
Structure–Activity Relationship Analysis of Rhosin, a RhoA GTPase Inhibitor, Reveals a New Class of Antiplatelet Agents
 , , , and
, , , and
Abstract
: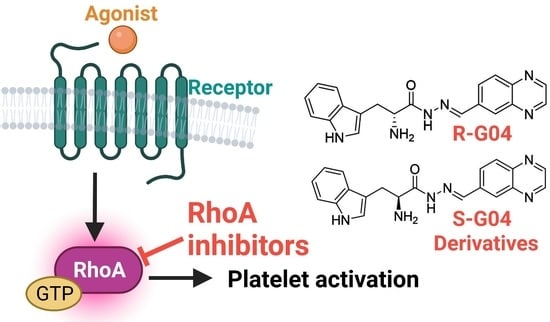
1. Introduction
2. Results and Discussion
3. Materials and Methods
3.1. Experimental Section
3.2. Virtual Screening
3.3. Ultra-High-Performance Liquid Chromatography Coupled with High-Resolution Mass Spectrometry Analysis (UHPLC-HRMS)
3.4. Collection of Blood and Preparation of Washed Human Platelet Suspensions
3.5. Lactate Dehydrogenase (LDH)-Based Cytotoxicity Assay
3.6. Antiplatelet Aggregation Activity and Secretion of ATP
3.7. RhoA GTPase Assay and Phosphorylation of MLC
3.8. In Vitro Complex-Formation Assay
3.9. Thromboxane B2 ELISA Assay
3.10. Statistical Analysis
4. Conclusions and Future Directions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Versteeg, H.H.; Heemskerk, J.W.; Levi, M.; Reitsma, P.H. New fundamentals in hemostasis. Physiol. Rev. 2013, 93, 327–358. [Google Scholar] [CrossRef] [Green Version]
- Kakouros, N.; Rade, J.J.; Kourliouros, A.; Resar, J.R. Platelet function in patients with diabetes mellitus: From a theoretical to a practical perspective. Int. J. Endocrinol. 2011, 2011, 742719. [Google Scholar] [CrossRef]
- Lievens, D.; von Hundelshausen, P. Platelets in atherosclerosis. Thromb. Haemost. 2011, 106, 827–838. [Google Scholar] [PubMed]
- Lip, G.Y.H. Hypertension, Platelets, and the Endothelium. Hypertension 2003, 41, 199–200. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fuster, V.; Sweeny, J.M. Aspirin. Circulation 2011, 123, 768–778. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Degrauwe, S.; Pilgrim, T.; Aminian, A.; Noble, S.; Meier, P.; Iglesias, J.F. Dual antiplatelet therapy for secondary prevention of coronary artery disease. Open Heart 2017, 4, e000651. [Google Scholar] [CrossRef] [Green Version]
- Eikelboom, J.W.; Hirsh, J.; Spencer, F.A.; Baglin, T.P.; Weitz, J.I. Antiplatelet drugs: Antithrombotic Therapy and Prevention of Thrombosis, 9th ed: American College of Chest Physicians Evidence-Based Clinical Practice Guidelines. Chest 2012, 141 (Suppl. 2), e89S–e119S. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iqbal, A.M.; Lopez, R.A.; Hai, O. Antiplatelet Medications; StatPearls: Treasure Island, FL, USA, 2022. [Google Scholar]
- Thachil, J. Antiplatelet therapy—A summary for the general physicians. Clin. Med. 2016, 16, 152–160. [Google Scholar] [CrossRef] [Green Version]
- Angiolillo, D.J. Variability in responsiveness to oral antiplatelet therapy. Am. J. Cardiol. 2009, 103 (Suppl. 3), 27A–34A. [Google Scholar] [CrossRef]
- Di Minno, M.N.; Guida, A.; Camera, M.; Colli, S.; Di Minno, G.; Tremoli, E. Overcoming limitations of current antiplatelet drugs: A concerted effort for more profitable strategies of intervention. Ann. Med. 2011, 43, 531–544. [Google Scholar] [CrossRef] [PubMed]
