
The Effects of Interstitial Lung Diseases on Alveolar Extracellular Vesicles Profile: A Multicenter Study

 , , , , , , , , and
, , , , , , , , and
Abstract
:1. Introduction
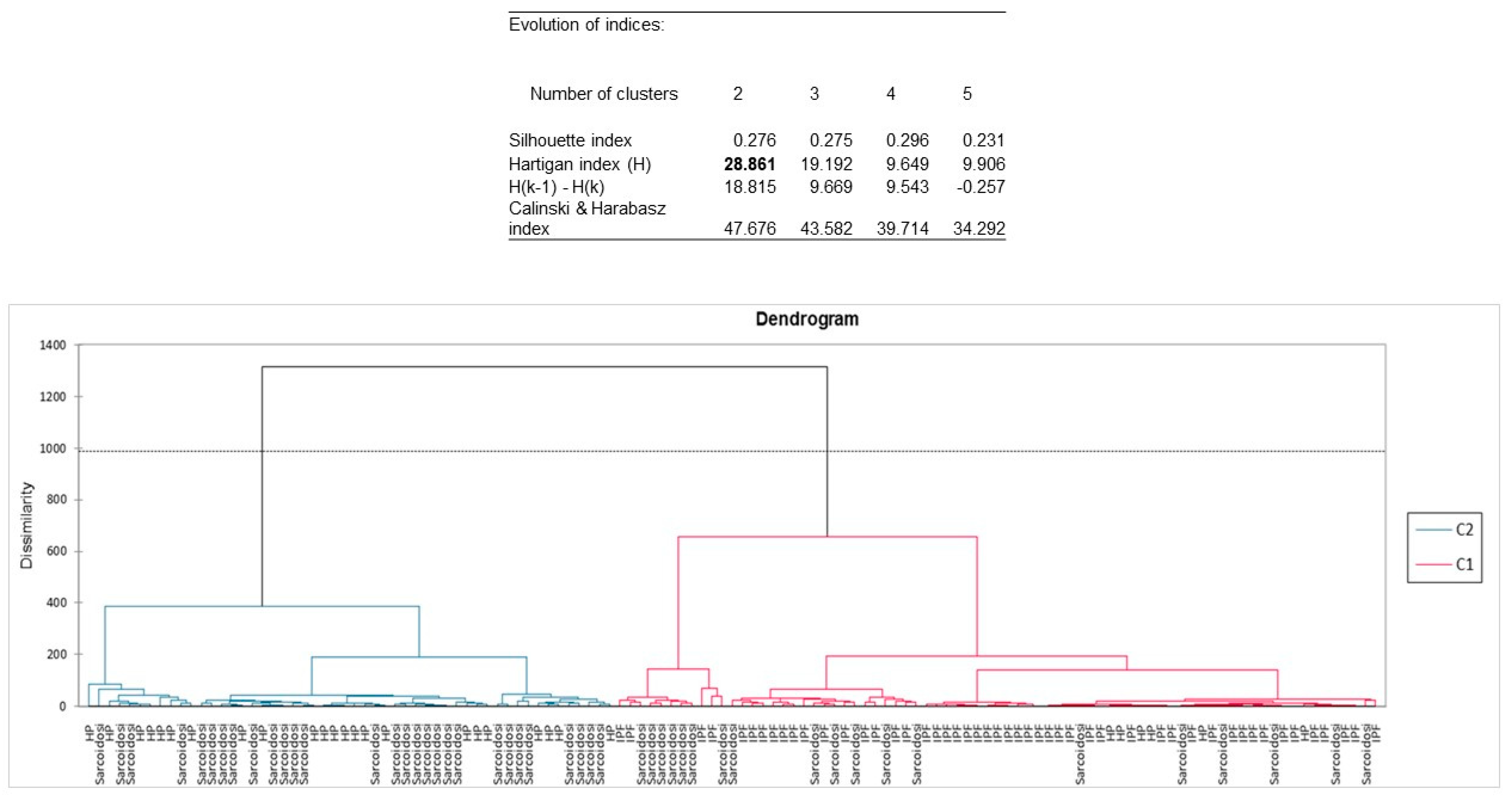
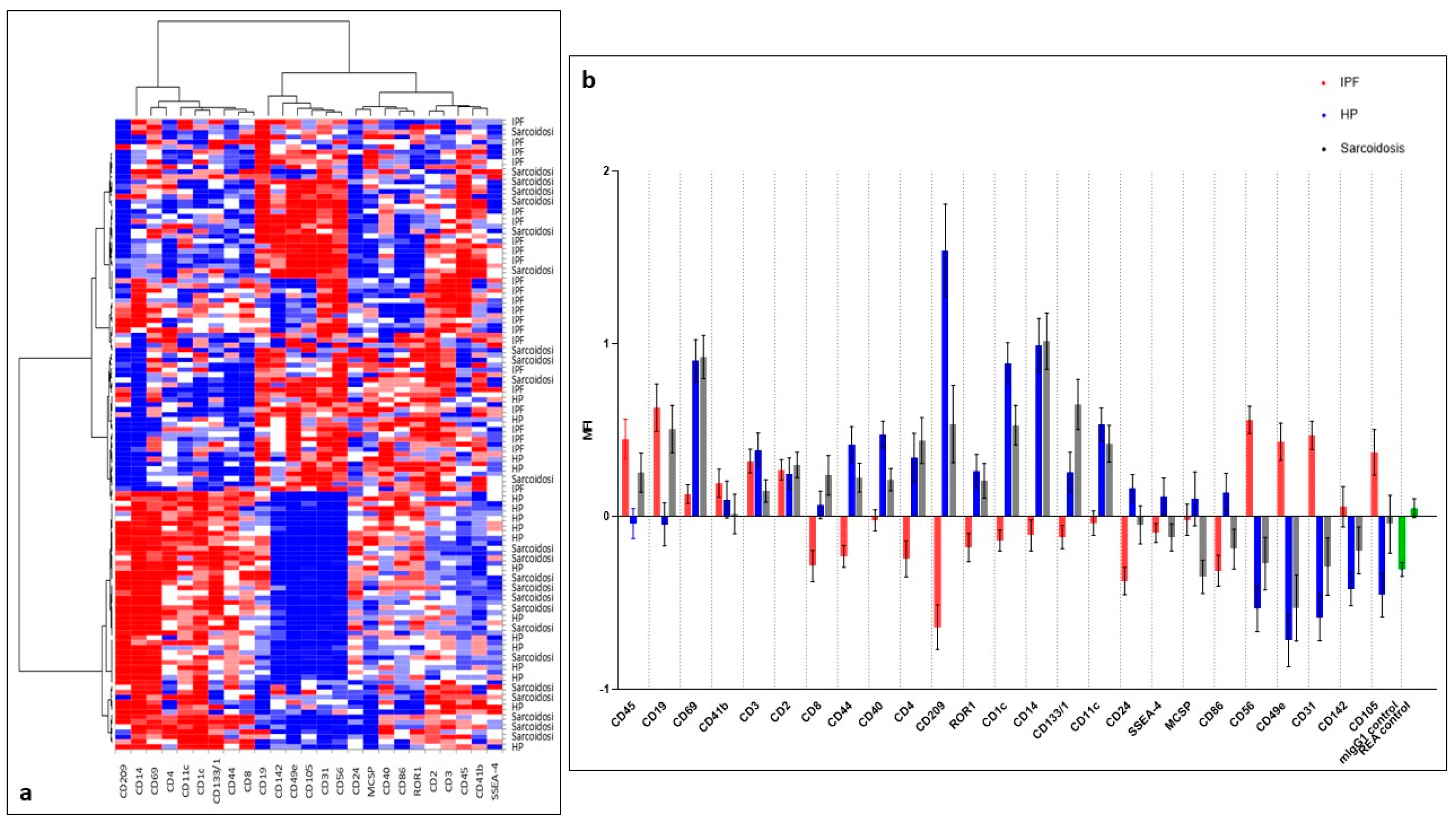
2. Results
2.1. Patients
2.2. EVs Surface Markers
3. Discussion
4. Materials and Methods
4.1. Study Cohort
4.2. Validation Cohort
4.3. Pulmonary Function Test Parameters
4.4. Bronchoscopy
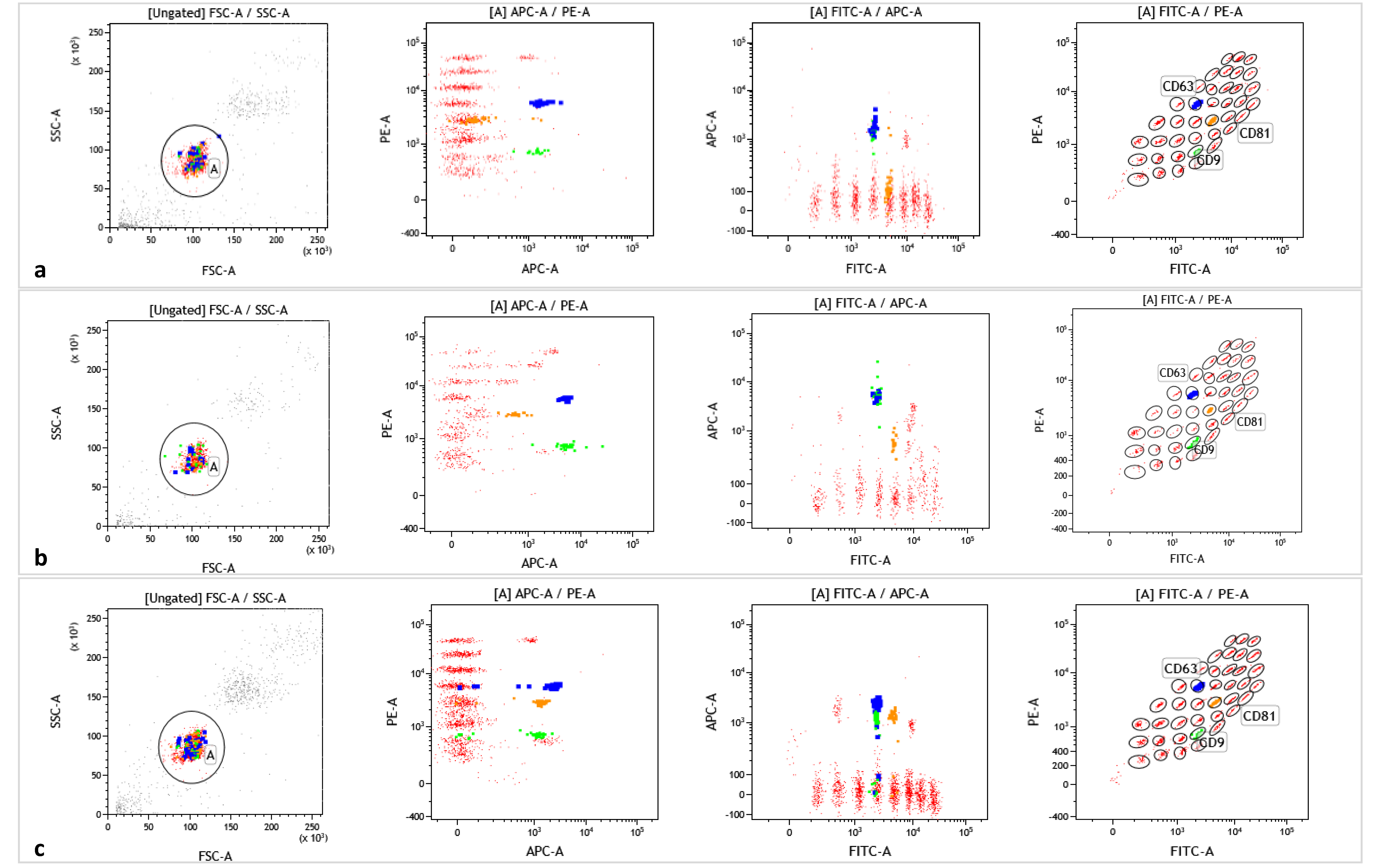
4.5. EVs Isolation and Characterization
4.6. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
References
- Meyer, K.C.; Raghu, G.; Baughman, R.P.; Brown, K.K.; Costabel, U.; du Bois, R.M.; Drent, M.; Haslam, P.L.; Kim, D.S.; Nagai, S.; et al. An official American Thoracic Society clinical practice guideline: The clinical utility of bronchoalveolar lavage cellular analysis in interstitial lung disease. Am. J. Respir. Crit. Care Med. 2012, 185, 1004–1014. [Google Scholar] [CrossRef] [PubMed]
- Behr, J. Approach to the diagnosis of interstitial lung disease. Clin. Chest Med. 2012, 33, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Raghu, G.; Remy-Jardin, M.; Richeldi, L.; Thomson, C.C.; Inoue, Y.; Johkoh, T.; Kreuter, M.; Lynch, D.A.; Maher, T.M.; Martinez, F.J.; et al. Idiopathic Pulmonary Fibrosis (an Update) and Progressive Pulmonary Fibrosis in Adults: An Official ATS/ERS/JRS/ALAT Clinical Practice Guideline. Am. J. Respir. Crit. Care Med. 2022, 205, e18–e47. [Google Scholar] [CrossRef] [PubMed]
- Crouser, E.D.; Maier, L.A.; Wilson, K.C.; Bonham, C.A.; Morgenthau, A.S.; Patterson, K.C.; Abston, E.; Bernstein, R.C.; Blankstein, R.; Chen, E.S.; et al. Diagnosis and Detection of Sarcoidosis. An Official American Thoracic Society Clinical Practice Guideline. Am. J. Respir. Crit. Care Med. 2020, 201, e26–e51. [Google Scholar] [CrossRef]
