e1032. [Google Scholar] [CrossRef]
- Menendez, J.T. The Mechanism of Action of LCZ696. Card Fail. Rev. 2016, 2, 40–46. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amore, L.; Alghisi, F.; Pancaldi, E.; Pascariello, G.; Cersosimo, A.; Cimino, G.; Bernardi, N.; Calvi, E.; Lombardi, C.M.; Sciatti, E.; et al. Study of endothelial function and vascular stiffness in patients affected by dilated cardiomyopathy on treatment with sacubitril/valsartan. Am. J. Cardiovasc. Dis. 2022, 12, 125–135. [Google Scholar] [CrossRef] [PubMed]
- Li, B.H.; Fang, K.F.; Lin, P.H.; Zhang, Y.H.; Huang, Y.X.; Jie, H. Effect of sacubitril valsartan on cardiac function and endothelial function in patients with chronic heart failure with reduced ejection fraction. Clin. Hemorheol. Microcirc. 2021, 77, 425–433. [Google Scholar] [CrossRef] [PubMed]
- Nathaniel, S.; McGinty, S.; Witman, M.A.H.; Edwards, D.G.; Farquhar, W.B.; Hosmane, V.; Wenner, M.M. Impact of angiotensin receptor-neprilysin inhibition on vascular function in heart failure with reduced ejection fraction: A pilot study. Physiol. Rep. 2022, 10, e15209. [Google Scholar] [CrossRef]
- Angadi, S.S.; Mookadam, F.; Lee, C.D.; Tucker, W.J.; Haykowsky, M.J.; Gaesser, G.A. High-intensity interval training vs. moderate-intensity continuous exercise training in heart failure with preserved ejection fraction: A pilot study. J. Appl. Physiol. (1985) 2015, 119, 753–758. [Google Scholar] [CrossRef]
- Winzer, E.B.; Gaida, P.; Hollriegel, R.; Fischer, T.; Linke, A.; Schuler, G.; Adams, V.; Erbs, S. Impact of Rosuvastatin Treatment on HDL-Induced PKC-betaII and eNOS Phosphorylation in Endothelial Cells and Its Relation to Flow-Mediated Dilatation in Patients with Chronic Heart Failure. Cardiol. Res. Pract. 2016, 2016, 4826102. [Google Scholar] [CrossRef] [Green Version]
- Ellis, G.R.; Nightingale, A.K.; Blackman, D.J.; Anderson, R.A.; Mumford, C.; Timmins, G.; Lang, D.; Jackson, S.K.; Penney, M.D.; Lewis, M.J.; et al. Addition of candesartan to angiotensin converting enzyme inhibitor therapy in patients with chronic heart failure does not reduce levels of oxidative stress. Eur. J. Heart Fail. 2002, 4, 193–199. [Google Scholar] [CrossRef] [Green Version]
- Nakamura, M.; Saito, S.; Yoshida, H.; Arakawa, N.; Sugawara, S.; Hiramori, K. Effects of chronic subdepressor dose of angiotensin II type 1 receptor antagonist on endothelium-dependent vasodilation in patients with congestive heart failure. J. Cardiovasc. Pharmacol. 2002, 40, 411–419. [Google Scholar] [CrossRef]
- Macdonald, J.E.; Kennedy, N.; Struthers, A.D. Effects of spironolactone on endothelial function, vascular angiotensin converting enzyme activity, and other prognostic markers in patients with mild heart failure already taking optimal treatment. Heart 2004, 90, 765–770. [Google Scholar] [CrossRef] [PubMed]
- Correale, M.; Mazzeo, P.; Mallardi, A.; Leopizzi, A.; Tricarico, L.; Fortunato, M.; Magnesa, M.; Tucci, S.; Maiellaro, P.; Pastore, G.; et al. Switch to SGLT2 Inhibitors and Improved Endothelial Function in Diabetic Patients with Chronic Heart Failure. Cardiovasc. Drugs Ther. 2022, 36, 1157–1164. [Google Scholar] [CrossRef] [PubMed]
- Sezai, A.; Sekino, H.; Unosawa, S.; Taoka, M.; Osaka, S.; Tanaka, M. Canagliflozin for Japanese patients with chronic heart failure and type II diabetes. Cardiovasc. Diabetol. 2019, 18, 76. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bunsawat, K.; Ratchford, S.M.; Alpenglow, J.K.; Park, S.H.; Jarrett, C.L.; Stehlik, J.; Smith, A.S.; Richardson, R.S.; Wray, D.W. Sacubitril-valsartan improves conduit vessel function and functional capacity and reduces inflammation in heart failure with reduced ejection fraction. J. Appl. Physiol. (1985) 2021, 130, 256–268. [Google Scholar] [CrossRef]
