
Molecular Dynamic Studies of Dye–Dye and Dye–DNA Interactions Governing Excitonic Coupling in Squaraine Aggregates Templated by DNA Holliday Junctions
 , , , and
, , , and
Abstract
:1. Introduction
2. Results
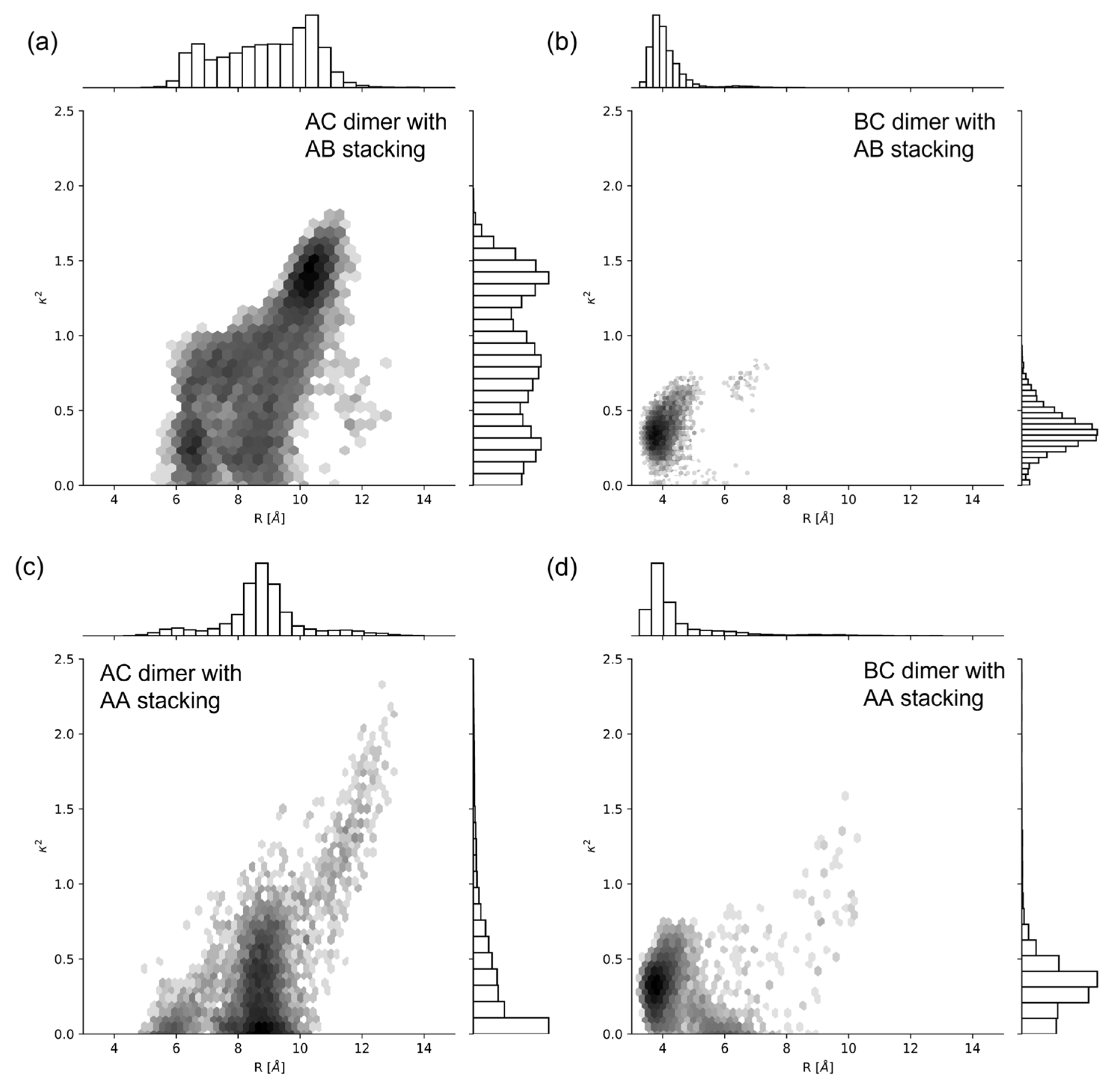
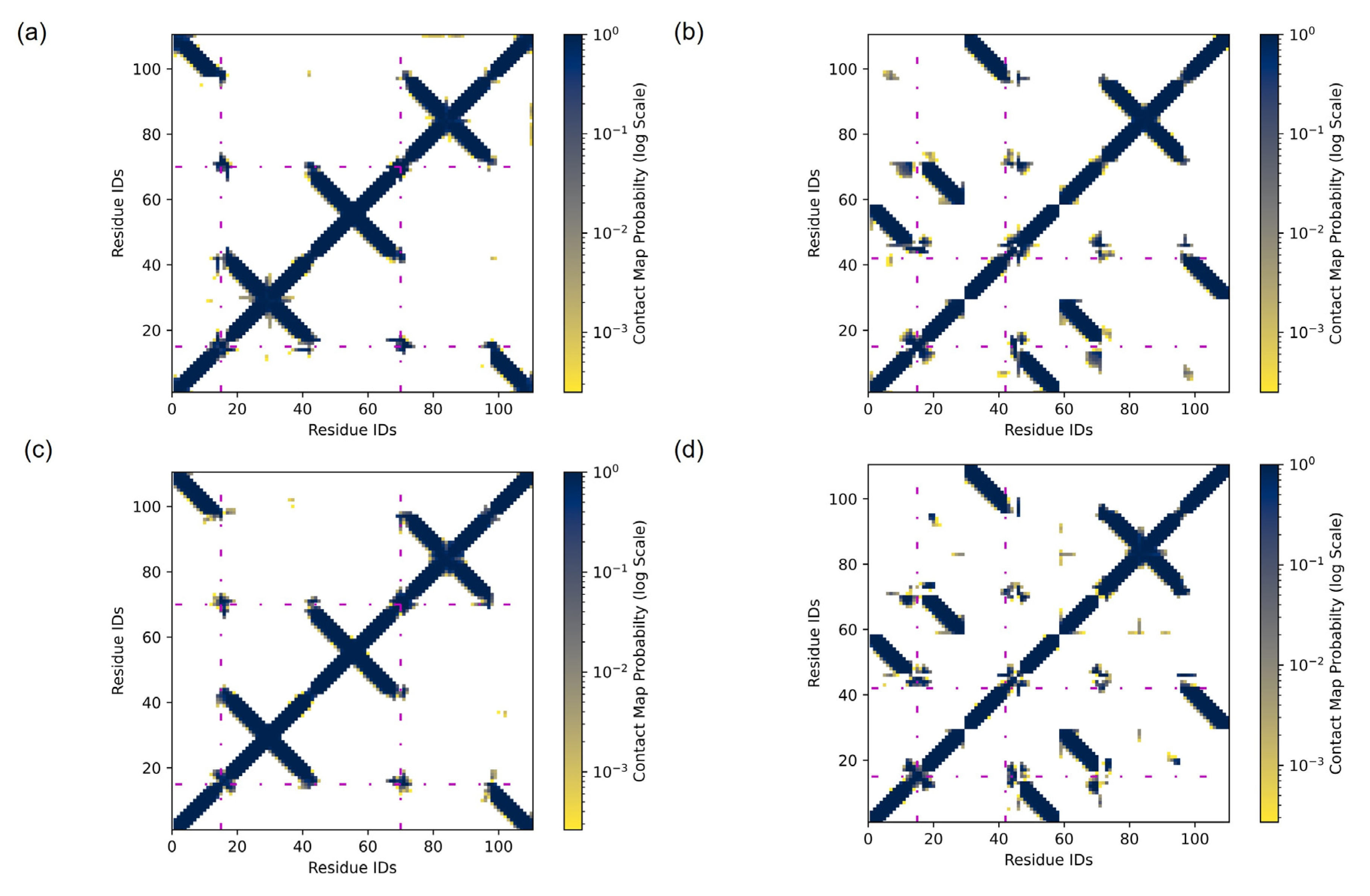
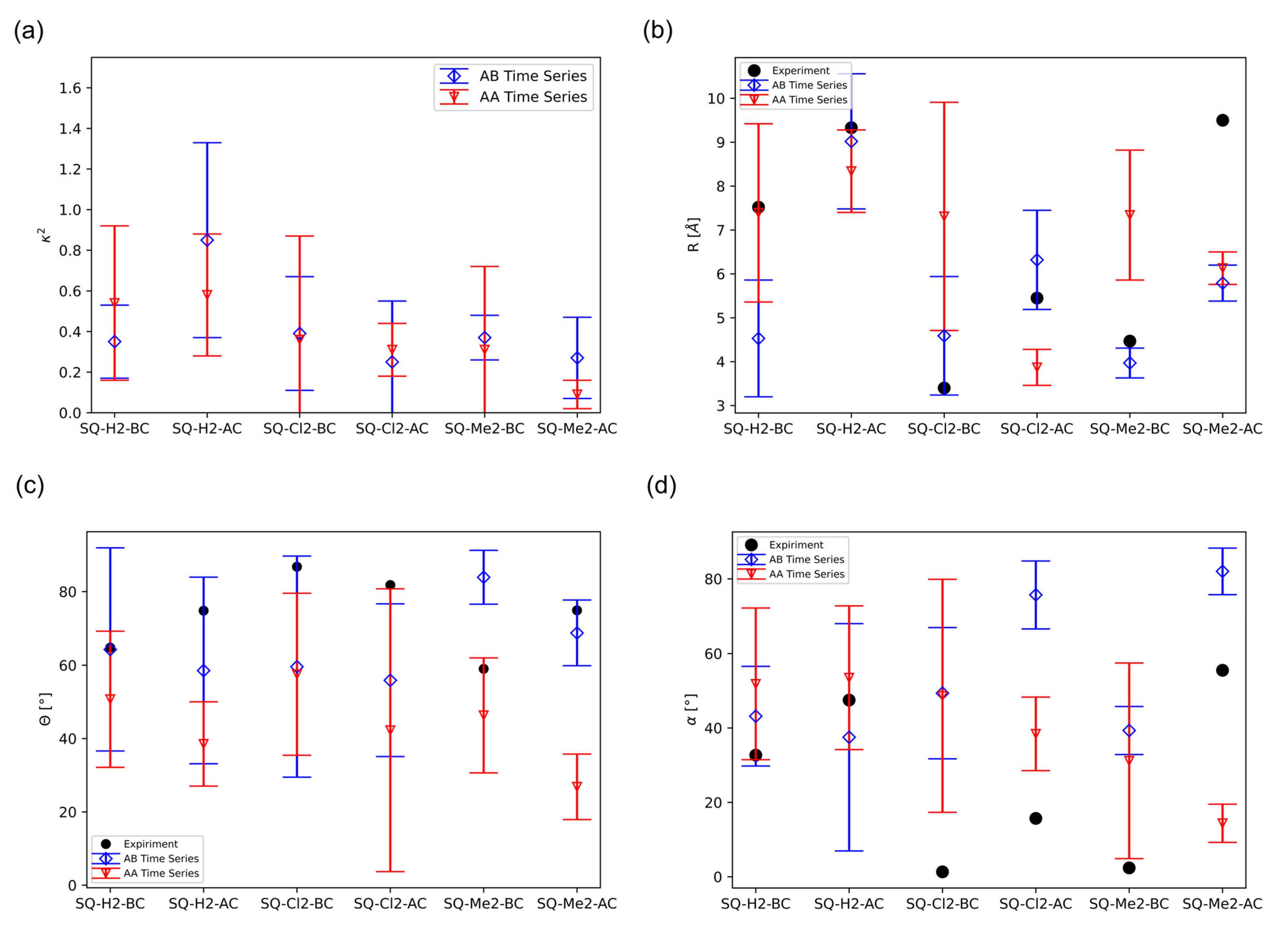
2.1. Dimer Orientation
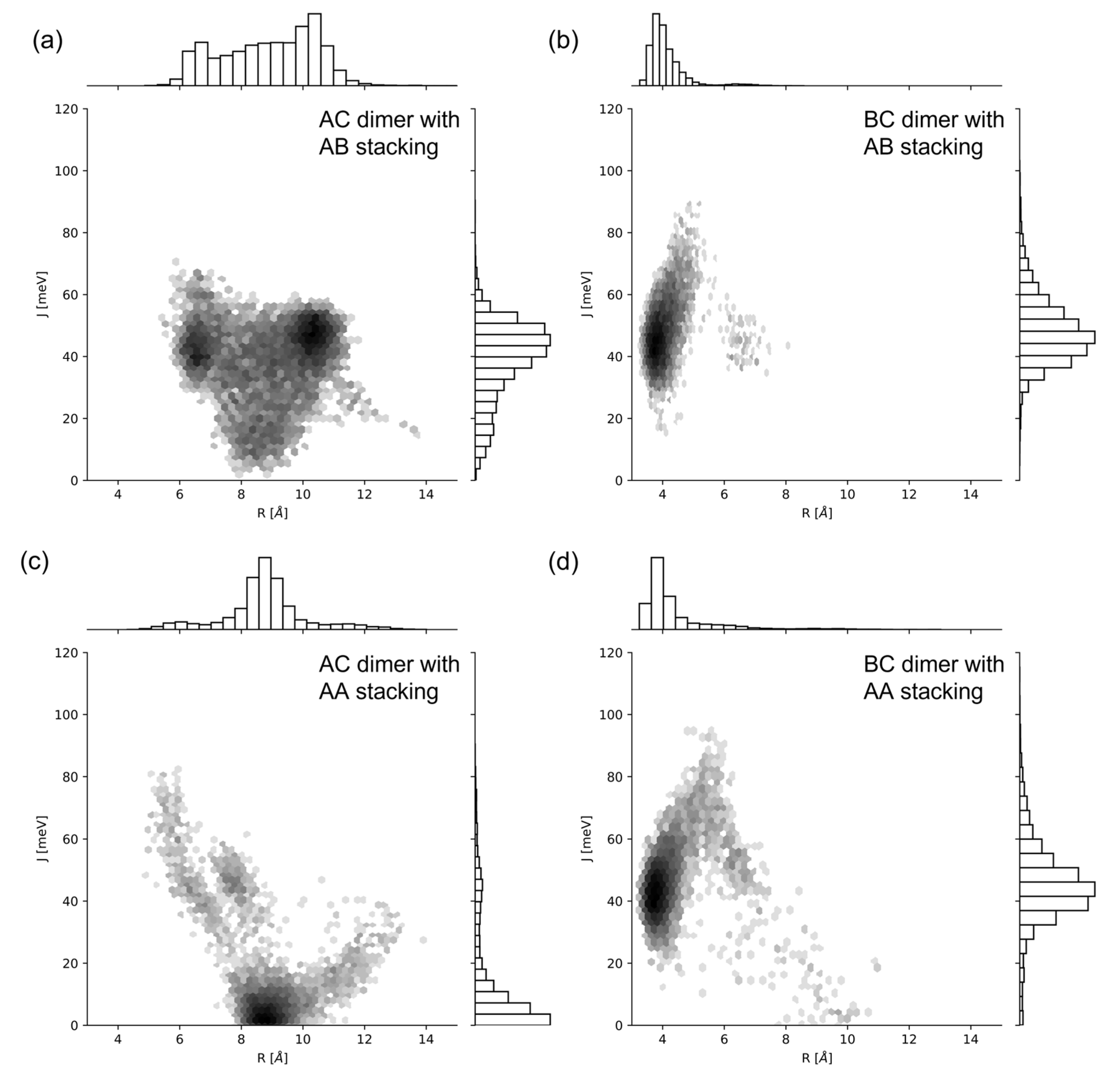
2.2. Excitonic Coupling
3. Discussion
4. Materials and Methods
4.1. Molecular Dynamics
4.2. Data Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Zheng, C.; Penmetcha, A.R.; Cona, B.; Spencer, S.D.; Zhu, B.; Heaphy, P.; Cody, J.A.; Collison, C.J. Contribution of Aggregate States and Energetic Disorder to a Squaraine System Targeted for Organic Photovoltaic Devices. Langmuir 2015, 31, 7717–7726. [Google Scholar] [CrossRef] [PubMed]
- Umezawa, K.; Citterio, D.; Suzuki, K. New Trends in Near-Infrared Fluorophores for Bioimaging. Anal. Sci. 2014, 30, 327–349. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sawaya, N.P.D.D.; Rappoport, D.; Tabor, D.P.; Aspuru-Guzik, A. Excitonics: A Set of Gates for Molecular Exciton Processing and Signaling. ACS Nano 2018, 12, 6410–6420. [Google Scholar] [CrossRef] [PubMed]
- Wasielewski, M.R.; Forbes, M.D.E.; Frank, N.L.; Kowalski, K.; Scholes, G.D.; Yuen-Zhou, J.; Baldo, M.A.; Freedman, D.E.; Goldsmith, R.H.; Goodson, T.; et al. Exploiting Chemistry and Molecular Systems for Quantum Information Science. Nat. Rev. Chem. 2020, 4, 490–504. [Google Scholar] [CrossRef]
- Castellanos, M.A.; Dodin, A.; Willard, A.P. On the Design of Molecular Excitonic Circuits for Quantum Computing: The Universal Quantum Gates. Phys. Chem. Chem. Phys. 2020, 22, 3048–3057. [Google Scholar] [CrossRef] [Green Version]
- Runeson, J.E.; Lawrence, J.E.; Mannouch, J.R.; Richardson, J.O. Explaining the Efficiency of Photosynthesis: Quantum Uncertainty or Classical Vibrations? J. Phys. Chem. Lett. 2022, 13, 3392–3399. [Google Scholar] [CrossRef]
- Engel, G.S.; Calhoun, T.R.; Read, E.L.; Ahn, T.K.; Mančal, T.; Cheng, Y.C.; Blankenship, R.E.; Fleming, G.R. Evidence for Wavelike Energy Transfer through Quantum Coherence in Photosynthetic Systems. Nature 2007, 446, 782–786. [Google Scholar] [CrossRef]
- Rafiq, S.; Scholes, G.D. From Fundamental Theories to Quantum Coherences in Electron Transfer. J. Am. Chem. Soc. 2018, 141, 708–722. [Google Scholar] [CrossRef]