- Shen, B.; Delaney, M.K.; Du, X. Inside-out, outside-in, and inside-outside-in: G protein signaling in integrin-mediated cell adhesion, spreading, and retraction. Curr. Opin. Cell. Biol. 2012, 24, 600–606. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Flevaris, P.; Li, Z.; Zhang, G.; Zheng, Y.; Liu, J.; Du, X. Two distinct roles of mitogen-activated protein kinases in platelets and a novel Rac1-MAPK-dependent integrin outside-in retractile signaling pathway. Blood 2009, 113, 893–901. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Akbar, H.; Shang, X.; Perveen, R.; Berryman, M.; Funk, K.; Johnson, J.F.; Tandon, N.N.; Zheng, Y. Gene targeting implicates Cdc42 GTPase in GPVI and non-GPVI mediated platelet filopodia formation, secretion and aggregation. PLoS ONE 2011, 6, e22117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Akbar, H.; Kim, J.; Funk, K.; Cancelas, J.A.; Shang, X.; Chen, L.; Johnson, J.F.; Williams, D.A.; Zheng, Y. Genetic and pharmacologic evidence that Rac1 GTPase is involved in regulation of platelet secretion and aggregation. J. Thromb. Haemost. 2007, 5, 1747–1755. [Google Scholar] [CrossRef]
- Pandey, D.; Goyal, P.; Dwivedi, S.; Siess, W. Unraveling a novel Rac1-mediated signaling pathway that regulates cofilin dephosphorylation and secretion in thrombin-stimulated platelets. Blood 2009, 114, 415–424. [Google Scholar] [CrossRef]
- Dwivedi, S.; Pandey, D.; Khandoga, A.L.; Brandl, R.; Siess, W. Rac1-mediated signaling plays a central role in secretion-dependent platelet aggregation in human blood stimulated by atherosclerotic plaque. J. Transl. Med. 2010, 8, 128. [Google Scholar] [CrossRef] [Green Version]
- Akbar, H.; Cancelas, J.; Williams, D.A.; Zheng, J.; Zheng, Y. Rational design and applications of a Rac GTPase-specific small molecule inhibitor. Methods Enzymol. 2006, 406, 554–565. [Google Scholar]
- Klages, B.; Brandt, U.; Simon, M.I.; Schultz, G.; Offermanns, S. Activation of G12/G13 results in shape change and Rho/Rho-kinase-mediated myosin light chain phosphorylation in mouse platelets. J. Cell. Biol. 1999, 144, 745–754. [Google Scholar] [CrossRef] [Green Version]
- Pleines, I.; Hagedorn, I.; Gupta, S.; May, F.; Chakarova, L.; van Hengel, J.; Offermanns, S.; Krohne, G.; Kleinschnitz, C.; Brakebusch, C.; et al. Megakaryocyte-specific RhoA deficiency causes macrothrombocytopenia and defective platelet activation in hemostasis and thrombosis. Blood 2012, 119, 1054–1063. [Google Scholar] [CrossRef] [Green Version]
- Offermanns, S. Activation of platelet function through G protein-coupled receptors. Circ. Res. 2006, 99, 1293–1304. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.S.; Kim, J.G.; Jeon, C.Y.; Won, H.Y.; Moon, M.Y.; Seo, J.Y.; Kim, J.I.; Kim, J.; Lee, J.Y.; Choi, S.Y.; et al. Downstream components of RhoA required for signal pathway of superoxide formation during phagocytosis of serum opsonized zymosans in macrophages. Exp. Mol. Med. 2005, 37, 575–587. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shang, X.; Marchioni, F.; Sipes, N.; Evelyn, C.R.; Jerabek-Willemsen, M.; Duhr, S.; Seibel, W.; Wortman, M.; Zheng, Y. Rational design of small molecule inhibitors targeting RhoA subfamily Rho GTPases. Chem. Biol. 2012, 19, 699–710. [Google Scholar] [CrossRef] [Green Version]