- Du, H.; Li, X.; Zhao, W.; Jiang, N. The Difference between Sacubitril Valsartan and Valsartan on Vascular Endothelial Function, APN, MMP-9, and BNP Levels in Patients with Hypertension and Chronic Heart Failure. J. Healthc. Eng. 2022, 2022, 9494981. [Google Scholar] [CrossRef]
- Karavolias, G.K.; Georgiadou, P.; Gkouziouta, A.; Kariofillis, P.; Karabela, G.; Tsiapras, D.; Sbarouni, E.; Chaidaroglou, A.; Degiannis, D.; Adamopoulos, S.; et al. Short and long term anti-inflammatory effects of bosentan therapy in patients with pulmonary arterial hypertension: Relation to clinical and hemodynamic responses. Expert Opin. Ther. Targets 2010, 14, 1283–1289. [Google Scholar] [CrossRef]
- Iannone, F.; Riccardi, M.T.; Guiducci, S.; Bizzoca, R.; Cinelli, M.; Matucci-Cerinic, M.; Lapadula, G. Bosentan regulates the expression of adhesion molecules on circulating T cells and serum soluble adhesion molecules in systemic sclerosis-associated pulmonary arterial hypertension. Ann. Rheum. Dis. 2008, 67, 1121–1126. [Google Scholar] [CrossRef]
- Sfikakis, P.P.; Papamichael, C.; Stamatelopoulos, K.S.; Tousoulis, D.; Fragiadaki, K.G.; Katsichti, P.; Stefanadis, C.; Mavrikakis, M. Improvement of vascular endothelial function using the oral endothelin receptor antagonist bosentan in patients with systemic sclerosis. Arthritis Rheum. 2007, 56, 1985–1993. [Google Scholar] [CrossRef]
- Eroglu, E.; Kocyigit, I.; Lindholm, B. The endothelin system as target for therapeutic interventions in cardiovascular and renal disease. Clin. Chim. Acta 2020, 506, 92–106. [Google Scholar] [CrossRef]
- Schneider, M.P.; Boesen, E.I.; Pollock, D.M. Contrasting actions of endothelin ET(A) and ET(B) receptors in cardiovascular disease. Annu. Rev. Pharmacol. Toxicol. 2007, 47, 731–759. [Google Scholar] [CrossRef] [Green Version]
- Price, L.C.; Howard, L.S. Endothelin receptor antagonists for pulmonary arterial hypertension: Rationale and place in therapy. Am. J. Cardiovasc. Drugs 2008, 8, 171–185. [Google Scholar] [CrossRef] [PubMed]
- Bisserier, M.; Pradhan, N.; Hadri, L. Current and emerging therapeutic approaches to pulmonary hypertension. Rev. Cardiovasc. Med. 2020, 21, 163–179. [Google Scholar] [CrossRef] [PubMed]
- Ilsar, R.; Levitt, J.; Ng, M.K.; Kritharides, L.; Adams, M.R.; Celermajer, D.S. Bosentan and improved pulmonary endothelial function in pulmonary arterial hypertension. Eur. Respir. J. 2010, 36, 1483–1485. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pearl, J.M.; Wellmann, S.A.; McNamara, J.L.; Lombardi, J.P.; Wagner, C.J.; Raake, J.L.; Nelson, D.P. Bosentan prevents hypoxia-reoxygenation-induced pulmonary hypertension and improves pulmonary function. Ann. Thorac. Surg. 1999, 68, 1714–1721, discussion 1712–1721. [Google Scholar] [CrossRef]
- Yagi, S.; Akaike, M.; Iwase, T.; Kusunose, K.; Niki, T.; Yamaguchi, K.; Koshiba, K.; Yoshida, S.; Sumitomo-Ueda, Y.; Aihara, K.; et al. Bosentan ameliorated exercise-induced pulmonary arterial hypertension complicated with systemic sclerosis. Intern. Med. 2010, 49, 2309–2312. [Google Scholar] [CrossRef] [Green Version]
- Cella, G.; Vianello, F.; Cozzi, F.; Marotta, H.; Tona, F.; Saggiorato, G.; Iqbal, O.; Fareed, J. Effect of bosentan on plasma markers of endothelial cell activity in patients with secondary pulmonary hypertension related to connective tissue diseases. J. Rheumatol. 2009, 36, 760–767. [Google Scholar] [CrossRef]


{kind=link}
| Study | Study Design | Key Findings |
|---|---|---|
| Exercise Training (ET) | ||
| Sandri M et al., 2016 [198] | RCT; 60 patients with chronic HF vs. 60 controls. | ET vs. control group improved FMD, CD34/KDR+ EPCs, and migratory capacity of cultured mononuclear cells. |
| Chen J et al., 2021 [199] | RCT; 80 patients with chronic HF and ΕΤ. | ET improved LVEF and LVFS. Additionally, ET had higher EPCs levels and proliferation ability and lower BNP levels and EPCs apoptosis rate. |