- Brookes, J.C. Quantum Effects in Biology: Golden Rule in Enzymes, Olfaction, Photosynthesis and Magnetodetection. Proc. R. Soc. A Math. Phys. Eng. Sci. 2017, 473, 20160822. [Google Scholar] [CrossRef] [Green Version]
- Frenkel, J. On the Transformation of Light into Heat in Solids. Phys. Rev. 1931, 37, 17–44. [Google Scholar] [CrossRef]
- Davydov, A.S. Theory of Absorption Spectra of Molecular Crystals. Zh. Eksp. Teor. Fiz. 1948, 18, 210–218. [Google Scholar]
- Kasha, M. Energy Transfer, Charge Transfer, and Proton Transfer in Molecular Composite Systems. In Physical and Chemical Mechanisms in Molecular Radiation Biology; Springer: Boston, MA, USA, 1991; pp. 231–255. [Google Scholar] [CrossRef]
- Kasha, M. Energy Transfer Mechanisms and the Molecular Exciton Model for Molecular Aggregates. Radiat. Res. 1963, 20, 55–70. [Google Scholar] [CrossRef]
- Mass, O.A.; Wilson, C.K.; Barcenas, G.; Terpetschnig, E.A.; Obukhova, O.M.; Kolosova, O.S.; Tatarets, A.L.; Li, L.; Yurke, B.; Knowlton, W.B.; et al. Influence of Hydrophobicity on Excitonic Coupling in DNA-Templated Indolenine Squaraine Dye Aggregates. J. Phys. Chem. C 2022, 126, 3475–3488. [Google Scholar] [CrossRef]
- Abramavicius, D.; Palmieri, B.; Mukamel, S. Extracting Single and Two-Exciton Couplings in Photosynthetic Complexes by Coherent Two-Dimensional Electronic Spectra. Chem. Phys. 2009, 357, 79–84. [Google Scholar] [CrossRef] [Green Version]
- Madjet, M.E.; Abdurahman, A.; Renger, T. Intermolecular Coulomb Couplings from Ab Initio Electrostatic Potentials: Application to Optical Transitions of Strongly Coupled Pigments in Photosynthetic Antennae and Reaction Centers. J. Phys. Chem. B 2006, 110, 17268–17281. [Google Scholar] [CrossRef]
- Davydov, A.S.S.A.S. The Theory of Molecular Excitons. Uspekhi Fiz. Nauk. 1964, 82, 393–448. [Google Scholar] [CrossRef] [Green Version]
- Czikklely, V.; Forsterling, H.D.D.; Kuhn, H. Extended Dipole Model for Aggregates of Dye Molecules. Chem. Phys. Lett. 1970, 6, 207–210. [Google Scholar] [CrossRef]
- Kuzyk, A.; Jungmann, R.; Acuna, G.P.; Liu, N. DNA Origami Route for Nanophotonics. ACS Photonics 2018, 5, 1151–1163. [Google Scholar] [CrossRef] [Green Version]
- Dutta, P.K.; Varghese, R.; Nangreave, J.; Lin, S.; Yan, H.; Liu, Y. DNA-Directed Artificial Light-Harvesting Antenna. J. Am. Chem. Soc. 2011, 133, 11985–11993. [Google Scholar] [CrossRef]
- Albinsson, B.; Hannestad, J.K.; Börjesson, K. Functionalized DNA Nanostructures for Light Harvesting and Charge Separation. Coord. Chem. Rev. 2012, 256, 2399–2413. [Google Scholar] [CrossRef]
