
Fibrosis: Types, Effects, Markers, Mechanisms for Disease Progression, and Its Relation with Oxidative Stress, Immunity, and Inflammation


Abstract
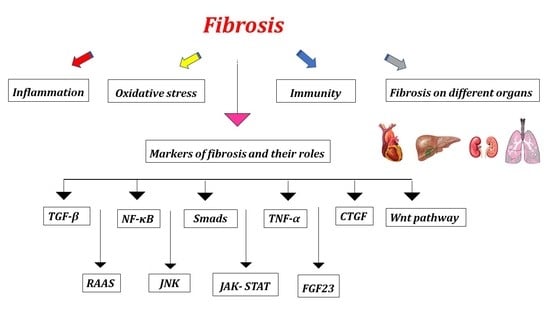
:
1. Introduction
2. Markers of Fibrosis and Their Roles
2.1. TGF-β’s Function in Mediating Fibrosis
2.2. Role of Smads in Fibrosis
2.3. Role of Fibroblast Growth Factor23 (FGF23) in Fibrosis
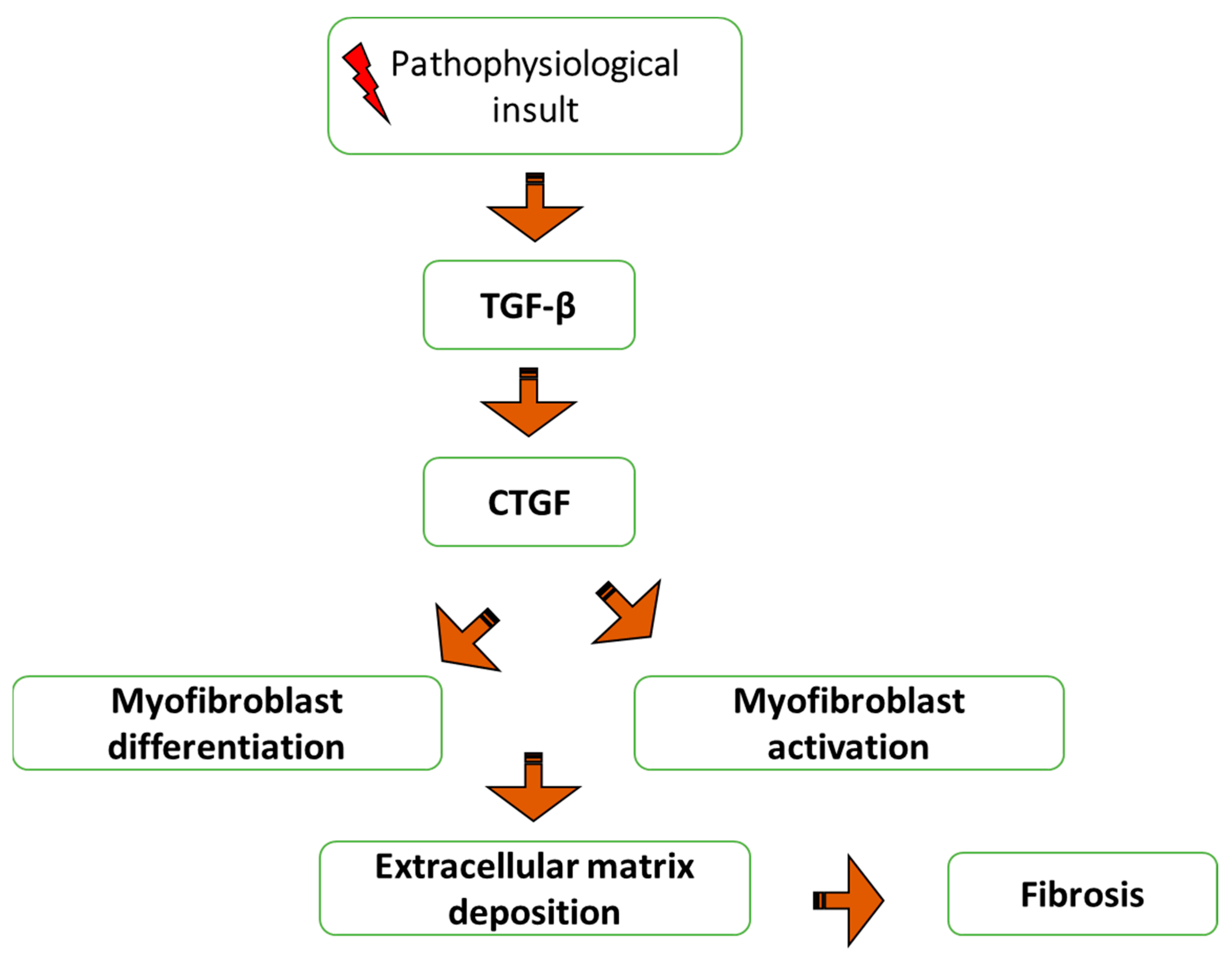
2.4. Role of Connective Tissue Growth Factor (CTGF) in Fibrosis
2.5. Role of Nuclear Erythroid 2-Related Factor 2 (Nrf2) in Fibrosis
2.6. Role of Renin-Angiotensin-Aldosterone System (RAAS) in Fibrosis
3. Oxidative Stress and Fibrosis
4. Antioxidant
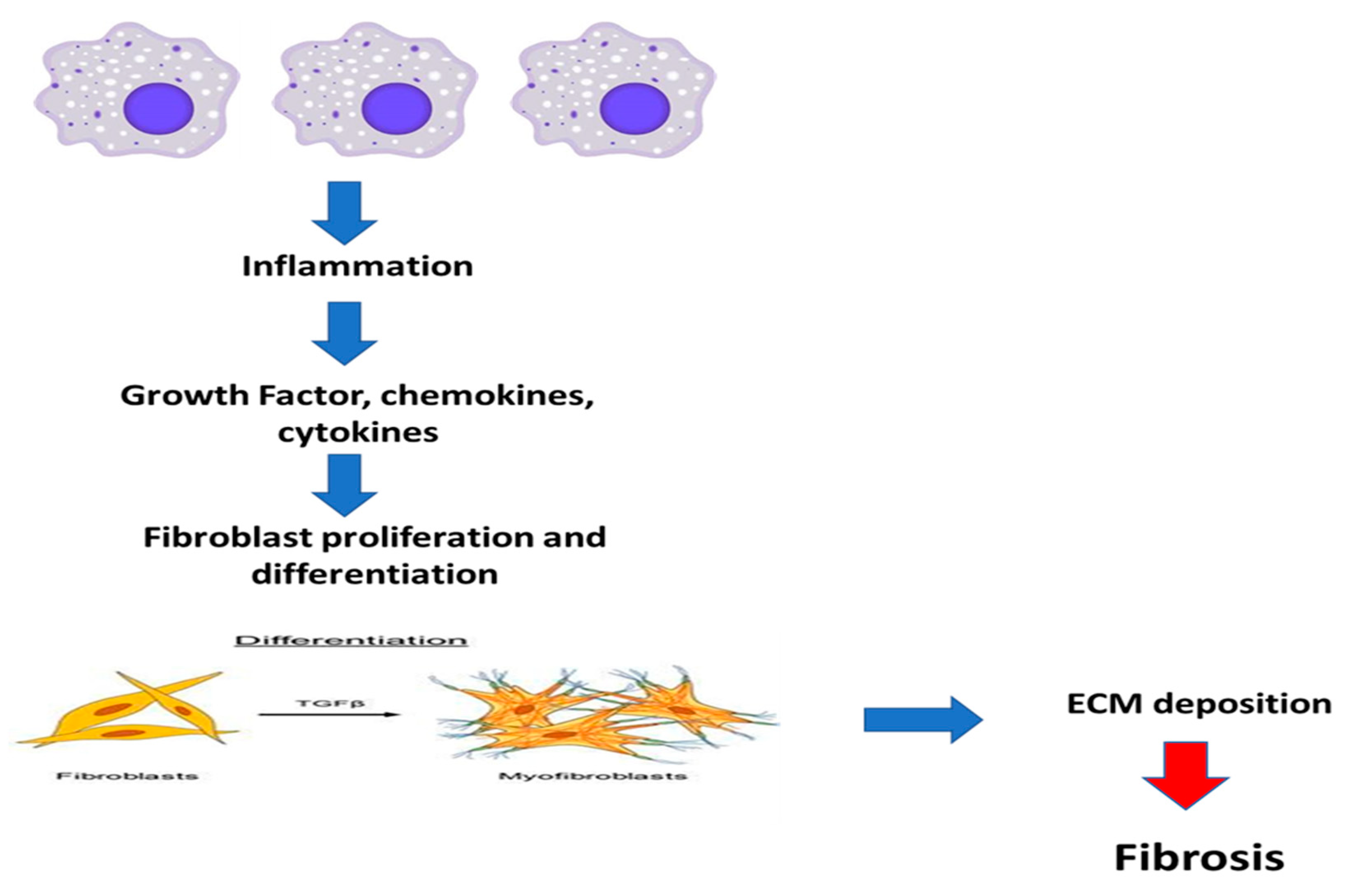
5. Inflammation and Fibrosis
5.1. Role of TNF-α in Fibrosis
5.2. Fibrosis and the NF-κB Pathway
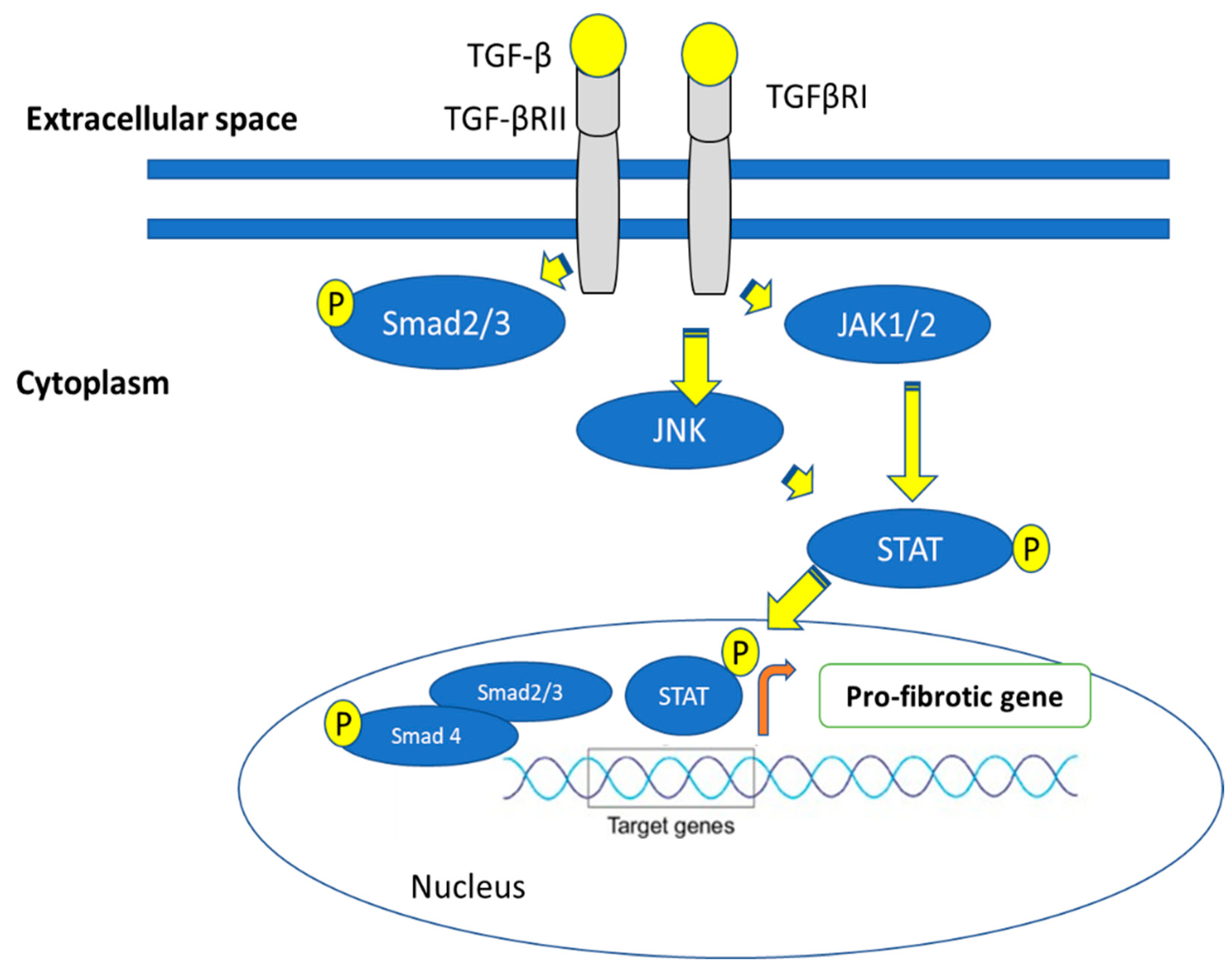
5.3. Role of Jun N-Terminal Kinase (JNK) in Fibrosis
6. Fibrosis Is Mediated via the Janus Kinase (JAK)-Signal Transducer and Activator of Transcription (STAT) Pathways
7. Role of Wnt Pathway in Fibrosis
8. Immunity and Fibrosis
8.1. T Cell Function and Fibrosis Pathways
8.1.1. Fibrosis and Th1 Cells
8.1.2. Fibrosis and Th9 Cells
8.1.3. Fibrosis and Cytotoxic T Cells (CTLs, CD8+ T Cells)
8.1.4. Tfh Cells and Fibrosis
9. Effect of Fibrosis on Different Organs
9.1. Cardiac Fibrosis
9.2. Kidney Fibrosis
9.3. Liver Fibrosis
9.4. Idiopathic Pulmonary Fibrosis (IPF)
9.5. Muscle Fibrosis
10. Novel Antifibrotic Drugs
11. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| Ang II | Angiotensin II |
| α-SMA | Alpha-smooth muscle actin |
| CKD | Chronic kidney disease |
| CTGF | Connective tissue growth factor |
| ECM | Extracellular matrix |
| EMT | Epithelial-mesenchymal transition |
| ESRD | End stage renal disease |
| FGF23 | Fibroblast growth factor23 |
| H2O2 | Hydrogen peroxide |
| IFN-γ | Interferon-γ |
| IL-6 | Interleukin-6 |
| iNOS | Inducible nitric oxide synthase |
| IPF | Idiopathic pulmonary fibrosis |
| LAP | Latency-associated peptide |
| LTBP | Latent TGF-β binding protein |
| MCP-1 | Monocyte chemoattractant protein |
| NF-κB | Nuclear factor kappa B |
| PAI1 | Plasminogen activator inhibitor-1 |
| RAAS | Renin–angiotensin–aldosterone system |
| RNS | reactive nitrogen species |
| ROS | Reactive oxygen species |
| TBR | TGF-β1 receptors |
| TGF-β1 | Transforming growth factor β1 |
| TIMP-1 | Tissue inhibitor of metalloproteinase-1 |
| TNF-α | Tumor necrosis factor-alpha |
| MMPs | Matrix metalloproteinases |
| OPN | Osteopontin |
References
- Conte, E.J. Targeting monocytes/macrophages in fibrosis and cancer diseases: Therapeutic approaches. Pharmacol. Ther. 2021, 234, 108031. [Google Scholar] [CrossRef] [PubMed]
- Davidson, M.D.; Burdick, J.A.; Wells, R.G. Engineered biomaterial platforms to study fibrosis. Adv. Healthc. Mater. 2020, 9, 1901682. [Google Scholar]
- Al-Hattab, D.S.; Chattopadhyaya, S.; Czubryt, M.P. Canadian Contributions in Fibroblast Biology. Cells 2022, 11, 2272. [Google Scholar] [CrossRef] [PubMed]
- Schuster, R.; Rockel, J.S.; Kapoor, M.; Hinz, B. The inflammatory speech of fibroblasts. Immunol. Rev. 2021, 302, 126–146. [Google Scholar] [CrossRef]
- Lafuse, W.P.; Wozniak, D.J.; Rajaram, M.V. Role of cardiac macrophages on cardiac inflammation, fibrosis and tissue repair. Cells 2020, 10, 51. [Google Scholar] [CrossRef]
- Meng, Q.; Bhandary, B.; Bhuiyan, M.S.; James, J.; Osinska, H.; Valiente-Alandi, I.; Shay-Winkler, K.; Gulick, J.; Molkentin, J.D.; Blaxall, B.C. Myofibroblast-specific TGFβ receptor II signaling in the fibrotic response to cardiac myosin binding protein C-induced cardiomyopathy. Circ. Res. 2018, 123, 1285–1297. [Google Scholar] [CrossRef]
- Distler, J.H.; Györfi, A.-H.; Ramanujam, M.; Whitfield, M.L.; Königshoff, M.; Lafyatis, R. Shared and distinct mechanisms of fibrosis. Nat. Rev. Rheumatol. 2019, 15, 705–730. [Google Scholar] [CrossRef]
- Xu, X.; Zheng, L.; Yuan, Q.; Zhen, G.; Crane, J.L.; Zhou, X.; Cao, X. Transforming growth factor-β in stem cells and tissue homeostasis. Bone Res. 2018, 6, 2. [Google Scholar] [CrossRef] [Green Version]
- Peng, D.; Fu, M.; Wang, M.; Wei, Y.; Wei, X. Targeting TGF-β signal transduction for fibrosis and cancer therapy. Mol. Cancer 2022, 21, 104. [Google Scholar] [CrossRef]
- Goteri, G.; Altobelli, E.; Tossetta, G.; Zizzi, A.; Avellini, C.; Licini, C.; Lorenzi, T.; Castellucci, M.; Ciavattini, A.; Marzioni, D. High temperature requirement A1, transforming growth factor beta 1, phosphoSmad2 and Ki67 in eutopic and ectopic endometrium of women with endometriosis. Eur. J. Histochem. 2015, 59, 2570. [Google Scholar] [CrossRef] [Green Version]
- Licini, C.; Tossetta, G.; Avellini, C.; Ciarmela, P.; Lorenzi, T.; Toti, P.; Gesuita, R.; Voltolini, C.; Petraglia, F.; Castellucci, M. Analysis of cell-cell junctions in human amnion and chorionic plate affected by chorioamnionitis. Histol. Histopathol. 2016, 31, 759–767. [Google Scholar] [PubMed]
- Yu, X.-Y.; Sun, Q.; Zhang, Y.-M.; Zou, L.; Zhao, Y.-Y. TGF-β/Smad signaling pathway in tubulointerstitial fibrosis. Front. Pharmacol. 2022, 13, 860588. [Google Scholar] [CrossRef] [PubMed]
- Lodyga, M.; Hinz, B. TGF-β1–a truly transforming growth factor in fibrosis and immunity. In Seminars in Cell & Developmental Biology; Academic Press: Cambridge, MA, USA, 2020; pp. 123–139. [Google Scholar]
- Yue, Y.; Meng, K.; Pu, Y.; Zhang, X. Transforming growth factor beta (TGF-β) mediates cardiac fibrosis and induces diabetic cardiomyopathy. Diabetes Res. Clin. Pract. 2017, 133, 124–130. [Google Scholar] [CrossRef] [PubMed]