- Shang, X.; Zheng, Y. Rational design of Rho GTPase-targeting inhibitors. Methods Mol. Biol. 2012, 928, 29–38. [Google Scholar]
- Akbar, H.; Duan, X.; Saleem, S.; Davis, A.K.; Zheng, Y. RhoA and Rac1 GTPases Differentially Regulate Agonist-Receptor Mediated Reactive Oxygen Species Generation in Platelets. PLoS ONE 2016, 11, e0163227. [Google Scholar] [CrossRef] [Green Version]
- Brito, F.C.; Kummerle, A.E.; Lugnier, C.; Fraga, C.A.; Barreiro, E.J.; Miranda, A.L. Novel thienylacylhydrazone derivatives inhibit platelet aggregation through cyclic nucleotides modulation and thromboxane A2 synthesis inhibition. Eur. J. Pharmacol. 2010, 638, 5–12. [Google Scholar] [CrossRef] [PubMed]
- Chelucci, R.C.; Dutra, L.A.; Lopes Pires, M.E.; de Melo, T.R.; Bosquesi, P.L.; Chung, M.C.; Dos Santos, J.L. Antiplatelet and antithrombotic activities of non-steroidal anti-inflammatory drugs containing an N-acyl hydrazone subunit. Molecules 2014, 19, 2089–2099. [Google Scholar] [CrossRef]
- Haj Mohammad Ebrahim Tehrani, K.; Sardari, S.; Mashayekhi, V.; Esfahani Zadeh, M.; Azerang, P.; Kobarfard, F. One Pot Synthesis and Biological Activity Evaluation of Novel Schiff Bases Derived from 2-Hydrazinyl-1,3,4-thiadiazole. Chem. Pharm. Bull. 2013, 61, 160–166. [Google Scholar] [CrossRef] [Green Version]
- Klawans, H.L.; Ringel, S.P.; Shenker, D.M. Failure of vitamin B6 to reverse the L-dopa effect in patients on a dopa decarboxylase inhibitor. J. Neurol. Neurosurg. Psychiatry 1971, 34, 682–686. [Google Scholar] [CrossRef] [Green Version]
- Mashayekhi, V.; Haj Mohammad Ebrahim Tehrani, K.; Amidi, S.; Kobarfard, F. Synthesis of Novel Indole Hydrazone Derivatives and Evaluation of Their Antiplatelet Aggregation Activity. Chem. Pharm. Bull. 2013, 61, 144–150. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McBride, B.C.; Van der Hoeven, J.S. Role of interbacterial adherence in colonization of the oral cavities of gnotobiotic rats infected with Streptococcus mutans and Veillonella alcalescens. Infect. Immun. 1981, 33, 467–472. [Google Scholar] [CrossRef] [Green Version]
- Park, M.K.; Rhee, Y.H.; Lee, H.J.; Lee, E.O.; Kim, K.H.; Park, M.J.; Jeon, B.H.; Shim, B.S.; Jung, C.H.; Choi, S.H.; et al. Antiplatelet and antithrombotic activity of indole-3-carbinol in vitro and in vivo. Phytother. Res. 2008, 22, 58–64. [Google Scholar] [CrossRef]
- Tavili, N.; Mokhtari, S.; Salehabadi, H.; Esfahanizadeh, M.; Mohebbi, S. Novel N-substituted indole hydrazones as potential antiplatelet agents: Synthesis, biological evaluations, and molecular docking studies. Res. Pharm. Sci. 2022, 17, 53–65. [Google Scholar] [PubMed]
- Sterling, T.; Irwin, J.J. ZINC 15—Ligand Discovery for Everyone. J. Chem. Inf. Model. 2015, 55, 2324–2337. [Google Scholar] [CrossRef] [PubMed]
- PubChem National Library of Medicine (US), National Center for Biotechnology Information. PubChem Compound Summary for CID 9552914, Rhosin. 2004. Available online: https://pubchem.ncbi.nlm.nih.gov/compound/Rhosin (accessed on 22 August 2022).