- Ageely, G.; Souza, C.; De Boer, K.; Zahra, S.; Gomes, M.; Voduc, N. The Impact of Multidisciplinary Discussion (MDD) in the Diagnosis and Management of Fibrotic Interstitial Lung Diseases. Can. Respir. J. 2020, 2020, 9026171. [Google Scholar] [CrossRef]
- Costabel, U.; Guzman, J. Bronchoalveolar lavage in interstitial lung disease. Curr. Opin. Pulm. Med. 2001, 7, 255–261. [Google Scholar] [CrossRef]
- Stanzel, F. “Bronchoalveolar Lavage”. In Principles and Practice of Interventional Pulmonology; Ernst, A., Herth, F.J., Eds.; Springer: New York, NY, USA, 2013; pp. 165–176. [Google Scholar] [CrossRef]
- Mohan, A.; Agarwal, S.; Clauss, M.; Britt, N.S.; Dhillon, N.K. Extracellular vesicles: Novel communicators in lung diseases. Respir. Res. 2020, 21, 175. [Google Scholar] [CrossRef]
- d’Alessandro, M.; Soccio, P.; Bergantini, L.; Cameli, P.; Scioscia, G.; Foschino Barbaro, M.P.; Lacedonia, D.; Bargagli, E. Extracellular Vesicle Surface Signatures in IPF Patients: A Multiplex Bead-Based Flow Cytometry Approach. Cells 2021, 10, 1045. [Google Scholar] [CrossRef]
- Kubo, H. Extracellular Vesicles in Lung Disease. Chest 2018, 153, 210–216. [Google Scholar] [CrossRef]
- d’Alessandro, M.; Bergantini, L.; Bargagli, E.; Vidal, S. Extracellular Vesicles in Pulmonary Fibrosis Models and Biological Fluids of Interstitial Lung Disease Patients: A Scoping Review. Life 2021, 11, 1401. [Google Scholar] [CrossRef]
- Ziegler, S.F.; Ramsdell, F.; Alderson, M.R. The activation antigen CD69. Stem Cells 1994, 12, 456–465. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Oldham, J.M.; Ma, S.-F.; Unterman, A.; Liao, S.-Y.; Barros, A.J.; Bonham, C.A.; Kim, J.S.; Vij, R.; Adegunsoye, A.; et al. Blood Transcriptomics Predicts Progression of Pulmonary Fibrosis and Associated Natural Killer Cells. Am. J. Respir. Crit. Care Med. 2021, 204, 197–208. [Google Scholar] [CrossRef] [PubMed]