- Kringle, L.; Sawaya, N.P.D.; Widom, J.; Adams, C.; Raymer, M.G.; Aspuru-Guzik, A.; Marcus, A.H. Temperature-Dependent Conformations of Exciton-Coupled Cy3 Dimers in Double-Stranded DNA. J. Chem. Phys. 2018, 148, 085101. [Google Scholar] [CrossRef] [PubMed]
- Jaekel, A.; Lill, P.; Whitelam, S.; Saccà, B. Insights into the Structure and Energy of DNA Nanoassemblies. Molecules 2020, 25, 5466. [Google Scholar] [CrossRef] [PubMed]
- Mazuski, R.J.; Díaz, S.A.; Wood, R.E.; Lloyd, L.T.; Klein, W.P.; Mathur, D.; Melinger, J.S.; Engel, G.S.; Medintz, I.L. Ultrafast Excitation Transfer in Cy5 DNA Photonic Wires Displays Dye Conjugation and Excitation Energy Dependency. J. Phys. Chem. Lett. 2020, 11, 4163–4172. [Google Scholar] [CrossRef]
- Asanuma, H.; Shirasuka, K.; Takarada, T.; Kashida, H.; Komiyama, M. DNA-Dye Conjugates for Controllable H* Aggregation. J. Am. Chem. Soc. 2003, 125, 2217–2223. [Google Scholar] [CrossRef] [PubMed]
- Mathur, D.; Kim, Y.C.; Díaz, S.A.; Cunningham, P.D.; Rolczynski, B.S.; Ancona, M.G.; Medintz, I.L.; Melinger, J.S. Can a DNA Origami Structure Constrain the Position and Orientation of an Attached Dye Molecule? J. Phys. Chem. C 2021, 125, 1509–1522. [Google Scholar] [CrossRef]
- Nicoli, F.; Roos, M.K.; Hemmig, E.A.; Di Antonio, M.; de Vivie-Riedle, R.; Liedl, T. Proximity-Induced H-Aggregation of Cyanine Dyes on DNA-Duplexes. J. Phys. Chem. A 2016, 120, 9941–9947. [Google Scholar] [CrossRef]
- Roy, S.K.; Mass, O.A.; Kellis, D.L.; Wilson, C.K.; Hall, J.A.; Yurke, B.; Knowlton, W.B. Exciton Delocalization and Scaffold Stability in Bridged Nucleotide-Substituted, DNA Duplex-Templated Cyanine Aggregates. J. Phys. Chem. B 2021, 125, 13670–13684. [Google Scholar] [CrossRef]
- Ruedas-Rama, M.J.; Orte, A.; Martin-Domingo, M.C.; Castello, F.; Talavera, E.M.; Alvarez-Pez, J.M. Interaction of YOYO-3 with Different DNA Templates to Form H-Aggregates. J. Phys. Chem. B 2014, 118, 6098–6106. [Google Scholar] [CrossRef]
- Buckhout-White, S.; Spillmann, C.M.; Algar, W.R.; Khachatrian, A.; Melinger, J.S.; Goldman, E.R.; Ancona, M.G.; Medintz, I.L. Assembling Programmable FRET-Based Photonic Networks Using Designer DNA Scaffolds. Nat. Commun. 2014, 5, 5615. [Google Scholar] [CrossRef] [Green Version]
- Wang, M.; Silva, G.L.; Armitage, B.A. DNA-Templated Formation of a Helical Cyanine Dye J-Aggregate. J. Am. Chem. Soc. 2000, 122, 9977–9986. [Google Scholar] [CrossRef]