- Born, G. Aggregation of Blood Platelets by Adenosine Diphosphate and Its Reversal; Nature: London, UK, 1962; p. 927. [Google Scholar]
- Baumgartner, H.R.; Haudenschild, C. Adhesion of platelets to subendothelium. Ann. N. Y. Acad. Sci. 1972, 201, 22–36. [Google Scholar] [CrossRef] [PubMed]
- Morton, L.F.; Peachey, A.R.; Barnes, M.J. Platelet-reactive sites in collagens type I and type III. Evidence for separate adhesion and aggregatory sites. Biochem. J. 1989, 258, 157–163. [Google Scholar] [CrossRef] [Green Version]
- Poole, A.W.; Watson, S.P. Regulation of cytosolic calcium by collagen in single human platelets. Br. J. Pharmacol. 1995, 115, 101–106. [Google Scholar] [CrossRef] [Green Version]
- Taylor, M.L.; Misso, N.L.; Stewart, G.A.; Thompson, P.J. The effects of varying doses of aspirin on human platelet activation induced by PAF, collagen and arachidonic acid. Br. J. Clin. Pharmacol. 1992, 33, 25–31. [Google Scholar] [CrossRef] [Green Version]
- Skovronsky, D.M.; Lee, V.M.; Pratico, D. Amyloid precursor protein and amyloid beta peptide in human platelets. Role of cyclooxygenase and protein kinase C. J. Biol. Chem. 2001, 276, 17036–17043. [Google Scholar] [CrossRef] [Green Version]
- Angiolillo, D.J.; Schneider, D.J.; Bhatt, D.L.; French, W.J.; Price, M.J.; Saucedo, J.F.; Shaburishvili, T.; Huber, K.; Prats, J.; Liu, T.; et al. Pharmacodynamic effects of cangrelor and clopidogrel: The platelet function substudy from the cangrelor versus standard therapy to achieve optimal management of platelet inhibition (CHAMPION) trials. J. Thromb. Thrombolysis 2012, 34, 44–55. [Google Scholar] [CrossRef]
- Crook, D.; Collins, A.J. Comparison of effects of aspirin and indomethacin on human platelet prostaglandin synthetase. Ann Rheum. Dis. 1977, 36, 459–463. [Google Scholar] [CrossRef] [Green Version]
- Ferreiro, J.L.; Ueno, M.; Angiolillo, D.J. Cangrelor: A review on its mechanism of action and clinical development. Expert. Rev. Cardiovasc. Ther. 2009, 7, 1195–1201. [Google Scholar] [CrossRef]
- Hule, V. Isoenzymes of lactic dehydrogenase in human platelets. Clin. Chim. Acta 1966, 13, 431–434. [Google Scholar] [CrossRef]
- Ravishankar, D.; Salamah, M.; Akimbaev, A.; Williams, H.F.; Albadawi, D.A.I.; Vaiyapuri, R.; Greco, F.; Osborn, H.M.I.; Vaiyapuri, S. Impact of specific functional groups in flavonoids on the modulation of platelet activation. Sci. Rep. 2018, 8, 9528. [Google Scholar] [CrossRef] [Green Version]
- Cunha, A.C.; Figueiredo, J.M.; Tributino, J.L.; Miranda, A.L.; Castro, H.C.; Zingali, R.B.; Fraga, C.A.; de Souza, M.C.; Ferreira, V.F.; Barreiro, E.J. Antiplatelet properties of novel N-substituted-phenyl-1,2,3-triazole-4-acylhydrazone derivatives. Bioorg. Med. Chem. 2003, 11, 2051–2059. [Google Scholar] [CrossRef]
- Fraga, A.G.M.; Rodrigues, C.R.; de Miranda, A.L.P.; Barreiro, E.J.; Fraga, C.A.M. Synthesis and pharmacological evaluation of novel heterotricyclic acylhydrazone derivatives, designed as PAF antagonists. Eur. J. Pharm. Sci. 2000, 11, 285–290. [Google Scholar] [CrossRef]