- Bergantini, L.; Cameli, P.; d’Alessandro, M.; Vagaggini, C.; Refini, R.M.; Landi, C.; Pieroni, M.G.; Spalletti, M.; Sestini, P.; Bargagli, E. NK and NKT-like cells in granulomatous and fibrotic lung diseases. Clin. Exp. Med. 2019, 19, 487–494. [Google Scholar] [CrossRef] [PubMed]
- Tzouvelekis, A.; Kaminski, N. Epigenetics in idiopathic pulmonary fibrosis. Biochem. Cell Biol. 2015, 93, 159–170. [Google Scholar] [CrossRef]
- Paolocci, G.; Folletti, I.; Torén, K.; Ekström, M.; Dell’Omo, M.; Muzi, G.; Murgia, N. Occupational risk factors for idiopathic pulmonary fibrosis in Southern Europe: A case-control study. BMC Pulm. Med. 2018, 18, 75. [Google Scholar] [CrossRef] [Green Version]
- Ranzieri, S.; Illica Magrini, E.; Mozzoni, P.; Andreoli, R.; Pelà, G.; Bertorelli, G.; Corradi, M. Idiopathic pulmonary fibrosis and occupational risk factors. Med. Lav. 2019, 110, 407–436. [Google Scholar] [CrossRef]
- Camelo, A.; Dunmore, R.; Sleeman, M.; Clarke, D. The epithelium in idiopathic pulmonary fibrosis: Breaking the barrier. Front. Pharmacol. 2014, 4, 173. Available online: https://www.frontiersin.org/article/10.3389/fphar.2013.00173 (accessed on 26 January 2022). [CrossRef] [Green Version]
- Selman, M.; Pardo, A. Idiopathic pulmonary fibrosis: An epithelial/fibroblastic cross-talk disorder. Respir. Res. 2002, 3, 3. [Google Scholar] [CrossRef]
- Cong, X.; Hubmayr, R.D.; Li, C.; Zhao, X. Plasma membrane wounding and repair in pulmonary diseases. Am. J. Physiol. Lung Cell. Mol. Physiol. 2017, 312, L371–L391. [Google Scholar] [CrossRef] [Green Version]
- Hanumegowda, C.; Farkas, L.; Kolb, M. Angiogenesis in pulmonary fibrosis: Too much or not enough? Chest 2012, 142, 200–207. [Google Scholar] [CrossRef]
- Sehgal, M.; Jakhete, S.M.; Manekar, A.G.; Sasikumar, S. Specific epigenetic regulators serve as potential therapeutic targets in idiopathic pulmonary fibrosis. Heliyon 2022, 8, e09773. [Google Scholar] [CrossRef] [PubMed]