- Lichtman, M.K.; Otero-Vinas, M.; Falanga, V. Transforming growth factor beta (TGF-β) isoforms in wound healing and fibrosis. Wound Repair Regen. 2016, 24, 215–222. [Google Scholar] [CrossRef]
- Hazem, R.M.; Antar, S.A.; Nafea, Y.K.; Al-Karmalawy, A.A.; Saleh, M.A.; El-Azab, M.F. Pirfenidone and vitamin D mitigate renal fibrosis induced by doxorubicin in mice with Ehrlich solid tumor. Life Sci. 2022, 288, 120185. [Google Scholar] [CrossRef]
- Parichatikanond, W.; Luangmonkong, T.; Mangmool, S.; Kurose, H. Therapeutic targets for the treatment of cardiac fibrosis and cancer: Focusing on TGF-β signaling. Front. Cardiovasc. Med. 2020, 7, 34. [Google Scholar] [CrossRef]
- Hu, H.-H.; Chen, D.-Q.; Wang, Y.-N.; Feng, Y.-L.; Cao, G.; Vaziri, N.D.; Zhao, Y.-Y. New insights into TGF-β/Smad signaling in tissue fibrosis. Chem. Biol. Interact. 2018, 292, 76–83. [Google Scholar] [CrossRef] [Green Version]
- Kaivo-oja, N.; Jeffery, L.A.; Ritvos, O.; Mottershead, D.G. Smad signalling in the ovary. Reprod. Biol. Endocrinol. 2006, 4, 21. [Google Scholar] [CrossRef]
- Zhang, Z.-H.; Li, M.-H.; Liu, D.; Chen, H.; Chen, D.-Q.; Tan, N.-H.; Ma, S.-C.; Zhao, Y.-Y. Rhubarb protect against tubulointerstitial fibrosis by inhibiting TGF-β/Smad pathway and improving abnormal metabolome in chronic kidney disease. Front. Pharmacol. 2018, 9, 1029. [Google Scholar] [CrossRef] [Green Version]
- Qing, Z.; Yuan, W.; Wang, J.; Song, W.; Luo, J.; Wu, X.; Lu, Q.; Li, Y.; Zeng, M. Verapamil inhibited the development of ureteral stricture by blocking CaMK II-mediated STAT3 and Smad3/JunD pathways. Int. Urol. Nephrol. 2022, 54, 2855–2866. [Google Scholar] [CrossRef]
- Li, S.-N.; Wu, J.-F. TGF-β/SMAD signaling regulation of mesenchymal stem cells in adipocyte commitment. Stem Cell Res. Ther. 2020, 11, 41. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, L.; Yang, T.; Lu, D.-W.; Zhao, H.; Feng, Y.-L.; Chen, H.; Chen, D.-Q.; Vaziri, N.D.; Zhao, Y.-Y. Central role of dysregulation of TGF-β/Smad in CKD progression and potential targets of its treatment. Biomed. Pharmacother. 2018, 101, 670–681. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soji, K.; Doi, S.; Nakashima, A.; Sasaki, K.; Doi, T.; Masaki, T. Deubiquitinase inhibitor PR-619 reduces Smad4 expression and suppresses renal fibrosis in mice with unilateral ureteral obstruction. PLoS ONE 2018, 13, e0202409. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, J.; Kong, D.; Qiu, J.; Xie, Y.; Lu, Z.; Zhou, C.; Liu, X.; Zhang, R.; Wang, Y. Praziquantel ameliorates CCl4-induced liver fibrosis in mice by inhibiting TGF-β/Smad signalling via up-regulating Smad7 in hepatic stellate cells. Br. J. Pharmacol. 2019, 176, 4666–4680. [Google Scholar] [CrossRef] [PubMed]
- Song, M.-K.; Lee, J.-H.; Ryoo, I.-g.; Lee, S.-h.; Ku, S.-K.; Kwak, M.-K. Bardoxolone ameliorates TGF-β1-associated renal fibrosis through Nrf2/Smad7 elevation. Free Radic. Biol. Med. 2019, 138, 33–42. [Google Scholar] [CrossRef] [PubMed]
- Lan, H.Y. Diverse roles of TGF-β/Smads in renal fibrosis and inflammation. Int. J. Biol. Sci. 2011, 7, 1056. [Google Scholar] [CrossRef] [Green Version]
- Saleh, M.A.; Antar, S.A.; Hazem, R.M.; El-Azab, M.F. Pirfenidone and vitamin D ameliorate cardiac fibrosis induced by doxorubicin in Ehrlich ascites carcinoma bearing mice: Modulation of monocyte chemoattractant protein-1 and Jun N-terminal kinase-1 pathways. Pharmaceuticals 2020, 13, 348. [Google Scholar] [CrossRef]
- Erben, R.G. Effects of FGF23 in the distal nephron. In Fibroblast Growth Factor 23; Elsevier: Amsterdam, The Netherlands, 2021; pp. 23–30. [Google Scholar]
- Dastghaib, S.; Koohpeyma, F.; Shams, M.; Saki, F.; Alizadeh, A. New concepts in regulation and function of the FGF23. Clin. Exp. Med. 2022, 1–12. [Google Scholar] [CrossRef]
- Clinkenbeard, E.L.; Noonan, M.L.; Thomas, J.C.; Ni, P.; Hum, J.M.; Aref, M.; Swallow, E.A.; Moe, S.M.; Allen, M.R.; White, K.E. Increased FGF23 protects against detrimental cardio-renal consequences during elevated blood phosphate in CKD. JCI Insight 2019, 4, e123817. [Google Scholar] [CrossRef]
- Methatham, T.; Tomida, S.; Kimura, N.; Imai, Y.; Aizawa, K. Inhibition of the canonical Wnt signaling pathway by a β-catenin/CBP inhibitor prevents heart failure by ameliorating cardiac hypertrophy and fibrosis. Sci. Rep. 2021, 11, 14886. [Google Scholar] [CrossRef]
- Chen, W.X.; Liu, H.H.; Li, R.X.; Mammadov, G.; Wang, J.J.; Liu, F.F.; Samadli, S.; Wu, Y.F.; Zhang, D.D.; Luo, H.H.; et al. C-type natriuretic peptide stimulates osteoblastic proliferation and collagen-X expression but suppresses fibroblast growth factor-23 expression in vitro. Pediatr. Rheumatol. 2020, 18, 46. [Google Scholar] [CrossRef] [PubMed]
- Barnes, J.W.; Duncan, D.; Helton, S.; Hutcheson, S.; Kurundkar, D.; Logsdon, N.J.; Locy, M.; Garth, J.; Denson, R.; Farver, C.; et al. Role of fibroblast growth factor 23 and klotho cross talk in idiopathic pulmonary fibrosis. Am. J. Physiol. Lung Cell. Mol. Physiol. 2019, 317, L141–L154. [Google Scholar] [PubMed]
- Kaasbøll, O.J.; Gadicherla, A.K.; Wang, J.-H.; Monsen, V.T.; Hagelin, E.M.V.; Dong, M.-Q.; Attramadal, H. Connective tissue growth factor (CCN2) is a matricellular preproprotein controlled by proteolytic activation. J. Biol. Chem. 2018, 293, 17953–17970. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schnieder, J.; Mamazhakypov, A.; Birnhuber, A.; Wilhelm, J.; Kwapiszewska, G.; Ruppert, C.; Markart, P.; Wujak, L.; Rubio, K.; Barreto, G. Loss of LRP1 promotes acquisition of contractile-myofibroblast phenotype and release of active TGF-β1 from ECM stores. Matrix Biol. 2020, 88, 69–88. [Google Scholar] [CrossRef] [PubMed]