- Boulais, É.; Schlau-Cohen, G.S.; Woodbury, N.W.; Yan, H.; Aspuru-Guzik, A.; Bathe, M.; Sawaya, N.P.D.; Veneziano, R.; Andreoni, A.; Banal, J.L.; et al. Programmed Coherent Coupling in a Synthetic DNA-Based Excitonic Circuit. Nat. Mater. 2018, 17, 159–166. [Google Scholar] [CrossRef]
- Fujii, T.; Kashida, H.; Asanuma, H. Analysis of Coherent Heteroclustering of Different Dyes by Use of Threoninol Nucleotides for Comparison with the Molecular Exciton Theory. Chem. A Eur. J. 2009, 15, 10092–10102. [Google Scholar] [CrossRef]
- Rauch, S.; Dickinson, B.C. Programmable RNA Binding Proteins for Imaging and Therapeutics. Biochemistry 2017, 57, 363–364. [Google Scholar] [CrossRef] [Green Version]
- de Alba Ortíz, A.P.; Vreede, J.; Ensing, B. Sequence Dependence of Transient Hoogsteen Base Pairing in DNA. PLoS Comput. Biol. 2022, 18, e1010113. [Google Scholar] [CrossRef]
- Hart, S.M.; Banal, J.L.; Castellanos, M.A.; Markova, L.; Vyborna, Y.; Gorman, J.; Häner, R.; Willard, A.P.; Bathe, M.; Schlau-Cohen, G.S. Activating Charge-Transfer State Formation in Strongly-Coupled Dimers Using DNA Scaffolds. Chem. Sci. 2022, 13, 13020–13031. [Google Scholar] [CrossRef]
- Adendorff, M.R.; Tang, G.Q.; Millar, D.P.; Bathe, M.; Bricker, W.P. Computational Investigation of the Impact of Core Sequence on Immobile DNA Four-Way Junction Structure and Dynamics. Nucleic Acids Res. 2022, 50, 717–730. [Google Scholar] [CrossRef]
- Seeman, N.C. Nucleic Acid Junctions and Lattices. J. Theor. Biol. 1982, 99, 237–247. [Google Scholar] [CrossRef]
- Kallenbach, N.R.; Ma, R.-I.; Seeman, N.C. An Immobile Nucleic Acid Junction Constructed from Oligonucleotides. Nature 1983, 305, 829–831. [Google Scholar] [CrossRef]
- Cannon, B.L.; Kellis, D.L.; Davis, P.H.; Lee, J.; Kuang, W.; Hughes, W.L.; Graugnard, E.; Yurke, B.; Knowlton, W.B. Excitonic AND Logic Gates on DNA Brick Nanobreadboards. ACS Photonics 2015, 2, 398–404. [Google Scholar] [CrossRef] [Green Version]
- Huff, J.S.; Turner, D.B.; Mass, O.A.; Patten, L.K.; Wilson, C.K.; Roy, S.K.; Barclay, M.S.; Yurke, B.; Knowlton, W.B.; Davis, P.H.; et al. Excited-State Lifetimes of DNA-Templated Cyanine Dimer, Trimer, and Tetramer Aggregates: The Role of Exciton Delocalization, Dye Separation, and DNA Heterogeneity. J. Phys. Chem. B 2021, 125, 10240–10259. [Google Scholar] [CrossRef]