- Kendall, R.T.; Feghali-Bostwick, C.A. Fibroblasts in fibrosis: Novel roles and mediators. Front. Pharmacol. 2014, 5, 123. [Google Scholar] [CrossRef] [Green Version]
- DeLeon-Pennell, K.Y.; Barker, T.H.; Lindsey, M.L. Fibroblasts: The arbiters of extracellular matrix remodeling. Matrix Biol. 2020, 91–92, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Morrissey, J.H. Tissue factor: A key molecule in hemostatic and nonhemostatic systems. Int. J. Hematol. 2004, 79, 103–108. [Google Scholar] [CrossRef] [PubMed]
- Crooks, M.G.; Hart, S.P. Coagulation and anticoagulation in idiopathic pulmonary fibrosis. Eur. Respir. Rev. 2015, 24, 392–399. [Google Scholar] [CrossRef]
- Schick, P.K.; Wojenski, C.M.; He, X.; Walker, J.; Marcinkiewicz, C.; Niewiarowski, S. Integrins involved in the adhesion of megakaryocytes to fibronectin and fibrinogen. Blood 1998, 92, 2650–2656. [Google Scholar] [CrossRef]
- Schoonderwoerd, M.J.A.; Goumans, M.-J.T.H.; Hawinkels, L.J.A.C. Endoglin: Beyond the Endothelium. Biomolecules 2020, 10, 289. [Google Scholar] [CrossRef] [Green Version]
- Kitagawa, M.; Yoshida, S.; Ishige, I.; Minami, J.; Kuwata, T.; Tanizawa, T.; Kamiyama, R. Immunolocalization of platelet-derived growth factor, transforming growth factor-beta, and fibronectin in acute megakaryoblastic leukemia manifesting tumor formation. Hum. Pathol. 1994, 25, 723–726. [Google Scholar] [CrossRef]
- Berman, M.E.; Xie, Y.; Muller, W.A. Roles of platelet/endothelial cell adhesion molecule-1 (PECAM-1, CD31) in natural killer cell transendothelial migration and beta 2 integrin activation. J. Immunol. 1996, 156, 1515–1524. [Google Scholar] [CrossRef]
- Kono, M.; Miyashita, K.; Hirama, R.; Oshima, Y.; Takeda, K.; Mochizuka, Y.; Tsutsumi, A.; Miwa, H.; Miki, Y.; Hashimoto, D.; et al. Prognostic significance of bronchoalveolar lavage cellular analysis in patients with acute exacerbation of interstitial lung disease. Respir. Med. 2021, 186, 106534. [Google Scholar] [CrossRef]