- Coeyman, S.J.; Richardson, W.J.; Bradshaw, A.D. Mechanics & Matrix: Positive Feedback Loops between Fibroblasts and ECM Drive Interstitial Cardiac Fibrosis. Curr. Opin. Physiol. 2022, 28, 100560. [Google Scholar]
- Valle-Tenney, R.; Rebolledo, D.L.; Lipson, K.E.; Brandan, E. Role of hypoxia in skeletal muscle fibrosis: Synergism between hypoxia and TGF-β signaling upregulates CCN2/CTGF expression specifically in muscle fibers. Matrix Biol. 2020, 87, 48–65. [Google Scholar] [CrossRef] [PubMed]
- Yanagihara, T.; Tsubouchi, K.; Gholiof, M.; Chong, S.G.; Lipson, K.E.; Zhou, Q.; Scallan, C.; Upagupta, C.; Tikkanen, J.; Keshavjee, S.; et al. Connective-Tissue Growth Factor Contributes to TGF-β1–induced Lung Fibrosis. Am. J. Respir. Cell Mol. Biol. 2022, 66, 260–270. [Google Scholar] [CrossRef]
- Strycharz, J.; Rygielska, Z.; Swiderska, E.; Drzewoski, J.; Szemraj, J.; Szmigiero, L.; Sliwinska, A. SIRT1 as a therapeutic target in diabetic complications. Curr. Med. Chem. 2018, 25, 1002–1035. [Google Scholar]
- Zhu, Z.; Hu, R.; Li, J.; Xing, X.; Chen, J.; Zhou, Q.; Sun, J. Alpinetin exerts anti-inflammatory, anti-oxidative and anti-angiogenic effects through activating the Nrf2 pathway and inhibiting NLRP3 pathway in carbon tetrachloride-induced liver fibrosis. Int. Immunopharmacol. 2021, 96, 107660. [Google Scholar] [CrossRef]
- Baird, L.; Llères, D.; Swift, S.; Dinkova-Kostova, A.T. Regulatory flexibility in the Nrf2-mediated stress response is conferred by conformational cycling of the Keap1-Nrf2 protein complex. Proc. Natl. Acad. Sci. USA 2013, 110, 15259–15264. [Google Scholar] [CrossRef] [Green Version]
- Marzioni, D.; Mazzucchelli, R.; Fantone, S.; Tossetta, G. NRF2 modulation in TRAMP mice: An in vivo model of prostate cancer. Mol. Biol. Rep. 2022, 50, 873–881. [Google Scholar] [CrossRef] [PubMed]
- Tossetta, G.; Marzioni, D. Natural and synthetic compounds in Ovarian Cancer: A focus on NRF2/KEAP1 pathway. Pharmacol. Res. 2022, 183, 106365. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Xu, H.; Zhang, L.; Zhang, C.; Yang, L.; Ma, E.; Liu, L.; Li, Y. Salvianolic acid B inhibits myofibroblast transdifferentiation in experimental pulmonary fibrosis via the up-regulation of Nrf2. Biochem. Biophys. Res. Commun. 2018, 495, 325–331. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; He, X.; Caldwell, L.; Goru, S.K.; Severino, L.U.; Tolosa, M.F.; Misra, P.S.; McEvoy, C.M.; Christova, T.; Liu, Y. NUAK1 promotes organ fibrosis via YAP and TGF-β/SMAD signaling. Sci. Transl. Med. 2022, 14, eaaz4028. [Google Scholar] [CrossRef]
- Kamel, A.K.A.; Hozayen, W.; El-kawi, S.H.A.; Hashem, K.S. Galaxaura elongata Extract (GE) Modulates Vanadyl Sulfate-Induced Renal Damage via Regulating TGF-β/Smads and Nrf2/NF-κB Pathways. Biol. Trace Elem. Res. 2022, 200, 3187–3204. [Google Scholar] [CrossRef]
- AlQudah, M.; Hale, T.M.; Czubryt, M.P. Targeting the renin-angiotensin-aldosterone system in fibrosis. Matrix Biol. 2020, 91, 92–108. [Google Scholar] [CrossRef]
- Chen, R.-R.; Fan, X.-H.; Chen, G.; Zeng, G.-W.; Xue, Y.-G.; Liu, X.-T.; Wang, C.-Y. Irisin attenuates angiotensin II-induced cardiac fibrosis via Nrf2 mediated inhibition of ROS/TGFβ1/Smad2/3 signaling axis. Chem. Biol. Interact. 2019, 302, 11–21. [Google Scholar] [CrossRef]
- Balakumar, P.; Sambathkumar, R.; Mahadevan, N.; Muhsinah, A.B.; Alsayari, A.; Venkateswaramurthy, N.; Jagadeesh, G. A potential role of the renin-angiotensin-aldosterone system in epithelial-to-mesenchymal transition-induced renal abnormalities: Mechanisms and therapeutic implications. Pharmacol. Res. 2019, 146, 104314. [Google Scholar] [CrossRef]
- Kapoor, D.; Singh, S.; Kumar, V.; Romero, R.; Prasad, R.; Singh, J. Antioxidant enzymes regulation in plants in reference to reactive oxygen species (ROS) and reactive nitrogen species (RNS). Plant Gene 2019, 19, 100182. [Google Scholar] [CrossRef]
- Antar, S.A.; El-Gammal, M.A.; Hazem, R.M.; Moustafa, Y.M. Etanercept Mitigates Cadmium Chloride-induced Testicular Damage in Rats “An Insight into Autophagy, Apoptosis, Oxidative Stress and Inflammation”. Environ. Sci. Pollut. Res. 2022, 29, 28194–28207. [Google Scholar] [CrossRef]
- Aziz, M.A.; Shehab, W.S.; Al-Karmalawy, A.A.; EL-Farargy, A.F.; Abdellattif, M.H. Design, Synthesis, Biological Evaluation, 2D-QSAR Modeling, and Molecular Docking Studies of Novel 1H-3-Indolyl Derivatives as Significant Antioxidants. Int. J. Mol. Sci. 2021, 22, 10396. [Google Scholar] [CrossRef] [PubMed]
- Casas, A.I.; Nogales, C.; Mucke, H.A.; Petraina, A.; Cuadrado, A.; Rojo, A.I.; Ghezzi, P.; Jaquet, V.; Augsburger, F.; Dufrasne, F.; et al. On the clinical pharmacology of reactive oxygen species. Pharmacol. Rev. 2020, 72, 801–828. [Google Scholar] [CrossRef] [PubMed]
- Antar, S.A.; Abdo, W.; Taha, R.S.; Farage, A.E.; El-Moselhy, L.E.; Amer, M.E.; Monsef, A.S.A.; Hamid, A.M.A.; Kamel, E.M.; Ahmeda, A.F. Telmisartan attenuates diabetic nephropathy by mitigating oxidative stress and inflammation, and upregulating Nrf2/HO-1 signaling in diabetic rats. Life Sci. 2022, 291, 120260. [Google Scholar] [CrossRef] [PubMed]
- Richter, K.; Kietzmann, T. Reactive oxygen species and fibrosis: Further evidence of a significant liaison. Cell Tissue Res. 2016, 365, 591–605. [Google Scholar] [CrossRef] [Green Version]
- Kato, K.; Hecker, L. NADPH oxidases: Pathophysiology and therapeutic potential in age-associated pulmonary fibrosis. Redox Biol. 2020, 33, 101541. [Google Scholar] [CrossRef]