| Isaksen K et al., 2019 [132] | Controlled prospective trial; 30 patients with ischemic HF and ICD. | ET vs. control group improved peak VO2 and endothelial function. |
| Kitzman DW et al., 2013 [202] | RCT; 63 patients with HFpEF. | ET improved peak VO2 and quality of life but no endothelial function. |
| Angadi SS et al., 2015 [247] | RCT; 19 patients with HFpEF. | High-intensity interval training improved peak VO2, LV diastolic dysfunction. No effect on endothelial function was demonstrated. |
| Statins | ||
| Oikonomou E et al., 2015 [205] | RCT; 26 patients with stable HF; evaluation of atorvastatin 10 mg/day vs. atorvastatin 40 mg/day for 4 weeks. | 40 mg/day Atorvastatin demonstrated higher EPCs and FMD values and decreased TNF-α levels in both compared groups. |
| Tousoulis D et al., 2011 [206] | RCT; 60 patients with systolic HF; administration of rosuvastatin 10 mg/day vs. allopurinol 300 mg/day or placebo. | Rosuvastatin group had increased CD34/KDR+, CD34/CD133/KDR+ and EPCs levels. Additionally, improvement of endothelial was observed. |
| Erbs S et al., 2011 [207] | RCT; 42 patients with chronic HF randomized to 12 weeks of oral rosuvastatin (40 mg/d) or placebo. | Rosuvastatin 40 mg/day increased VEGF levels, CD34+ stem cell count, number of CD34/KDR+ EPCs, EPC integrative capacity, and FMD. |
| Tousoulis D et al., 2005 [208] | RCT; 38 patients with HF: Group 1: atorvastatin 10 mg/day (n = 19), group 2: control (n = 19), duration of treatment: 4 weeks. | Atorvastatin 10 mg/day improved forearm vasodilatory response to reactive hyperemia; decreased levels of IL-6, TNF-α, and sVCAM-1. |
| Tousoulis D et al., 2013 [209] | RCT; atorvastatin in 22 patients with ischemic HF. | Atorvastatin 40 mg/day vs. 10 mg/day significantly improved FMD and AIx values. |
| Winzer EB et al., 2016 [248] | RCT; 18 patients with chronic HF; randomized to 12 weeks of rosuvastatin vs. placebo | Rosuvastatin improved FMD and LDL cholesterol levels. Moreover, deterioration of FMD and LDL after cessation of therapy was detected. |
| Angiotensin-converting enzyme inhibitors (ACEi)/Angiotensin Receptor Inhibitors (ARBs) | ||
| Gibbs CR et al., 2001 [222] | Cross-sectional study; 120 patients with chronic HF and sinus rhythm; n = 20 on lisinopril vs. n = 20 on β blocker (carvedilol or bisoprolol) | Initiation of beta-blocker therapy revealed no significant changes in hemorheological, endothelial function, and platelet indices. Moreover, initiation of ACEi decreased fibrinogen and vWF levels. |
| Hryniewicz K et al., 2005 [225] | RCT, 64 patients with chronic HF; randomization in placebo vs. 10 mg ramipril vs. 50 mg sildenafil vs. combination of ramipril/sildenafil. | Ramipril vs. placebo increased FMD at 4 h. Moreover, Sildenafil vs. placebo increased FMD at 1, 2, and 4 h. Combination of sildenafil/ramipril vs. placebo increased FMD at 1, 2, and 4 h. |
| Safonova JI et al., 2022 [226] | Cross-sectional study; 40 patients with HF (n = 20 patients with HFpEF; n = 20 patients with HFmrEF); administration of 12-months perindopril. | Perindopril increased in phase shift in both HFpEF and HFmrEF; increase in occlusion index in both HFpEF and HFmrEF; decreased E-selectin in both HFpEF and HFmrEF; ET-1 levels significantly decreased only in HFpEF |
| Ellis GR et al., 2002 [249] | RCT; 28 patients with HF on ACEi; randomization to candesartan vs. placebo | Candesartan vs. placebo has no effects on brachial artery FMD, exercise capacity (peak VO2), and biomarkers of oxidative stress. |
| Nakamura M et al., 2002 [250] | RCT; 26 patients with congestive HF that randomized to losartan vs. placebo. | Losartan group revealed increased forearm blood flow in response to intra-arterial infusion of acetylcholine. |
| Mineralocorticoid Receptor Antagonists (MRAs) | ||