- Song, H.; Sun, D.; Ban, C.; Liu, Y.; Zhu, M.; Ye, Q.; Yan, W.; Ren, Y.; Dai, H. Independent Clinical Factors Relevant to Prognosis of Patients with Idiopathic Pulmonary Fibrosis. Med. Sci. Monit. 2019, 25, 4193–4201. [Google Scholar] [CrossRef] [PubMed]
- Bergantini, L.; d’Alessandro, M.; Cameli, P.; Carleo, A.; Landi, C.; Vietri, L.; Lanzarone, N.; Pieroni, M.; Sestini, P.; Bargagli, E. Antithrombin III as predictive indicator of survival in idiopathic pulmonary fibrosis (IPF) patients treated with nintedanib: A preliminary study. Intern. Med. J. 2021, 51, 705–711. [Google Scholar] [CrossRef] [PubMed]
- Kinder, B.W.; Brown, K.K.; Schwarz, M.I.; Ix, J.H.; Kervitsky, A.; King, T.E. Baseline BAL neutrophilia predicts early mortality in idiopathic pulmonary fibrosis. Chest 2008, 133, 226–232. [Google Scholar] [CrossRef] [PubMed]
- Bringardner, B.D.; Baran, C.P.; Eubank, T.D.; Marsh, C.L.A.Y.B. The Role of Inflammation in the Pathogenesis of Idiopathic Pulmonary Fibrosis. Antioxid. Redox Signal. 2008, 10, 287–301. [Google Scholar] [CrossRef] [Green Version]
- d’Alessandro, M.; Bergantini, L.; Cameli, P.; Lanzarone, N.; Perillo, F.; Perrone, A.; Bargagli, E. BAL and serum multiplex lipid profiling in idiopathic pulmonary fibrosis and fibrotic hypersensitivity pneumonitis. Life Sci. 2020, 256, 117995. [Google Scholar] [CrossRef]
- Lepzien, R.; Liu, S.; Czarnewski, P.; Nie, M.; Österberg, B.; Baharom, F.; Pourazar, J.; Rankin, G.; Eklund, A.; Bottai, M.; et al. Monocytes in sarcoidosis are potent tumour necrosis factor producers and predict disease outcome. Eur. Respir. J. 2021, 58, 2003468. [Google Scholar] [CrossRef]
- d’Alessandro, M.; Bergantini, L.; Cameli, P.; Mezzasalma, F.; Refini, R.M.; Pieroni, M.; Sestini, P.; Bargagli, E. Adaptive immune system in pulmonary sarcoidosis-Comparison of peripheral and alveolar biomarkers. Clin. Exp. Immunol. 2021, 205, 406–416. [Google Scholar] [CrossRef]