- Lilley, D.M.J.; Norman, D.G. The Holliday Junction Is Finally Seen with Crystal Clarity. Nat. Struct. Biol. 1999, 6, 897–899. [Google Scholar] [CrossRef] [PubMed]
- Simmons, C.R.R.; MacCulloch, T.; Krepl, M.; Matthies, M.; Buchberger, A.; Crawford, I.; Šponer, J.; Šulc, P.; Stephanopoulos, N.; Yan, H. The Influence of Holliday Junction Sequence and Dynamics on DNA Crystal Self-Assembly. Nat. Commun. 2022, 13, 3112. [Google Scholar] [CrossRef] [PubMed]
- Stadler, A.L.; Renikuntla, B.R.; Yaron, D.; Fang, A.S.; Armitage, B.A. Substituent Effects on the Assembly of Helical Cyanine Dye Aggregates in the Minor Groove of a DNA Template. Langmuir 2011, 27, 1472–1479. [Google Scholar] [CrossRef] [PubMed]
- Mass, O.A.; Wilson, C.K.; Roy, S.K.; Barclay, M.S.; Patten, L.K.; Terpetschnig, E.A.; Lee, J.; Pensack, R.D.; Yurke, B.; Knowlton, W.B. Exciton Delocalization in Indolenine Squaraine Aggregates Templated by DNA Holliday Junction Scaffolds. J. Phys. Chem. B 2020, 124, 9636–9647. [Google Scholar] [CrossRef]
- Kolosova, O.S.; Shishkina, S.V.; Marks, V.; Gellerman, G.; Hovor, I.V.; Tatarets, A.L.; Terpetschnig, E.A.; Patsenker, L.D. Molecular Structure and Spectral Properties of Indolenine Based Norsquaraines versus Squaraines. Dye. Pigment. 2019, 163, 318–329. [Google Scholar] [CrossRef]
- Kuster, S.; Geiger, T. Coupled π-Conjugated Chromophores: Squaraine Dye Dimers as Two Connected Pendulums. Dye. Pigment. 2015, 113, 110–116. [Google Scholar] [CrossRef]
- Ilina, K.; MacCuaig, W.M.; Laramie, M.; Jeouty, J.N.; Mcnally, L.R.; Henary, M. Squaraine Dyes: Molecular Design for Different Applications and Remaining Challenges. Bioconjug. Chem. 2020, 31, 194–213. [Google Scholar] [CrossRef]
- Biaggne, A.; Knowlton, W.B.; Yurke, B.; Lee, J.; Li, L. Substituent Effects on the Solubility and Electronic Properties of the Cyanine Dye Cy5: Density Functional and Time-Dependent Density Functional Theory Calculations. Molecules 2021, 26, 524. [Google Scholar] [CrossRef]
- Barcenas, G.; Biaggne, A.; Mass, O.A.; Wilson, C.K.; Obukhova, O.M.; Kolosova, O.S.; Tatarets, A.L.; Terpetschnig, E.; Pensack, R.D.; Lee, J.; et al. First-Principles Studies of Substituent Effects on Squaraine Dyes. RSC Adv. 2021, 11, 19029–19040. [Google Scholar] [CrossRef]
- Barclay, M.S.; Roy, S.K.; Huff, J.S.; Mass, O.A.; Turner, D.B.; Wilson, C.K.; Kellis, D.L.; Terpetschnig, E.A.; Lee, J.; Davis, P.H.; et al. Rotaxane Rings Promote Oblique Packing and Extended Lifetimes in DNA-Templated Molecular Dye Aggregates. Commun. Chem. 2021, 4, 19. [Google Scholar] [CrossRef]