- Hecker, L.; Logsdon, N.J.; Kurundkar, D.; Kurundkar, A.; Bernard, K.; Hock, T.; Meldrum, E.; Sanders, Y.Y.; Thannickal, V.J. Reversal of persistent fibrosis in aging by targeting Nox4-Nrf2 redox imbalance. Sci. Transl. Med. 2014, 6, 231ra47. [Google Scholar] [CrossRef] [Green Version]
- Firuzi, O.; Miri, R.; Tavakkoli, M.; Saso, L. Antioxidant therapy: Current status and future prospects. Curr. Med. Chem. 2011, 18, 3871–3888. [Google Scholar] [CrossRef]
- Day, B.J. Antioxidants as potential therapeutics for lung fibrosis. Antioxid. Redox Signal. 2008, 10, 355–370. [Google Scholar] [CrossRef] [Green Version]
- Mansour, D.F.; Saleh, D.O.; Ahmed-Farid, O.A.; Rady, M.; Bakeer, R.M.; Hashad, I.M. Ginkgo biloba extract (EGb 761) mitigates methotrexate-induced testicular insult in rats: Targeting oxidative stress, energy deficit and spermatogenesis. Biomed. Pharmacother. 2021, 143, 112201. [Google Scholar] [CrossRef]
- Sies, H.; Jones, D.P. Reactive oxygen species (ROS) as pleiotropic physiological signalling agents. Nat. Rev. Mol. Cell Biol. 2020, 21, 363–383. [Google Scholar] [CrossRef]
- Surai, P.F.; Kochish, I.I.; Fisinin, V.I.; Kidd, M.T. Antioxidant defence systems and oxidative stress in poultry biology: An update. Antioxidants 2019, 8, 235. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dhawan, V. Reactive oxygen and nitrogen species: General considerations. In Studies on Respiratory Disorders; Springer: Berlin/Heidelberg, Germany, 2014; pp. 27–47. [Google Scholar]
- Della Latta, V.; Cecchettini, A.; Del Ry, S.; Morales, M.A. Bleomycin in the setting of lung fibrosis induction: From biological mechanisms to counteractions. Pharmacol. Res. 2015, 97, 122–130. [Google Scholar] [CrossRef] [PubMed]
- Kandhare, A.D.; Mukherjee, A.; Ghosh, P.; Bodhankar, S.L. Efficacy of antioxidant in idiopathic pulmonary fibrosis: A systematic review and meta-analysis. EXCLI J. 2016, 15, 636. [Google Scholar] [PubMed]
- Cheresh, P.; Kim, S.-J.; Tulasiram, S.; Kamp, D.W. Oxidative stress and pulmonary fibrosis. Biochim. Et Biophys. Acta BBA-Mol. Basis Dis. 2013, 1832, 1028–1040. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Estornut, C.; Milara, J.; Bayarri, M.A.; Belhadj, N.; Cortijo, J. Targeting Oxidative Stress as a Therapeutic Approach for Idiopathic Pulmonary Fibrosis. Front. Pharmacol. 2021, 12, 3873. [Google Scholar] [CrossRef]
- Valdivia, A.; Perez-Alvarez, S.; Aroca-Aguilar, J.; Ikuta, I.; Jordan, J. Superoxide dismutases: A physiopharmacological update. J. Physiol. Biochem. 2009, 65, 195–208. [Google Scholar] [CrossRef]
- Hybertson, B.M.; Gao, B.; Bose, S.K.; McCord, J.M. Oxidative stress in health and disease: The therapeutic potential of Nrf2 activation. Mol. Asp. Med. 2011, 32, 234–246. [Google Scholar] [CrossRef]
- Ciarcià, G.; Bianchi, S.; Tomasello, B.; Acquaviva, R.; Malfa, G.A.; Naletova, I.; La Mantia, A.; Di Giacomo, C. Vitamin E and Non-Communicable Diseases: A Review. Biomedicines 2022, 10, 2473. [Google Scholar] [CrossRef]
- Xia, X.; Zhang, X.; Liu, M.; Duan, M.; Zhang, S.; Wei, X.; Liu, X. Toward improved human health: Efficacy of dietary selenium on immunity at the cellular level. Food Funct. 2021, 12, 976–989. [Google Scholar] [CrossRef]
- Zhitkovich, A. Nuclear and cytoplasmic functions of vitamin C. Chem. Res. Toxicol. 2020, 33, 2515–2526. [Google Scholar] [CrossRef]
- Bratovcic, A. Antioxidant enzymes and their role in preventing cell damage. Acta Sci. Nutr. Health 2020, 4, 01–07. [Google Scholar] [CrossRef]
- Gao, J.; Peng, S.; Shan, X.; Deng, G.; Shen, L.; Sun, J.; Jiang, C.; Yang, X.; Chang, Z.; Sun, X.; et al. Inhibition of AIM2 inflammasome-mediated pyroptosis by Andrographolide contributes to amelioration of radiation-induced lung inflammation and fibrosis. Cell Death Dis. 2019, 10, 957. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ibrahim, M. Design, Synthesis, Molecular Docking and Biological Evaluation of Some Novel Quinazolin-4 (3h)-One Derivatives As Anti-Inflammatory Agents. Al-Azhar J. Pharm. Sci. 2012, 46, 185–203. [Google Scholar] [CrossRef]
- Toda, N.; Mukoyama, M.; Yanagita, M.; Yokoi, H. CTGF in kidney fibrosis and glomerulonephritis. Inflamm. Regen. 2018, 38, 14. [Google Scholar] [CrossRef]
- Dellepiane, S.; Leventhal, J.S.; Cravedi, P. T cells and acute kidney injury: A two-way relationship. Front. Immunol. 2020, 11, 1546. [Google Scholar] [CrossRef]
- Zhang, Z.-q.; Tian, H.-t.; Liu, H.; Xie, R. The role of macrophage-derived TGF-β1 on SiO2-induced pulmonary fibrosis: A review. Toxicol. Ind. Health 2021, 37, 240–250. [Google Scholar] [CrossRef]
- Binatti, E.; Gerussi, A.; Barisani, D.; Invernizzi, P. The Role of Macrophages in Liver Fibrosis: New Therapeutic Opportunities. Int. J. Mol. Sci. 2022, 23, 6649. [Google Scholar] [CrossRef]
- Liu, J.-R.; Miao, H.; Deng, D.-Q.; Vaziri, N.D.; Li, P.; Zhao, Y.-Y. Gut microbiota-derived tryptophan metabolism mediates renal fibrosis by aryl hydrocarbon receptor signaling activation. Cell. Mol. Life Sci. 2021, 78, 909–922. [Google Scholar] [CrossRef]
- ElMahdy, M.K.; Antar, S.A.; Elmahallawy, E.K.; Abdo, W.; Hijazy, H.H.A.; Albrakati, A.; Khodir, A.E. A Novel Role of Dapagliflozin in Mitigation of Acetic Acid-Induced Ulcerative Colitis by Modulation of Monocyte Chemoattractant Protein 1 (MCP-1)/Nuclear Factor-Kappa B (NF-κB)/Interleukin-18 (IL-18). Biomedicines 2021, 10, 40. [Google Scholar] [CrossRef]
- Chen, T.; Zhang, X.; Zhu, G.; Liu, H.; Chen, J.; Wang, Y.; He, X. Quercetin inhibits TNF-α induced HUVECs apoptosis and inflammation via downregulating NF-kB and AP-1 signaling pathway in vitro. Medicine 2020, 99, e22241. [Google Scholar] [CrossRef]