| Farquharson et al., 2000 [232] | RCT; 10 patients with chronic HF on standard diuretic/ACEi therapy were randomized to 50 mg/day spironolactone vs. placebo for 1 month. | Spironolactone improved forearm blood flow response to acetylcholine increased NO bioactivity, and inhibition of vascular angiotensin I/angiotensin II conversion. |
| Abiose AK et al., 2004 [233] | Cross-sectional; n = 20 patients with congestive HF; administration of spironolactone. | Administration of spironolactone improved FMD at 4 and 8 weeks. |
| Macdonald JE et al., 2004 [251] | RCT; 43 patients with congestive HF under ACEi and beta blockers; administration of 12.5-50 mg/day spironolactone vs. placebo for 3 months. | Administration of spironolactone increased acetylcholine-mediated vasodilatation and vascular ACE activity. Moreover, spironolactone decreased BNP and procollagen III N-terminal peptide. |
| Sodium-glucose cotransporter 2 (SGLT-2) inhibitors | ||
| Correale M et al., 2021 [252] | Cross-sectional study; 22 patients with chronic HF and type 2 diabetes mellitus vs. n = 23 controls treated with other antidiabetic drugs | SGLT2 i administration improved endothelial function and arterial stiffness. |
| Sezai A et al., 2019 [253] | Prospective cohort study; 35 Japanese patients with chronic HF; administration of canagliflozin for 12 months. | Administration of canagliflozin decreased fat content at 12 months. Moreover, significant decrease in natriuretic peptides, improvement of renal function, FMD, E/e’ and oxidized LDL levels were reported after canagliflozin administration. |
| Sacubitril/Valsartan | ||
| Li BH et al., 2021 [245] | RCT; 80 patients with HFrEF; randomized to observation group (n = 40, sacubitril/valsartan plus conventional treatment) vs. control group (n = 40, perindopril plus conventional treatment) for 12 weeks. | Sacubitril valsartan improves endothelial function while increasing cardiac function in HFrEF patients. |
| Bunsawat K et al., 2021 [254] | Prospective cohort study; 11 patients with HFrEF under optimal treatment; administration of sacubitril-valsartan for 3 months. | Administration of sacubitril/valsartan improved FMD at 1 month and at 2-3 months. Moreover, decreased levels of TNF-α and IL-18 were reported. |
| Du H et al., 2022 [255] | RCT; 60 patients with chronic HF and hypertension; randomly divided into observation group (n = 30, sacubitril/valsartan) and control group (n = 30, valsartan) for 6 months. | Sacubitril/valsartan subjects reported improvement in endothelium-dependent vasodilation, serum NO, and decreased ET-1 levels. |
| Nathaniel S et al., 2022 [246] | Case-control study; 20 HFrEF patients (n = 10 on sacubitril/valsartan vs. n = 10 on conventional treatment with ACEi/ARBs) for 12 weeks. | Sacubitril/valsartan decreased PWV and improved FMD. |
| Amore L et al., 2022 [244] | Prospective cohort study; 15 patients with dilated cardiomyopathy and reduced LVEF; administration of sacubitril/valsartan for 6 months. | Administration of sacubitril/valsartan shows an increase in reactive hyperemia index and AIx six months after first administration. |
| Endothelin receptor antagonists (ERAs) | ||
| Karavolias GK et al., 2010 [256] | Cross-sectional study; 16 patients with moderate-severe idiopathic PAH under conventional treatment; administration of bosentan (62.5 mg twice daily for 1 month followed by 125 mg twice daily for 11 months). | Bosentan therapy modifies endothelial cell activation by down-regulating the levels of ICAM-1 at 2 months. |
| Iannone F et al., 2008 [257] | Cross-sectional study; 35 patients with systemic sclerosis (n = 10 with isolated PAH) vs. n = 25 healthy subjects; administration of bosentan in patients with isolated PAH. | Administration of bosentan for 12 months down-regulated endothelial activation and reduced ICAM-1, VCAM-1, P-selectin, PECAM-1, CD3 LFA-1 T, and CD3-L-selectin T-cell levels. |