- Barclay, M.S.; Wilson, C.K.; Roy, S.K.; Mass, O.A.; Obukhova, O.M.; Svoiakov, R.P.; Tatarets, A.L.; Chowdhury, A.U.; Huff, J.S.; Turner, D.B.; et al. Oblique Packing and Tunable Excitonic Coupling in DNA-Templated Squaraine Rotaxane Dimer Aggregates. ChemPhotoChem 2022, 6, e202200039. [Google Scholar] [CrossRef]
- Jang, Y.H.; Hwang, S.; Kim, Y.H.; Jang, S.S.; Goddard, W.A. Density Functional Theory Studies of the [2]Rotaxane Component of the Stoddart-Heath Molecular Switch. J. Am. Chem. Soc. 2004, 126, 12636–12645. [Google Scholar] [CrossRef] [Green Version]
- Fothergill, J.W.; Hernandez, A.C.; Knowlton, W.B.; Yurke, B.; Li, L. Ab Initio Studies of Exciton Interactions of Cy5 Dyes. J. Phys. Chem. A 2018, 122, 8989–8997. [Google Scholar] [CrossRef]
- Stennett, E.M.S.; Ma, N.; van der Vaart, A.; Levitus, M. Photophysical and Dynamical Properties of Doubly Linked Cy3–DNA Constructs. J. Phys. Chem. B 2014, 118, 152–163. [Google Scholar] [CrossRef]
- Cunningham, P.D.; Kim, Y.C.; Sebastián, S.; Díaz, S.A.; Buckhout-White, S.; Mathur, D.; Medintz, I.L.; Melinger, J.S. Optical Properties of Vibronically Coupled Cy3 Dimers on DNA Scaffolds. J. Phys. Chem. B 2018, 122, 5020–5029. [Google Scholar] [CrossRef]
- Biaggne, A.; Kim, Y.C.; Melinger, J.S.; Knowlton, W.B.; Yurke, B.; Li, L. Molecular Dynamics Simulations of Cyanine Dimers Attached to DNA Holliday Junctions. RSC Adv. 2022, 12, 28063–28078. [Google Scholar] [CrossRef]
- Biaggne, A.; Spear, L.; Ketteridge, M.; Kim, Y.C.; Melinger, J.S.; Knowlton, W.B.; Yurke, B.; Li, L. Data-Driven and Multiscale Modeling of DNA-Templated Dye Aggregates. Molecules 2022, 27, 3456. [Google Scholar] [CrossRef]
- Iyer, M.; Li, Z.; Jaroszewski, L.; Sedova, M.; Godzik, A. Difference Contact Maps: From What to Why in the Analysis of the Conformational Flexibility of Proteins. PLoS ONE 2020, 15, e0226702. [Google Scholar] [CrossRef] [Green Version]
- Bhattacharya, S.; Bhattacharya, D. Evaluating the Significance of Contact Maps in Low-Homology Protein Modeling Using Contact-Assisted Threading. Sci. Rep. 2020, 10, 2908. [Google Scholar] [CrossRef] [Green Version]
- Best, R.B.; Hummer, G.; Eaton, W.A. Native Contacts Determine Protein Folding Mechanisms in Atomistic Simulations. Proc. Natl. Acad. Sci. USA 2013, 110, 17874–17879. [Google Scholar] [CrossRef] [Green Version]