- Bargagli, E.; Magi, B.; Olivieri, C.; Bianchi, N.; Landi, C.; Rottoli, P. Analysis of serum amyloid A in sarcoidosis patients. Respir. Med. 2011, 105, 775–780. [Google Scholar] [CrossRef] [Green Version]
- Ten Berge, B.; Kleinjan, A.; Muskens, F.; Hammad, H.; Hoogsteden, H.C.; Hendriks, R.W.; Lambrecht, B.N.; Van den Blink, B. Evidence for local dendritic cell activation in pulmonary sarcoidosis. Respir. Res. 2012, 13, 33. [Google Scholar] [CrossRef] [Green Version]
- Hayashi, Y.; Ishii, Y.; Hata-Suzuki, M.; Arai, R.; Chibana, K.; Takemasa, A.; Fukuda, T. Comparative analysis of circulating dendritic cell subsets in patients with atopic diseases and sarcoidosis. Respir. Res. 2013, 14, 29. [Google Scholar] [CrossRef] [Green Version]
- Wells, A.U. Sarcoidosis: A benign disease or a culture of neglect? Respir. Med. 2018, 144S, S1–S2. [Google Scholar] [CrossRef] [PubMed]
- Laman, J.D.; Claassen, E.; Noelle, R.J. Functions of CD40 and Its Ligand, gp39 (CD40L). Crit. Rev. Immunol. 2017, 37, 371–420. [Google Scholar] [CrossRef] [PubMed]
- Kudryavtsev, I.; Serebriakova, M.; Starshinova, A.; Zinchenko, Y.; Basantsova, N.; Malkova, A.; Soprun, L.; Churilov, L.P.; Toubi, E.; Yablonskiy, P.; et al. Imbalance in B cell and T Follicular Helper Cell Subsets in Pulmonary Sarcoidosis. Sci. Rep. 2020, 10, 1059. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Courtemanche, O.; Huppé, C.-A.; Blais Lecours, P.; Lerdu, O.; Roy, J.; Lauzon-Joset, J.-F.; Blanchet, M.-R.; Morissette, M.C.; Marsolais, D. Co-modulation of T cells and B cells enhances the inhibition of inflammation in experimental hypersensitivity pneumonitis. Respir. Res. 2022, 23, 275. [Google Scholar] [CrossRef]