- Khalid, W.; Badshah, A.; Khan, A.U.; Nadeem, H.; Ahmed, S. Synthesis, characterization, molecular docking evaluation, antiplatelet and anticoagulant actions of 1,2,4 triazole hydrazone and sulphonamide novel derivatives. Chem. Cent. J. 2018, 12, 11. [Google Scholar] [CrossRef] [Green Version]
- Lima, L.M.; Frattani, F.S.; Dos Santos, J.L.; Castro, H.C.; Fraga, C.A.; Zingali, R.B.; Barreiro, E.J. Synthesis and anti-platelet activity of novel arylsulfonate--acylhydrazone derivatives, designed as antithrombotic candidates. Eur. J. Med. Chem. 2008, 43, 348–356. [Google Scholar] [CrossRef] [PubMed]
- Faghih Akhlaghi, M.; Amidi, S.; Esfahanizadeh, M.; Daeihamed, M.; Kobarfard, F. Synthesis of N-arylmethyl Substituted Indole Derivatives as New Antiplatelet Aggregation Agents. Iran. J. Pharm. Res. 2014, 13, 35–42. [Google Scholar] [PubMed]
- Kristelly, R.; Gao, G.; Tesmer, J.J. Structural determinants of RhoA binding and nucleotide exchange in leukemia-associated Rho guanine-nucleotide exchange factor. J. Biol. Chem. 2004, 279, 47352–47362. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Catella, F.; Healy, D.; Lawson, J.A.; FitzGerald, G.A. 11-Dehydrothromboxane B2: A Quantitative Index of Thromboxane A<sub>2</sub> Formation in the Human Circulation. Proc. Natl. Acad. Sci. USA 1986, 83, 5861–5865. [Google Scholar] [PubMed] [Green Version]
- FitzGerald, G.A.; Healy, C.; Daugherty, J. Thromboxane A2 biosynthesis in human disease. Fed. Proc. 1987, 46, 154–158. [Google Scholar] [PubMed]
- Hamberg, M.; Svensson, J.; Samuelsson, B. Thromboxanes: A new group of biologically active compounds derived from prostaglandin endoperoxides. Proc. Natl. Acad. Sci. USA 1975, 72, 2994–2998. [Google Scholar] [CrossRef] [Green Version]
- Wei, Y.; Zhang, Y.; Derewenda, U.; Liu, X.; Minor, W.; Nakamoto, R.K.; Somlyo, A.V.; Somlyo, A.P.; Derewenda, Z.S. Crystal structure of RhoA-GDP and its functional implications. Nat. Struct. Biol. 1997, 4, 699–703. [Google Scholar] [CrossRef] [PubMed]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef] [Green Version]
- Pellegrini, E.; Bowleri, M.W. RhoA GDP with novel switch II conformation. Protein Data Bank. 2016. [Google Scholar] [CrossRef]
- Akbar, H.; Ardlie, N.G. Evidence that collagen releases human platelet constituents by two different mechanisms. Br. J. Hematol. 1976, 34, 137–146. [Google Scholar] [CrossRef]
- Akbar, H.; Duan, X.; Piatt, R.; Saleem, S.; Davis, A.K.; Tandon, N.N.; Bergmeier, W.; Zheng, Y. Small molecule targeting the Rac1-NOX2 interaction prevents collagen-related peptide and thrombin-induced reactive oxygen species generation and platelet activation. J. Thromb. Haemost. 2018, 16, 2083–2096. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mendez, D.; Urra, F.A.; Millas-Vargas, J.P.; Alarcon, M.; Rodriguez-Lavado, J.; Palomo, I.; Trostchansky, A.; Araya-Maturana, R.; Fuentes, E. Synthesis of antiplatelet ortho-carbonyl hydroquinones with differential action on platelet aggregation stimulated by collagen or TRAP-6. Eur. J. Med. Chem. 2020, 192, 112187. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Derivative | Structure | % Platelet Aggregation b | |
|---|---|---|---|
| Collagen (1 µg/mL) | Collagen (5 µg/mL) | ||
| UC-177629 |  | 7.3 ± 5.9 | 10.8 ± 3 |
| UC-391695 |  | 79 ± 8 | 84.8 ± 12.1 |