- Basu, S.; Cervantes-Salguero, K.; Yurke, B.; Knowlton, W.B.; Lee, J.; Mass, O.A. Photocrosslinking Probes Proximity of Thymine Modifiers Tethering Excitonically Coupled Dye Aggregates to DNA Holliday Junction. Molecules 2022, 27, 4006. [Google Scholar] [CrossRef] [PubMed]
- Uy, J.L.; Asbury, C.L.; Petersen, T.W.; Van Den Engh, G. The Polarization of Fluorescence of DNA Stains Depends on the Incorporation Density of the Dye Molecules. Cytom. Part A 2004, 61, 18–25. [Google Scholar] [CrossRef] [PubMed]
- Duarte, D.J.R.; Sosa, G.L.; Peruchena, N.M.; Alkorta, I. Halogen Bonding. The Role of the Polarizability of the Electron-Pair Donor. Phys. Chem. Chem. Phys. 2016, 18, 7300–7309. [Google Scholar] [CrossRef] [PubMed]
- Bresme, F.; Wynveen, A. On the Influence of Solute Polarizability on the Hydrophobic Interaction. J. Chem. Phys. 2007, 126, 044501. [Google Scholar] [CrossRef] [Green Version]
- Sugita, Y.; Okamoto, Y. Replica-Exchange Molecular Dynamics Method for Protein Folding. Chem. Phys. Lett. 1999, 314, 141–151. [Google Scholar] [CrossRef]
- Husic, B.E.; Pande, V.S. Markov State Models: From an Art to a Science. J. Am. Chem. Soc. 2018, 140, 2386–2396. [Google Scholar] [CrossRef]
- Bauer, P.; Hess, B.; Lindahl, E. GROMACS 2022.1 Manual. Zenodo 2022. [Google Scholar] [CrossRef]
- Bekker, H.; Benendsen, H.J.C.; Dijkstra, E.J.; Achterop, S.; Vondrumen, R.; Vanderspoel, D.; Sijbers, A.; Keegstra, H.; Renardus, M.R. Gromacs—A Parallel Computer for Molecular-Dynamics Simulations; Nadrchal, R.D., Nadrchal, J., Eds.; World Scientific Publishing: Singapore, 1993. [Google Scholar]
- Graen, T.; Hoefling, M.; Grubmü, H. AMBER-DYES: Characterization of Charge Fluctuations and Force Field Parameterization of Fluorescent Dyes for Molecular Dynamics Simulations. J. Chem. Theory Comput. 2014, 10, 5505–5512. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Wolf, R.M.; Caldwell, J.W.; Kollman, P.A.; Case, D.A. Development and Testing of a General Amber Force Field. J. Comput. Chem. 2004, 25, 1157–1174. [Google Scholar] [CrossRef]
- Bayly, C.I.; Cieplak, P.; Cornell, W.D.; Kollman, P.A. A Well-Behaved Electrostatic Potential Based Method Using Charge Restraints for Deriving Atomic Charges: The RESP Model. J. Phys. Chem. 1993, 97, 10269–10280. [Google Scholar] [CrossRef]
- Leland, B.; Paul, D.; Krueger, B.; Walker, R. Tutorial A1: Setting up an Advanced System (Including Basic Charge Derivation). Available online: https://ambermd.org/tutorials/advanced/tutorial1/index.php (accessed on 3 June 2022).