- Wang, J.; Lin, S.; Brown, J.M.; van Wagoner, D.; Fiocchi, C.; Rieder, F. Novel mechanisms and clinical trial endpoints in intestinal fibrosis. Immunol. Rev. 2021, 302, 211–227. [Google Scholar] [CrossRef] [PubMed]
- Sethi, J.K.; Hotamisligil, G.S. Metabolic Messengers: Tumour necrosis factor. Nat. Metab. 2021, 3, 1302–1312. [Google Scholar] [CrossRef] [PubMed]
- Waszczykowska, A.; Podgórski, M.; Waszczykowski, M.; Gerlicz-Kowalczuk, Z.; Jurowski, P. Matrix metalloproteinases MMP-2 and MMP-9, their inhibitors TIMP-1 and TIMP-2, vascular endothelial growth factor and sVEGFR-2 as predictive markers of ischemic retinopathy in patients with systemic sclerosis—Case series report. Int. J. Mol. Sci. 2020, 21, 8703. [Google Scholar] [CrossRef] [PubMed]
- Capece, D.; Verzella, D.; Flati, I.; Arboretto, P.; Cornice, J.; Franzoso, G. NF-κB: Blending metabolism, immunity, and inflammation. Trends Immunol. 2022, 43, 757–775. [Google Scholar] [CrossRef] [PubMed]
- Domino, M.; Jasinski, T.; Kautz, E.; Juszczuk-Kubiak, E.; Ferreira-Dias, G.; Zabielski, R.; Sady, M.; Gajewski, Z. Expression of genes involved in the NF-κB-dependent pathway of the fibrosis in the mare endometrium. Theriogenology 2020, 147, 18–24. [Google Scholar] [CrossRef] [PubMed]
- Zou, M.; Zou, J.; Hu, X.; Zheng, W.; Zhang, M.; Cheng, Z. Latent Transforming Growth Factor-β Binding Protein-2 Regulates Lung Fibroblast-to-Myofibroblast Differentiation in Pulmonary Fibrosis via NF-κB Signaling. Front. Pharmacol. 2021, 12, 788714. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Ma, D.; Liu, Y.; Liu, L.; Chen, Y.; Liu, H.; Zhang, L.; Lu, J.; Chen, K.; You, J.; et al. Extracts of Periplaneta americana alleviate hepatic fibrosis by affecting hepatic TGF-β and NF-κB expression in rats with pig serum-induced liver fibrosis. Folia Histochem. Et Cytobiol. 2022, 60, 125–135. [Google Scholar] [CrossRef]
- Dong, J.; Ma, Q. In vivo activation and pro-fibrotic function of NF-κB in fibroblastic cells during pulmonary inflammation and fibrosis induced by carbon nanotubes. Front. Pharmacol. 2019, 10, 1140. [Google Scholar] [CrossRef]
- Antar, S.; Al-Karmalawy, A.A.; Mourad, A.; Mourad, M.; Elbadry, M.; Saber, S.; Khodir, A. Protective effects of mirazid on gentamicin-induced nephrotoxicity in rats through antioxidant, anti-inflammatory, JNK1/iNOS, and apoptotic pathways; novel mechanistic insights. Pharm. Sci. 2022, 28, 525–540. [Google Scholar] [CrossRef]
- Murata, H.; Khine, C.C.; Nishikawa, A.; Yamamoto, K.-i.; Kinoshita, R.; Sakaguchi, M. c-Jun N-terminal kinase (JNK)-mediated phosphorylation of SARM1 regulates NAD+ cleavage activity to inhibit mitochondrial respiration. J. Biol. Chem. 2018, 293, 18933–18943. [Google Scholar] [CrossRef]
- Jiang, M.; Fan, J.; Qu, X.; Li, S.; Nilsson, S.K.; Sun, Y.B.Y.; Chen, Y.; Yu, D.; Liu, D.; Liu, B.-C.; et al. Combined blockade of Smad3 and JNK pathways ameliorates progressive fibrosis in folic acid nephropathy. Front. Pharmacol. 2019, 10, 880. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nikolic-Paterson, D.J.; Grynberg, K.; Ma, F.Y. JUN amino terminal kinase in cell death and inflammation in acute and chronic kidney disease. Integr. Med. Nephrol. Androl. 2021, 8, 10. [Google Scholar] [CrossRef]
- Grynberg, K.; Ma, F.Y.; Nikolic-Paterson, D.J. The JNK signaling pathway in renal fibrosis. Front. Physiol. 2017, 8, 829. [Google Scholar] [CrossRef] [PubMed]
- Geng, C.; Wei, J.; Wu, C. Mammalian STE20-like kinase 1 knockdown attenuates TNFα-mediated neurodegenerative disease by repressing the JNK pathway and mitochondrial stress. Neurochem. Res. 2019, 44, 1653–1664. [Google Scholar] [CrossRef]
- Mu, M.; Zuo, S.; Wu, R.-M.; Deng, K.-S.; Lu, S.; Zhu, J.-J.; Zou, G.-L.; Yang, J.; Cheng, M.-L.; Zhao, X.-K. Ferulic acid attenuates liver fibrosis and hepatic stellate cell activation via inhibition of TGF-β/Smad signaling pathway. Drug Des. Dev. Ther. 2018, 12, 4107. [Google Scholar] [CrossRef] [Green Version]
- Gai, L.; Zhu, Y.; Zhang, C.; Meng, X. Targeting canonical and non-canonical STAT signaling pathways in renal diseases. Cells 2021, 10, 1610. [Google Scholar] [CrossRef]
- Alunno, A.; Padjen, I.; Fanouriakis, A.; Boumpas, D.T. Pathogenic and therapeutic relevance of JAK/STAT signaling in systemic lupus erythematosus: Integration of distinct inflammatory pathways and the prospect of their inhibition with an oral agent. Cells 2019, 8, 898. [Google Scholar] [CrossRef] [Green Version]
- Tieyuan, Z.; Ying, Z.; Xinghua, Z.; Huimin, W.; Huagang, L. Piceatannol-mediated JAK2/STAT3 signaling pathway inhibition contributes to the alleviation of oxidative injury and collagen synthesis during pulmonary fibrosis. Int. Immunopharmacol. 2022, 111, 109107. [Google Scholar] [CrossRef]
- Dees, C.; Pötter, S.; Zhang, Y.; Bergmann, C.; Zhou, X.; Luber, M.; Wohlfahrt, T.; Karouzakis, E.; Ramming, A.; Gelse, K.; et al. TGF-β–induced epigenetic deregulation of SOCS3 facilitates STAT3 signaling to promote fibrosis. J. Clin. Investig. 2020, 130, 2347–2363. [Google Scholar] [CrossRef] [Green Version]
- Frenquelli, M.; Tonon, G. WNT signaling in hematological malignancies. Front. Oncol. 2020, 10, 615190. [Google Scholar] [CrossRef]
- Danek, P.; Kardosova, M.; Janeckova, L.; Karkoulia, E.; Vanickova, K.; Fabisik, M.; Lozano-Asencio, C.; Benoukraf, T.; Tirado-Magallanes, R.; Zhou, Q. β-Catenin–TCF/LEF signaling promotes steady-state and emergency granulopoiesis via G-CSF receptor upregulation. Blood 2020, 136, 2574–2587. [Google Scholar] [CrossRef] [PubMed]
- Griffin, M.F.; Huber, J.; Evan, F.J.; Quarto, N.; Longaker, M.T. The role of Wnt signaling in skin fibrosis. Med. Res. Rev. 2022, 42, 615–628. [Google Scholar] [CrossRef] [PubMed]