| UC-436352 |  | 84.7 ± 15.5 | 89 ± 2.5 |
| UC-392453 |  | 90.7 ± 8.5 | 94.7 ± 11.2 |
| UC-830013 |  | 93 ± 10.8 | 91.3 ± 14.7 |
| UC-984842 |  | 91.3 ± 10.5 | 95.9 ± 6.2 |
| UC-651009 |  | 86 ± 1.7 | 89.2 ± 10.6 |
| Indomethacin a | 12 ± 4.4 | 69 ± 12.3 | |
| Cangrelor a | 27.67 ± 2.52 | 63.33 ± 4.16 | |
 | ||||
| Derivative | R1 | R2 | % Platelet Aggregation b | |
| Collagen (1 µg/mL) | Collagen (5 µg/mL) | |||
| UC-177626 |  |  | 86 ± 14.4 | 87.6 ± 19.4 |
| UC-177629 |  |  | 7.3 ± 5.9 | 10.8 ± 3 |
| UC-177627 |  |  | 17.7 ± 11.4 | 19 ± 17.7 |
| UC-177628 |  |  | 8.7 ± 4.7 | 12.3 ± 9 |
| UC-177630 |  |  | 73 ± 21.6 | 85 ± 15.3 |
| UC-178838 |  |  | 7.3 ± 5.8 | 13.7 ± 7.9 |
| UC-177617 |  |  | 13 ± 10.4 | 26.8 ± 9 |
| UC-177619 |  |  | 9.3 ± 3.5 | 12 ± 2.1 |
| UC-177618 |  |  | 5 ± 1.7 | 10.2 ± 4.1 |
| UC-177631 |  |  | 87.7 ± 13.1 | 90.4 ± 3.7 |
| UC-177634 |  |  | 17 ± 2.6 | 24.5 ± 3.9 |
| UC-177632 |  |  | 86.7 ± 11 | 93.1 ± 12.8 |
| UC-177633 |  |  | 3.3 ± 3.2 | 10.1 ± 8.3 |
| UC-177635 |  |  | 79.1 ± 7 | 90.1 ± 8.3 |
| UC-177639 |  | 80 ± 17.3 | 90.4 ± 9.1 | |
| UC-390484 |  | 81 ± 14 | 88.1 ± 10 | |
| UC-406794 |  | 12.7 ± 7.2 | 25 ± 5.2 | |
| UC-612333 |  | 70 ± 5.6 | 78.6 ± 14.9 | |
| UC-936377 |  | 61.3 ± 4.5 | 71.7 ± 5.3 | |
| UC177623 |  | 84.3 ± 9 | 90.7 ± 6.2 | |
| UC-177613 |  | 84.3 ± 15.5 | 95.2 ± 4.9 | |
| UC-177614 |  | 87.3 ± 9.6 | 90 ± 3.1 | |
| UC-177615 |  | 9.7 ± 6 | 11.4 ± 9.9 | |
| UC-177616 |  | 11.7 ± 2.3 | 10.6 ± 3.6 | |
| Indomethacin a | 12 ± 4.4 | 69 ± 12.3 | ||
| Cangrelor a | 27.67 ± 2.52 | 63.33 ± 4.16 | ||
| Aryl Group |  |  |  | |
| Quinoline Group | ||||
 |  IC50 > 20 µM |  IC50 = 5.64 µM |  IC50 > 20 µM | |
 |  IC50 = 2.39 µM |  IC50 = 0.82 µM |  IC50 = 4.59 µM | |
 |  IC50 = 2.23 µM |  IC50 > 20 µM | ||
 |  IC50 = 2.84 µM |  IC50 = 1.5 µM |  IC50 = 2.34 µM | |
 |  IC50 > 20 µM |  IC50 > 20 µM | ||
 |  IC50 = 3.55 µM | |||
| Derivative | Structure | % Platelet Aggregation b | |
|---|---|---|---|
| Collagen (1 µg/mL) | Collagen (5 µg/mL) | ||
| Rhosin/ R-G04 |  | 70.33 ± 18.90 | 71.36 ± 15.01 |
| S-G04 |  | 9.33 ± 4.93 | 20 ± 5 |
| Indomethacin a | 12 ± 4.4 | 69 ± 12.3 | |
| Cangrelor a | 27.67 ± 2.52 | 63.33 ± 4.16 | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dandamudi, A.; Seibel, W.; Tourdot, B.; Cancelas, J.A.; Akbar, H.; Zheng, Y. Structure–Activity Relationship Analysis of Rhosin, a RhoA GTPase Inhibitor, Reveals a New Class of Antiplatelet Agents. Int. J. Mol. Sci. 2023, 24, 4167. https://doi.org/10.3390/ijms24044167
Dandamudi A, Seibel W, Tourdot B, Cancelas JA, Akbar H, Zheng Y. Structure–Activity Relationship Analysis of Rhosin, a RhoA GTPase Inhibitor, Reveals a New Class of Antiplatelet Agents. International Journal of Molecular Sciences. 2023; 24(4):4167. https://doi.org/10.3390/ijms24044167
Chicago/Turabian StyleDandamudi, Akhila, William Seibel, Benjamin Tourdot, Jose A. Cancelas, Huzoor Akbar, and Yi Zheng. 2023. "Structure–Activity Relationship Analysis of Rhosin, a RhoA GTPase Inhibitor, Reveals a New Class of Antiplatelet Agents" International Journal of Molecular Sciences 24, no. 4: 4167. https://doi.org/10.3390/ijms24044167