- Sims, M.; Smith, B.; Helfrich, B.; Sathaye, N.; Messick, E.; Moore, T.; Fish, R.; Rajagopalan, M.; Rotkiewicz, P.; Drexler, K.E.; et al. Nanoengineer-1—A CAD-Based Molecular Modeling Program for Structural DNA Nanotechnology. In Proceedings of the Foundations of Nanoscience: Self-Assembled Architectures and Devices 2008 (FNANO08), Snowbird, UT, USA, 22–25 April 2008; pp. 175–178. [Google Scholar]
- Rappé, A.K.; Casewit, C.J.; Colwell, K.S.; Goddard, W.A.; Skiff, W.M. UFF, a Full Periodic Table Force Field for Molecular Mechanics and Molecular Dynamics Simulations. J. Am. Chem. Soc. 1992, 114, 10024–10035. [Google Scholar] [CrossRef]
- Wheatley, E.G.; Pieniazek, S.N.; Mukerji, I.; Beveridge, D.L. Molecular Dynamics of a DNA Holliday Junction: The Inverted Repeat Sequence d(CCGGTACCGG) 4. Biophys. J. 2012, 102, 552–560. [Google Scholar] [CrossRef] [Green Version]
- Yu, J. Conformational Model of the Holliday Junction Transition Deduced from Molecular Dynamics Simulations. Nucleic Acids Res. 2004, 32, 6683–6695. [Google Scholar] [CrossRef] [Green Version]
- Watson, J.; Hays, F.A.; Ho, P.S. Definitions and Analysis of DNA Holliday Junction Geometry. Nucleic Acids Res. 2004, 32, 3017–3027. [Google Scholar] [CrossRef] [Green Version]
- Maier, J.A.; Martinez, C.; Kasavajhala, K.; Wickstrom, L.; Hauser, K.E.; Simmerling, C. Ff14SB: Improving the Accuracy of Protein Side Chain and Backbone Parameters from Ff99SB. J. Chem. Theory Comput. 2015, 11, 3696–3713. [Google Scholar] [CrossRef] [Green Version]
- Hess, B.; Bekker, H.; Berendsen, H.J.C.C.; Fraaije, J.G.E.M.E.M. LINCS: A Linear Constraint Solver for Molecular Simulations. J. Comput. Chem. 1997, 18, 1463–1472. [Google Scholar] [CrossRef]
- Goga, N.; Rzepiela, A.J.; De Vries, A.H.; Marrink, S.J.; Berendsen, H.J.C. Efficient Algorithms for Langevin and DPD Dynamics. J. Chem. Theory Comput. 2012, 8, 3637–3649. [Google Scholar] [CrossRef] [Green Version]
- Beyerle, E.R.; Dinpajooh, M.; Ji, H.; von Hippel, P.H.; Marcus, A.H.; Guenza, M.G. Dinucleotides as Simple Models of the Base Stacking-Unstacking Component of DNA ‘Breathing’ Mechanisms. Nucleic Acids Res. 2021, 49, 1872–1885. [Google Scholar] [CrossRef]
- Michaud-Agrawal, N.; Denning, E.J.; Woolf, T.B.; Beckstein, O. MDAnalysis: A Toolkit for the Analysis of Molecular Dynamics Simulations. J. Comput. Chem. 2011, 32, 2319–2327. [Google Scholar] [CrossRef] [Green Version]
- Gowers, R.; Linke, M.; Barnoud, J.; Reddy, T.; Melo, M.; Seyler, S.; Domański, J.; Dotson, D.; Buchoux, S.; Kenney, I.; et al. MDAnalysis: A Python Package for the Rapid Analysis of Molecular Dynamics Simulations. In Proceedings of the 15th Python in Science Conference, Austin, TX, USA, 11–17 July 2016; pp. 98–105. [Google Scholar] [CrossRef] [Green Version]
- Daura, X.; Gademann, K.; Jaun, B.; Seebach, D.; van Gunsteren, W.F.; Mark, A.E.; Rigault, A.; Siegel, J.; Harrowfield, J.; Chevrier, B.; et al. Peptide Folding: When Simulation Meets Experiment; Wiley: Hoboken, NJ, USA, 1998; Volume 31. [Google Scholar]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Dye | |µ| [D] |
|---|---|
| SQ-H2 | 12.99 |
| SQ-Cl2 | 13.41 |
| SQ-Me2 | 13.73 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Barcenas, G.; Biaggne, A.; Mass, O.A.; Knowlton, W.B.; Yurke, B.; Li, L. Molecular Dynamic Studies of Dye–Dye and Dye–DNA Interactions Governing Excitonic Coupling in Squaraine Aggregates Templated by DNA Holliday Junctions. Int. J. Mol. Sci. 2023, 24, 4059. https://doi.org/10.3390/ijms24044059
Barcenas G, Biaggne A, Mass OA, Knowlton WB, Yurke B, Li L. Molecular Dynamic Studies of Dye–Dye and Dye–DNA Interactions Governing Excitonic Coupling in Squaraine Aggregates Templated by DNA Holliday Junctions. International Journal of Molecular Sciences. 2023; 24(4):4059. https://doi.org/10.3390/ijms24044059
Chicago/Turabian StyleBarcenas, German, Austin Biaggne, Olga A. Mass, William B. Knowlton, Bernard Yurke, and Lan Li. 2023. "Molecular Dynamic Studies of Dye–Dye and Dye–DNA Interactions Governing Excitonic Coupling in Squaraine Aggregates Templated by DNA Holliday Junctions" International Journal of Molecular Sciences 24, no. 4: 4059. https://doi.org/10.3390/ijms24044059