- Katoh, S.; Matsubara, Y.; Taniguchi, H.; Fukushima, K.; Mukae, H.; Kadota, J.; Matsukura, S.; Kohno, S. Characterization of CD44 expressed on alveolar macrophages in patients with diffuse panbronchiolitis. Clin. Exp. Immunol. 2001, 126, 545–550. [Google Scholar] [CrossRef]
- Tarique, A.A.; Logan, J.; Thomas, E.; Holt, P.G.; Sly, P.D.; Fantino, E. Phenotypic, functional, and plasticity features of classical and alternatively activated human macrophages. Am. J. Respir. Cell. Mol. Biol. 2015, 53, 676–688. [Google Scholar] [CrossRef]
- Raghu, G.; Remy-Jardin, M.; Ryerson, C.J.; Myers, J.L.; Kreuter, M.; Vasakova, M.; Bargagli, E.; Chung, J.H.; Collins, B.F.; Bendstrup, E.; et al. Diagnosis of Hypersensitivity Pneumonitis in Adults. An Official ATS/JRS/ALAT Clinical Practice Guideline. Am. J. Respir. Crit. Care Med. 2020, 202, e36–e69. [Google Scholar] [CrossRef]
- Costabel, U.; Hunninghake, G.W. ATS/ERS/WASOG statement on sarcoidosis. Sarcoidosis Statement Committee. American Thoracic Society. European Respiratory Society. World Association for Sarcoidosis and Other Granulomatous Disorders. Eur. Respir. J. 1999, 14, 735–737. [Google Scholar] [CrossRef] [Green Version]
- Culver, B.H.; Graham, B.L.; Coates, A.L.; Wanger, J.; Berry, C.E.; Clarke, P.K.; Hallstrand, T.S.; Hankinson, J.L.; Kaminsky, D.A.; MacIntyre, N.R.; et al. Recommendations for a Standardized Pulmonary Function Report. An Official American Thoracic Society Technical Statement. Am. J. Respir. Crit. Care Med. 2017, 196, 1463–1472. [Google Scholar] [CrossRef]
- Haslam, P.L.; Baughman, R.P. Report of ERS Task Force: Guidelines for measurement of acellular components and standardization of BAL. Eur. Respir. J. 1999, 14, 245–248. [Google Scholar] [CrossRef] [Green Version]
- MACSPlex Exosome Kit, Human|Multiplex Assays|Kits and Support Reagents|MACS Flow Cytometry|Products|Miltenyi Biotec|Suomi. Available online: https://www.miltenyibiotec.com/FI-en/products/macsplex-exosome-kit-human.html#gref (accessed on 22 March 2021).