| Sfikakis PP et al., 2007 [258] | RCT; Cross-sectional study, 12 patients with systemic sclerosis/PAH who received bosentan for 4 weeks vs. n = 12 patients without bosentan. | Small doses of bosentan improve endothelial function without affecting hemodynamic parameters or endothelial activation-related processes. |
| Clinical Trial Identifier/Official Title | Study Design/Estimated Enrollment/Inclusion Criteria | Primary Outcome Measures | Secondary Outcome Measures |
|---|---|---|---|
| NCT04539093 “Assessment of Endothelial Function in Patients with Advanced Heart Failure Requiring Mechanical Circulatory Support” | Prospective cohort; n = 20 patients with end-stage HF scheduled for LVAD implantation | Evaluation of endothelial function (blood NO levels, FMD) Evaluation of microvascular function (contrast-enhanced ultrasound of the peripheral skeletal muscle of lower extremities) | Functional outcomes: Quality of Life (KCCQ); Mobility (6 MWT); Handgrip; Lower extremity strength; Ventilation and gas exchange (CPET) |
| NCT05230732 “Neuromodulation of Inflammation and Endothelial Function to Treat Elderly Patients With Systolic Heart Failure” | Prospective, randomized, double-blind study; n = 158 patients with systolic HF and EF < 40%; use of Low-level Tragus stimulation- vs. sham treatment for 1 h daily/12 weeks | 6 MWT | Quality of life (Minnesota living with heart failure questionnaire) FMD HRV Inflammatory cytokines |
| NCT02997462 “Monocyte Phenotypic Changes in Heart Failure” | Prospective cohort; n = 60 patients with HF admitted to the ICU or HF service vs. healthy, age-matched controls | Change in IL-6 levels between hospital admission and discharge | Evaluation of cell surface markers, cytokines, monocyte-gene expression, microRNA, monocyte/macrophage morphology, markers of oxidative stress and inflammation at admission, discharge, and at first post-discharge appointment |
| NCT04323371 “Cardiogenic Shock Integrated PHenotyping for Event Reduction” | Prospective observational study; n = 26 patients with acute decompensated HF complicated by cardiogenic shock | Markers of inflammation, endothelial permeability, endothelial glycocalyx perturbation, and metabolomics profile | |
| NCT05498584 “Targeting LOXL2 and Cardiac Fibrosis for Post-acute Heart Failure Treatment- A Prospective Study” | Prospective cohort study; n = 126 patients with HF (EF ≤ 40%) admitted for worsening HF in 3 years; administration of a cardiac rehabilitation program according to cardiopulmonary exercise test | All-cause mortality | HF readmission FMD (change from baseline to 6 months) KCCQ (change from baseline to 6 and 12 months) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tsigkou, V.; Oikonomou, E.; Anastasiou, A.; Lampsas, S.; Zakynthinos, G.E.; Kalogeras, K.; Katsioupa, M.; Kapsali, M.; Kourampi, I.; Pesiridis, T.; et al. Molecular Mechanisms and Therapeutic Implications of Endothelial Dysfunction in Patients with Heart Failure. Int. J. Mol. Sci. 2023, 24, 4321. https://doi.org/10.3390/ijms24054321
Tsigkou V, Oikonomou E, Anastasiou A, Lampsas S, Zakynthinos GE, Kalogeras K, Katsioupa M, Kapsali M, Kourampi I, Pesiridis T, et al. Molecular Mechanisms and Therapeutic Implications of Endothelial Dysfunction in Patients with Heart Failure. International Journal of Molecular Sciences. 2023; 24(5):4321. https://doi.org/10.3390/ijms24054321
Chicago/Turabian StyleTsigkou, Vasiliki, Evangelos Oikonomou, Artemis Anastasiou, Stamatios Lampsas, George E. Zakynthinos, Konstantinos Kalogeras, Maria Katsioupa, Maria Kapsali, Islam Kourampi, Theodoros Pesiridis, and et al. 2023. "Molecular Mechanisms and Therapeutic Implications of Endothelial Dysfunction in Patients with Heart Failure" International Journal of Molecular Sciences 24, no. 5: 4321. https://doi.org/10.3390/ijms24054321