
Transcriptome and DNA Methylome Analysis of Two Contrasting Rice Genotypes under Salt Stress during Germination

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
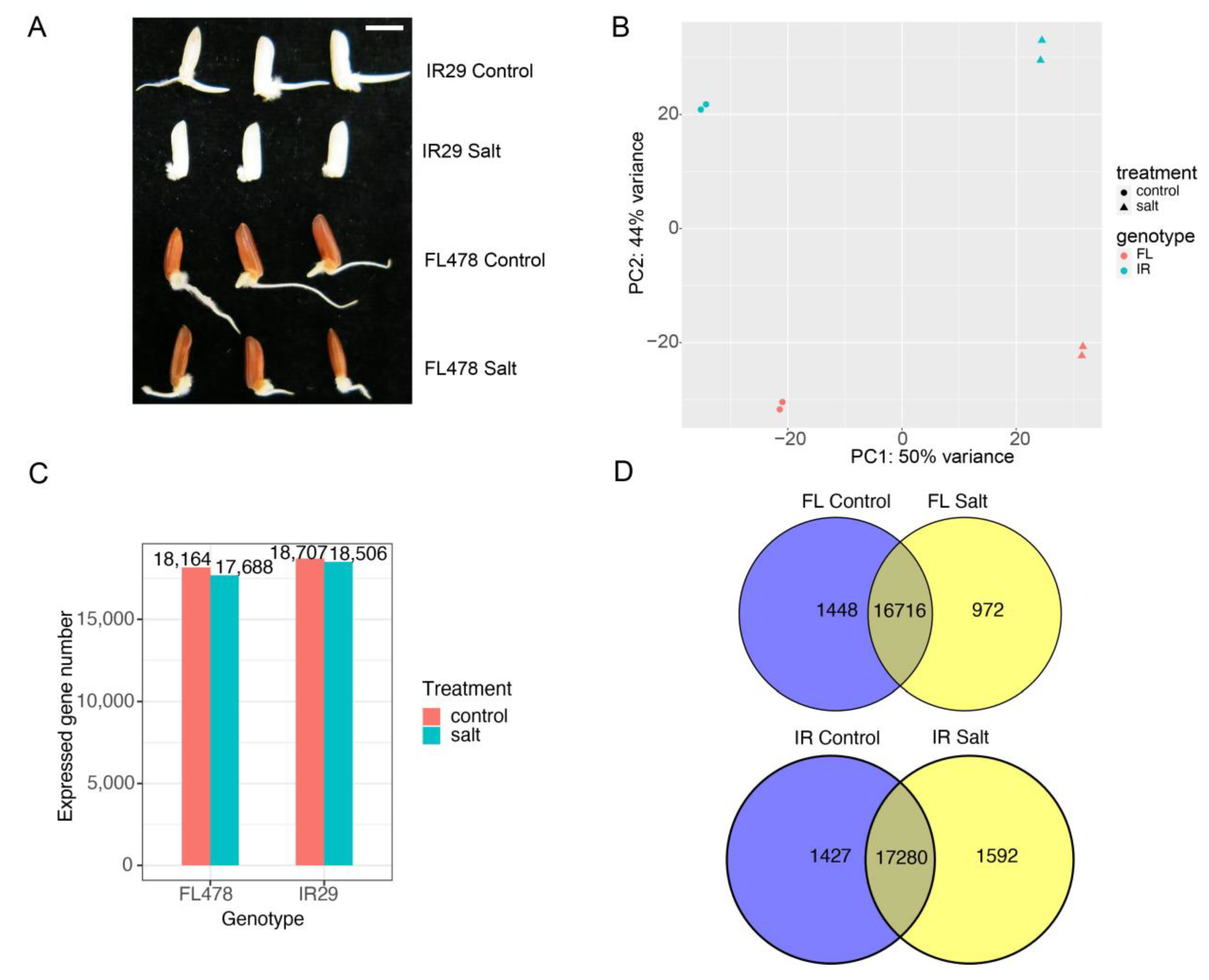
2.1. The Effects of Salinity Stress on Seed Germination in Two Contrasting Genotypes