- Hu, H.-H.; Cao, G.; Wu, X.-Q.; Vaziri, N.D.; Zhao, Y.-Y. Wnt signaling pathway in aging-related tissue fibrosis and therapies. Ageing Res. Rev. 2020, 60, 101063. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Shen, L.; Dreißigacker, K.; Zhu, H.; Trinh-Minh, T.; Meng, X.; Tran-Manh, C.; Dees, C.; Matei, A.-E.; Chen, C.-W. Targeting of canonical WNT signaling ameliorates experimental sclerodermatous chronic graft-versus-host disease. Blood 2021, 137, 2403–2416. [Google Scholar] [CrossRef]
- Duspara, K.; Bojanic, K.; Pejic, J.I.; Kuna, L.; Kolaric, T.O.; Nincevic, V.; Smolic, R.; Vcev, A.; Glasnovic, M.; Curcic, I.B.; et al. Targeting the Wnt Signaling Pathway in Liver Fibrosis for Drug Options: An Update. J. Clin. Transl. Hepatol. 2021, 9, 960. [Google Scholar] [CrossRef]
- Lara-Reyna, S.; Holbrook, J.; Jarosz-Griffiths, H.H.; Peckham, D.; McDermott, M.F. Dysregulated signalling pathways in innate immune cells with cystic fibrosis mutations. Cell. Mol. Life Sci. 2020, 77, 4485–4503. [Google Scholar] [CrossRef]
- Laurent, P.; Sisirak, V.; Lazaro, E.; Richez, C.; Duffau, P.; Blanco, P.; Truchetet, M.-E.; Contin-Bordes, C. Innate immunity in systemic sclerosis fibrosis: Recent advances. Front. Immunol. 2018, 9, 1702. [Google Scholar] [CrossRef] [Green Version]
- Liaskou, E.; Patel, S.R.; Webb, G.; Dimakou, D.B.; Akiror, S.; Krishna, M.; Mells, G.; Jones, D.E.; Bowman, S.J.; Barone, F. Increased sensitivity of Treg cells from patients with PBC to low dose IL-12 drives their differentiation into IFN-γ secreting cells. J. Autoimmun. 2018, 94, 143–155. [Google Scholar] [CrossRef] [Green Version]
- Huwait, E.A.; Al-Ghamdi, M.A.; Ghattas, M.H.; Hinnis, A.R.; El-Maaty, D.A.A.; Abo-Elmatty, D.M.; Abdel-Hamed, A.R. Role of heme oxygenase-1, cytokines, and vascular endothelial growth factor in murine Schistosoma mansoni. Int. J. Health Sci. 2021, 15, 22. [Google Scholar]
- Zhang, M.; Zhang, S. T cells in fibrosis and fibrotic diseases. Front. Immunol. 2020, 11, 1142. [Google Scholar] [CrossRef]
- Patra, M.C.; Shah, M.; Choi, S. Toll-like receptor-induced cytokines as immunotherapeutic targets in cancers and autoimmune diseases. In Seminars in Cancer Biology; Academic Press: Cambridge, MA, USA, 2020; pp. 61–82. [Google Scholar]
- Deng, K.M.; Yang, X.S.; Luo, Q.; She, Y.X.; Yu, Q.Y.; Tang, X.X. Deleterious Role of Th9 Cells in Pulmonary Fibrosis. Cells 2021, 10, 3209. [Google Scholar] [CrossRef] [PubMed]
- Guo, X.; Cen, Y.; Wang, J.; Jiang, H. CXCL10-induced IL-9 promotes liver fibrosis via Raf/MEK/ERK signaling pathway. Biomed. Pharmacother. 2018, 105, 282–289. [Google Scholar] [CrossRef] [PubMed]
- Gao, M.; Wang, J.; Zang, J.; An, Y.; Dong, Y. The Mechanism of CD8+ T Cells for Reducing Myofibroblasts Accumulation during Renal Fibrosis. Biomolecules 2021, 11, 990. [Google Scholar] [CrossRef] [PubMed]
- Diao, W.; Chen, W.; Cao, W.; Yuan, H.; Ji, H.; Wang, T.; Zhu, X.; Zhou, H.; Guo, H.; Zhao, X. Astaxanthin protects against renal fibrosis through inhibiting myofibroblast activation and promoting CD8+ T cell recruitment. Biochim. Biophys. Acta Gen. Subj. 2019, 1863, 1360–1370. [Google Scholar] [CrossRef] [PubMed]
- Ma, C.S.; Suryani, S.; Avery, D.T.; Chan, A.; Nanan, R.; Santner-Nanan, B.; Deenick, E.K.; Tangye, S.G. Early commitment of naïve human CD4+ T cells to the T follicular helper (TFH) cell lineage is induced by IL-12. Immunol. Cell Biol. 2009, 87, 590–600. [Google Scholar] [CrossRef] [PubMed]
- Zheng, B.; Zhang, J.; Chen, H.; Nie, H.; Miller, H.; Gong, Q.; Liu, C. T lymphocyte-mediated liver immunopathology of schistosomiasis. Front. Immunol. 2020, 11, 61. [Google Scholar] [CrossRef] [Green Version]
- Taylor, D.K.; Mittereder, N.; Kuta, E.; Delaney, T.; Burwell, T.; Dacosta, K.; Zhao, W.; Cheng, L.I.; Brown, C.; Boutrin, A. T follicular helper–like cells contribute to skin fibrosis. Sci. Transl. Med. 2018, 10, eaaf5307. [Google Scholar] [CrossRef] [Green Version]
- Long, Y.; Zhao, X.; Liu, C.; Xia, C.; Liu, C. Activated inducible co-stimulator-positive programmed cell death 1-positive follicular helper T cells indicate disease activity and severity in ulcerative colitis patients. Clin. Exp. Immunol. 2020, 202, 106–118. [Google Scholar] [CrossRef]
- Frangogiannis, N.G. Cardiac fibrosis. Cardiovasc. Res. 2021, 117, 1450–1488. [Google Scholar] [CrossRef]
- Tanno, B.; Novelli, F.; Leonardi, S.; Merla, C.; Babini, G.; Giardullo, P.; Kadhim, M.; Traynor, D.; Medipally, D.K.; Meade, A.D. MiRNA-Mediated Fibrosis in the Out-of-Target Heart following Partial-Body Irradiation. Cancers 2022, 14, 3463. [Google Scholar] [CrossRef]
- Triposkiadis, F.; Xanthopoulos, A.; Butler, J. Cardiovascular aging and heart failure: JACC review topic of the week. J. Am. Coll. Cardiol. 2019, 74, 804–813. [Google Scholar] [CrossRef] [PubMed]
- Tuleta, I.; Frangogiannis, N.G. Fibrosis of the diabetic heart: Clinical significance, molecular mechanisms, and therapeutic opportunities. Adv. Drug Deliv. Rev. 2021, 176, 113904. [Google Scholar] [CrossRef] [PubMed]
- Qin, W.; Cao, L.; Massey, I.Y. Role of PI3K/Akt signaling pathway in cardiac fibrosis. Mol. Cell. Biochem. 2021, 476, 4045–4059. [Google Scholar] [CrossRef]
- Yang, L. How acute kidney injury contributes to renal fibrosis. Ren. Fibros. Mech. Ther. 2019, 1165, 117–142. [Google Scholar]
- Menshikh, A.; Scarfe, L.; Delgado, R.; Finney, C.; Zhu, Y.; Yang, H.; de Caestecker, M.P. Capillary rarefaction is more closely associated with CKD progression after cisplatin, rhabdomyolysis, and ischemia-reperfusion-induced AKI than renal fibrosis. Am. J. Physiol. Ren. Physiol. 2019, 317, F1383–F1397. [Google Scholar] [CrossRef] [PubMed]