- Safford, H.R.; Bischel, H.N. Performance comparison of four commercially available cytometers using fluorescent, polystyrene, submicron-scale beads. Data Brief 2019, 24, 103872. [Google Scholar] [CrossRef] [PubMed]
- Jotschke, S.; Schulze, S.; Jaekel, N.; Ludwig-Kraus, B.; Engelmann, R.; Kraus, F.B.; Zahn, C.; Nedlitz, N.; Prange-Krex, G.; Mohm, J.; et al. Longitudinal Humoral and Cellular Immune Responses Following SARS-CoV-2 Vaccination in Patients with Myeloid and Lymphoid Neoplasms Compared to a Reference Cohort: Results of a Prospective Trial of the East German Study Group for Hematology and Oncology (OSHO). Cancers 2022, 14, 1544. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameters | Study Cohort | Validation Cohort | ||||
|---|---|---|---|---|---|---|
| IPF (n = 17) | HP (n = 24) | Sarcoidosis (n = 42) | IPF (n = 44) | HP (n = 11) | Sarcoidosis (n = 10) | |
| Age (years) | 64.7 ± 23.8 | 68.23 ± 11.9 | 52.2 ± 21.2 1 | 62.6 ± 19.8 | 67.11 ± 9.2 | 50.2 ± 20.5 1 |
| Gender (male/female) | 13/4 | 14/10 | 16/36 2 | 31/13 | 6/5 | 3/7 2 |
| Smoking habit (never/former) | 6/11 | 6/18 | 15/27 | 15/28 | 4/7 | 4/6 |
| BAL cellular pattern | ||||||
| Macrophages | 71.8 ± 23.4 | 76.5 ± 16.0 | 67.0 ± 19.5 | 59.7 ± 22.4 | 47.6 ± 18.4 | 40.0 ± 14.1 |
| Lymphocytes | 13.4 ± 16.6 | 14.5 ± 14.8 | 21.1 ± 14.8 | 10.1 ± 5.07 | 19.0 ± 5.48 | 33.0 ± 14.7 |
| Neutrophils | 11.4 ± 11.4 | 5.46 ± 5.03 | 11.2 ± 20.9 | 19.2 ± 19.1 | 28.0 ± 20.4 | 10.8 ± 5.68 |
| Pulmonary function test parameters | ||||||
| FVC % | 81.9 ± 27.5 | 69.1 ± 22.2 | 99.3 ± 19.5 | 79.6 ± 10.4 | 63.6 ± 13.2 | 93.8 ± 8.73 |
| FVC L | 2.44 ± 0.573 | 2.37 ± 0.992 | 3.51 ± 1.16 | 3.27 ± 0.598 | 2.47 ± 0.951 | 4.59 ± 0.667 |
| FEV1 % | 84.9 ± 22.0 | 72.0 ± 21.2 | 95.4 ± 20.3 | 81.6 ± 12.0 | 60.8 ± 10.6 | 90.0 ± 8.29 |
| FEV1 L | 2.04 ± 0.475 | 1.98 ± 0.757 | 5.83 ± 16.3 | 2.48 ± 0.570 | 1.76 ± 0.715 | 3.39 ± 0.472 |
| DLco % | 51.4 ± 21.2 3 | 58.4 ± 28.5 | 78.2 ± 20.9 | 52.6 ± 19.8 3 | 59.2 ± 32.3 | 75.7 ± 20.5 |
| DLco L | 3.82 ± 1.30 | 5.14 ± 21.2 | 7.62 ± 1.92 | 4.4 ± 4.93 | 6.7 ± 8.90 | 10.7 ± 6.93 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
d’Alessandro, M.; Gangi, S.; Soccio, P.; Cantó, E.; Osuna-Gómez, R.; Bergantini, L.; Cameli, P.; Fabbri, G.; Croce, S.; Scioscia, G.; et al. The Effects of Interstitial Lung Diseases on Alveolar Extracellular Vesicles Profile: A Multicenter Study. Int. J. Mol. Sci. 2023, 24, 4071. https://doi.org/10.3390/ijms24044071
d’Alessandro M, Gangi S, Soccio P, Cantó E, Osuna-Gómez R, Bergantini L, Cameli P, Fabbri G, Croce S, Scioscia G, et al. The Effects of Interstitial Lung Diseases on Alveolar Extracellular Vesicles Profile: A Multicenter Study. International Journal of Molecular Sciences. 2023; 24(4):4071. https://doi.org/10.3390/ijms24044071
Chicago/Turabian Styled’Alessandro, Miriana, Sara Gangi, Piera Soccio, Elisabet Cantó, Rubén Osuna-Gómez, Laura Bergantini, Paolo Cameli, Gaia Fabbri, Sara Croce, Giulia Scioscia, and et al. 2023. "The Effects of Interstitial Lung Diseases on Alveolar Extracellular Vesicles Profile: A Multicenter Study" International Journal of Molecular Sciences 24, no. 4: 4071. https://doi.org/10.3390/ijms24044071