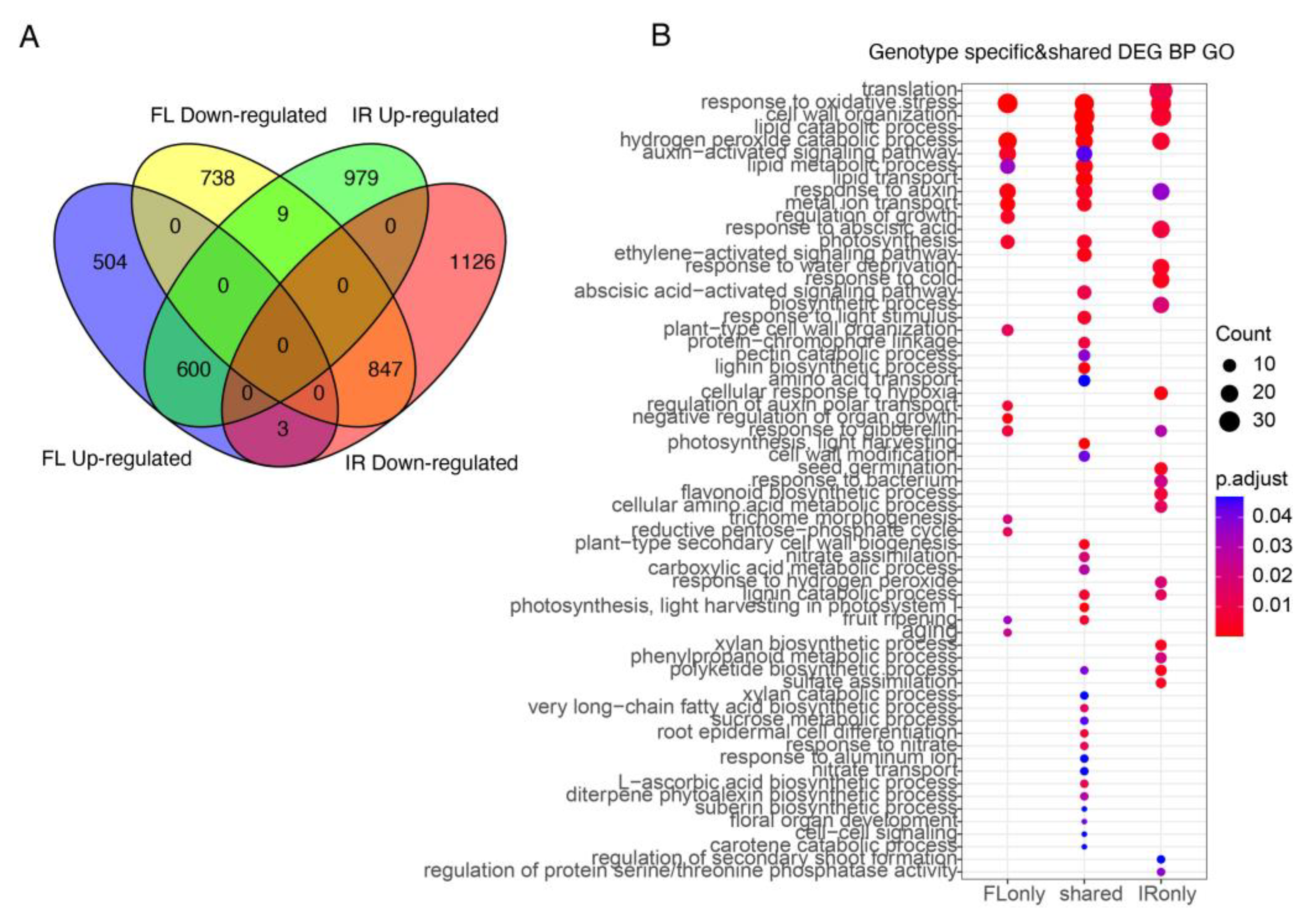
2.2. Differentially Expressed Genes and GO Enrichment Analysis
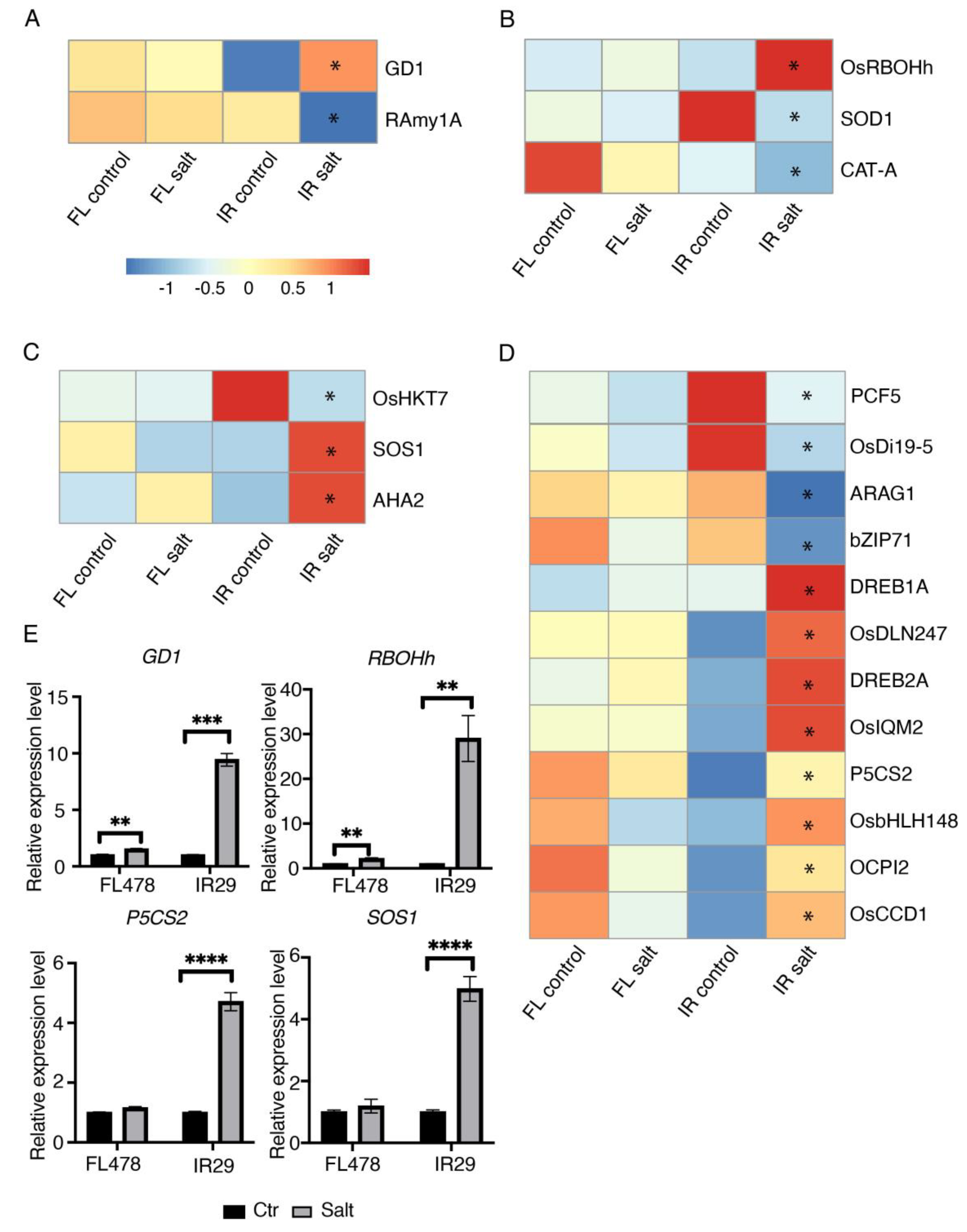