- Zhao, M.; Wang, L.; Wang, M.; Zhou, S.; Lu, Y.; Cui, H.; Racanelli, A.C.; Zhang, L.; Ye, T.; Ding, B.; et al. Targeting fibrosis: Mechanisms and clinical trials. Signal Transduct. Target. Ther. 2022, 7, 206. [Google Scholar] [CrossRef] [PubMed]
- Boyer-Diaz, Z.; Aristu-Zabalza, P.; Andrés-Rozas, M.; Robert, C.; Ortega-Ribera, M.; Fernández-Iglesias, A.; Broqua, P.; Junien, J.-L.; Wettstein, G.; Bosch, J. Pan-PPAR agonist lanifibranor improves portal hypertension and hepatic fibrosis in experimental advanced chronic liver disease. J. Hepatol. 2021, 74, 1188–1199. [Google Scholar] [CrossRef]
- Lee, C.; Kim, M.; Han, J.; Yoon, M.; Jung, Y. Mesenchymal stem cells influence activation of hepatic stellate cells, and constitute a promising therapy for liver fibrosis. Biomedicines 2021, 9, 1598. [Google Scholar] [CrossRef]
- Da Silva Meirelles, L.; Marson, R.F.; Solari, M.I.G.; Nardi, N.B. Are liver pericytes just precursors of myofibroblasts in hepatic diseases? Insights from the crosstalk between perivascular and inflammatory cells in liver injury and repair. Cells 2020, 9, 188. [Google Scholar] [CrossRef] [Green Version]
- Blokland, K.E.; Pouwels, S.D.; Schuliga, M.; Knight, D.A.; Burgess, J.K. Regulation of cellular senescence by extracellular matrix during chronic fibrotic diseases. Clin. Sci. 2020, 134, 2681–2706. [Google Scholar] [CrossRef]
- Samarelli, A.V.; Tonelli, R.; Marchioni, A.; Bruzzi, G.; Gozzi, F.; Andrisani, D.; Castaniere, I.; Manicardi, L.; Moretti, A.; Tabbì, L.; et al. Fibrotic idiopathic interstitial lung disease: The molecular and cellular key players. Int. J. Mol. Sci. 2021, 22, 8952. [Google Scholar] [CrossRef] [PubMed]
- Phan, T.H.G.; Paliogiannis, P.; Nasrallah, G.K.; Giordo, R.; Eid, A.H.; Fois, A.G.; Zinellu, A.; Mangoni, A.A.; Pintus, G. Emerging cellular and molecular determinants of idiopathic pulmonary fibrosis. Cell. Mol. Life Sci. 2021, 78, 2031–2057. [Google Scholar] [CrossRef] [PubMed]
- Mumby, S.; Chung, K.F.; Adcock, I.M. Transcriptional effects of ozone and impact on airway inflammation. Front. Immunol. 2019, 10, 1610. [Google Scholar] [CrossRef]
- Hosseinzadeh, A.; Javad-Moosavi, S.A.; Reiter, R.J.; Yarahmadi, R.; Ghaznavi, H.; Mehrzadi, S. Oxidative/nitrosative stress, autophagy and apoptosis as therapeutic targets of melatonin in idiopathic pulmonary fibrosis. Expert Opin. Ther. Targets 2018, 22, 1049–1061. [Google Scholar] [CrossRef] [PubMed]
- Meurer, S.K.; Karsdal, M.A.; Weiskirchen, R. Advances in the clinical use of collagen as biomarker of liver fibrosis. Expert Rev. Mol. Diagn. 2020, 20, 947–969. [Google Scholar] [CrossRef] [PubMed]
- Mahdy, M.A. Skeletal muscle fibrosis: An overview. Cell Tissue Res. 2019, 375, 575–588. [Google Scholar] [CrossRef] [PubMed]
- Forcina, L.; Miano, C.; Scicchitano, B.M.; Musarò, A. Signals from the niche: Insights into the role of IGF-1 and IL-6 in modulating skeletal muscle fibrosis. Cells 2019, 8, 232. [Google Scholar] [CrossRef] [Green Version]
- Palka, J.; Oscilowska, I.; Szoka, L. Collagen metabolism as a regulator of proline dehydrogenase/proline oxidase-dependent apoptosis/autophagy. Amino Acids 2021, 53, 1917–1925. [Google Scholar] [CrossRef]
- Busilacchi, E.M.; Costantini, A.; Mancini, G.; Tossetta, G.; Olivieri, J.; Poloni, A.; Viola, N.; Butini, L.; Campanati, A.; Goteri, G.; et al. Nilotinib Treatment of Patients Affected by Chronic Graft-versus-Host Disease Reduces Collagen Production and Skin Fibrosis by Downmodulating the TGF-β and p-SMAD Pathway. Biol. Blood Marrow Transplant. 2020, 26, 823–834. [Google Scholar] [CrossRef]
- Chang, C.-J.; Lin, C.-F.; Lee, C.-H.; Chuang, H.-C.; Shih, F.-C.; Wan, S.-W.; Tai, C.; Chen, C.-L. Overcoming interferon (IFN)-γ resistance ameliorates transforming growth factor (TGF)-β-mediated lung fibroblast-to-myofibroblast transition and bleomycin-induced pulmonary fibrosis. Biochem. Pharmacol. 2021, 183, 114356. [Google Scholar] [CrossRef]
- Lee, J.; Ko, J.; Kim, H.-J.; Im, H.; Kim, E.J.; Yi, J.Y. Imatinib mesylate elicits extracellular signal-related kinase (ERK) activation and enhances the survival of γ-irradiated epithelial cells. Biochem. Biophys. Res. Commun. 2018, 506, 939–943. [Google Scholar] [CrossRef] [PubMed]
- Kunimi, H.; Miwa, Y.; Inoue, H.; Tsubota, K.; Kurihara, T. A novel HIF inhibitor halofuginone prevents neurodegeneration in a murine model of retinal ischemia-reperfusion. Int. J. Mol. Sci. 2019, 20, 3171. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Valkovic, A.L.; Bathgate, R.A.; Samuel, C.S.; Kocan, M. Understanding relaxin signalling at the cellular level. Mol. Cell. Endocrinol. 2019, 487, 24–33. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Z.; Bian, M.; Wu, J.; Li, D.; Ding, L.; Zeng, Q. J Oltipraz prevents high glucose-induced oxidative stress and apoptosis in RSC96 cells through the Nrf2/NQO1 signalling pathway. BioMed Res. Int. 2020, 2020, 5939815. [Google Scholar] [CrossRef] [PubMed]
















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Role of Enzymatic Antioxidants | |
| Catalase | An H2O2 scavenger that is expressed in lung AEC and inflammatory cells can stop fibroblasts in IPF lung tissue from becoming activated by H2O2 [67]. |
| Glutathione (GSH) | One of the best small-molecule antioxidants and one of the most tested indicators is GSH. In mouse models of fibrosis, N-acetyl cysteine (NAC), a precursor to GSH, has been shown to have anti-fibrotic properties. NAC raises lung GSH levels and reduces bleomycin-induced fibrosis [68]. |