2.3. Salt Stress Induced GD1 Expression in Salt Sensitive IR29
2.4. The Expression Patterns of Important Salt Responsive Genes
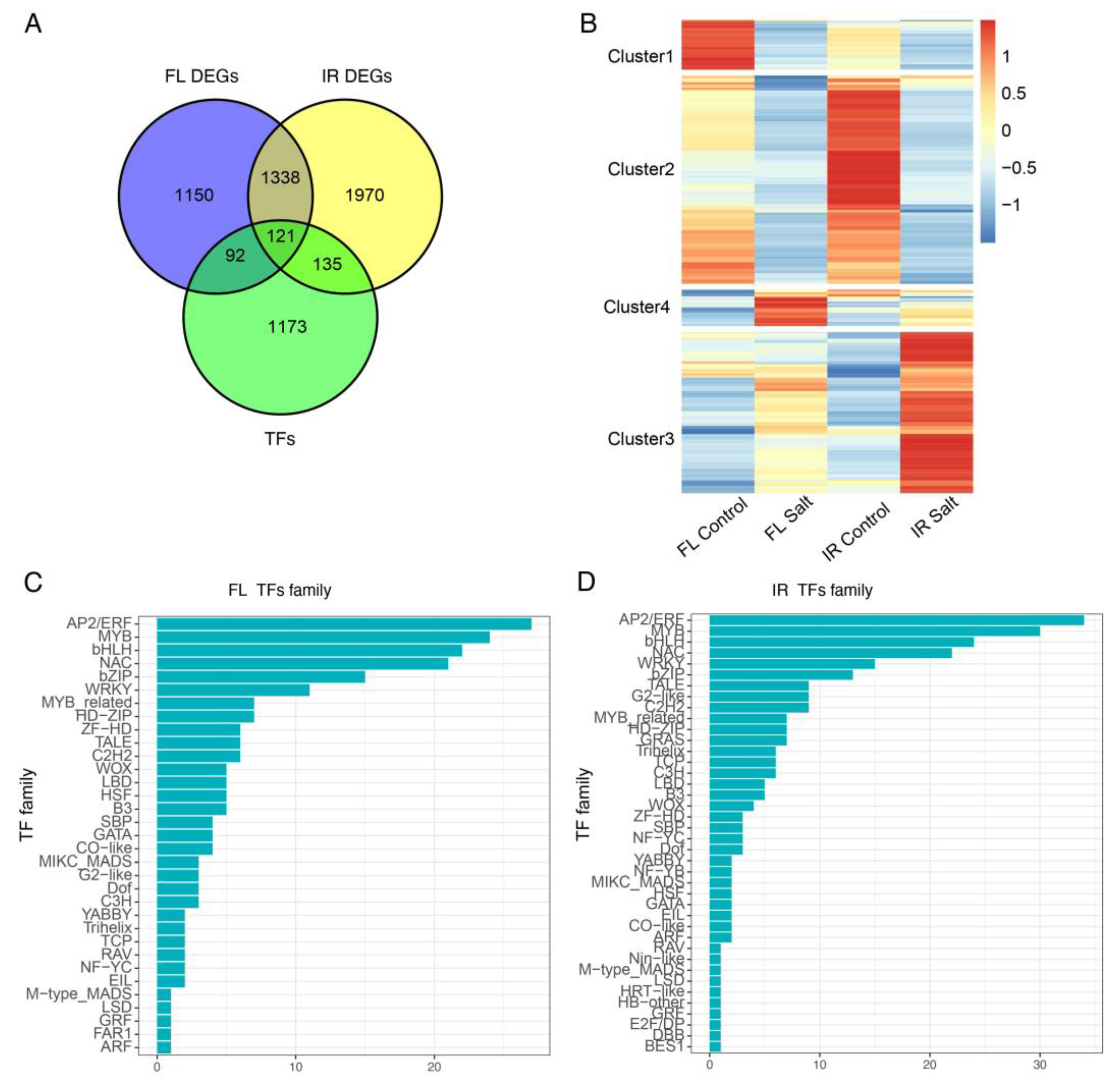