| Superoxide dismutase (SOD) | SOD converts superoxide radicals into H2O2. The distribution and expression of the three mammalian SOD isoforms—intracellular copper-zinc SOD, mitochondrial manganese SOD, and extracellular SOD [EC-SOD]—vary depending on the type of cell [69]. |
| Nuclear factor-erythroid 2-related factor 2 (Nrf2) | The “master regulator” of the antioxidant response is a transcription factor called Nrf2. The antioxidant response element controls hundreds of genes, including NAD(P)H quinone oxidoreductase 1, antioxidant-related genes involved in glutathione biosynthesis, Phase II detoxifying “stress response” genes, and genes regulating the inflammatory and fibrotic responses [70]. |
| Role of Non-Enzymatic Antioxidants | |
| Vitamin-E | As an antioxidant, its primary function is to scavenge loose electrons, or “free radicals”, which can harm cells. Additionally, it strengthens the immune system and prevents heart artery clots from forming [71]. |
| Selenium | This antioxidant assists the body in reducing oxidative stress, which lowers inflammation and improves immunity. Increased blood levels of selenium have been linked to improved immunological response [72]. |
| Vitamin C | It is a hydrophilic free radical scavenger and acts as a reducing and antioxidant agent. Vitamin C and vitamin E interact together synergistically to restore the antioxidant capabilities of oxidized vitamin E, which is necessary for the formation of collagen, carnitine, and neurotransmitters. The antioxidant and prooxidant reserves of ascorbic acid were reported previously [73]. |
| Vitamin A | Because of their ability to scavenge free radicals, carotenoids function as antioxidants. Dietary antioxidants reduce the effectiveness and negative effects of chemotherapy by squelching free radicals and other reactive oxygen species, primarily singlet oxygen species [74]. |
| Drug | Nature | Mechanism of Action |
|---|---|---|
| Nintedanib | Tyrosine kinase inhibitor | It is an efficient and well-tolerated tyrosine kinase inhibitor (TKI) that showed an important anti-fibrotic effect in patients with chronic graft-versus-host disease (GVHD). It is also used to treat idiopathic pulmonary fibrosis [143]. Focusing on upstream receptors necessary for the growth of fibrosis inhibits the proliferation, migration, and transformation of fibroblasts. It blocks the fibroblast growth factor receptor, vascular endothelial growth factor receptor, and platelet-derived growth factor receptor binding sites [144]. |
| Pirfenidone | Orally active (modified phenyl pyridine) | It is used to treat idiopathic pulmonary fibrosis and inhibits the production and activity of TGF-β. It can diminish fibroblast proliferation and inhibit collagen formation as well as the transcription of the TGF-1 gene and the expression of collagen type 1 mRNA [16]. |
| Imatinib mesylate | Tyrosine kinase inhibitor | It prevents the progression of fibrosis in systemic sclerosis patients and is used in the treatment of established fibrosis. It binds to the Abelson kinase (c-AblATP-binding) site and effectively inhibits its tyrosine kinase activity, which necessitates the conversion of ATP into ADP and the phosphorylation of target proteins. An essential TGF- and PDGF downstream signaling molecule is c-Abl [145]. |
| Halofuginone | Plant alkaloid (from Dichroa febrifuga) | It is used on patients with cutaneous cGvHD, a condition marked by significant skin fibrosis and contractures. It leads to skin integrity decline and a dose-dependent reduction in the skin’s collagen content [146]. |
| Relaxin | A polypeptide of the insulin/relaxin superfamily | It has antifibrotic effects in experimental models of renal fibrosis. It reduces collagen synthesis and encourages collagen breakdown by raising MMP levels and activity. A lot of relaxin’s effects are antagonistic to TGF’s effects. Relaxin’s receptor (RXFP1) was only recently discovered, which accelerated the development of novel antifibrotic relaxin discoveries. The effects of relaxin as an antifibrotic drug in cardiac, liver, kidney, lung and even cutaneous fibrosis are demonstrated by clinical investigations [147]. |
| Oltipraz | Cancer chemo-preventive agent | It is used for treatment in patients with liver fibrosis and cirrhosis. The inhibition of matrix synthesis and the number of target cytokines engaged in this process is increasing. Since TGF-β1 is the most powerful inducer of collagen I and other matrix components, blocking its effects continues to be a primary goal of liver antifibrotic initiatives [148]. |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Antar, S.A.; Ashour, N.A.; Marawan, M.E.; Al-Karmalawy, A.A. Fibrosis: Types, Effects, Markers, Mechanisms for Disease Progression, and Its Relation with Oxidative Stress, Immunity, and Inflammation. Int. J. Mol. Sci. 2023, 24, 4004. https://doi.org/10.3390/ijms24044004
Antar SA, Ashour NA, Marawan ME, Al-Karmalawy AA. Fibrosis: Types, Effects, Markers, Mechanisms for Disease Progression, and Its Relation with Oxidative Stress, Immunity, and Inflammation. International Journal of Molecular Sciences. 2023; 24(4):4004. https://doi.org/10.3390/ijms24044004
Chicago/Turabian StyleAntar, Samar A., Nada A. Ashour, Mohamed E. Marawan, and Ahmed A. Al-Karmalawy. 2023. "Fibrosis: Types, Effects, Markers, Mechanisms for Disease Progression, and Its Relation with Oxidative Stress, Immunity, and Inflammation" International Journal of Molecular Sciences 24, no. 4: 4004. https://doi.org/10.3390/ijms24044004