2.5. Salt-Responsive Transcription Factors during Germination
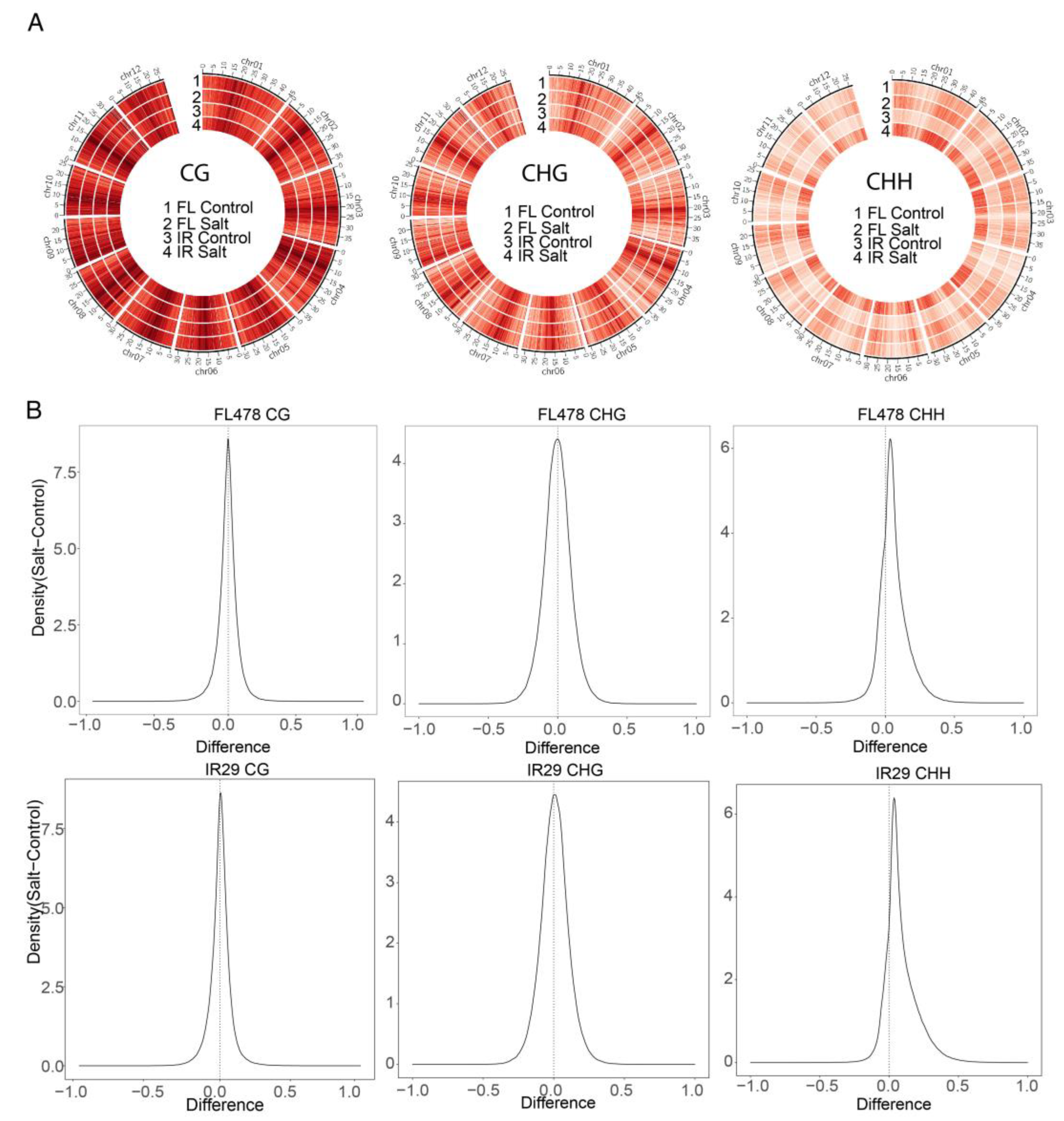
2.6. Global CHH DNA Methylation Increase under Salt Stress
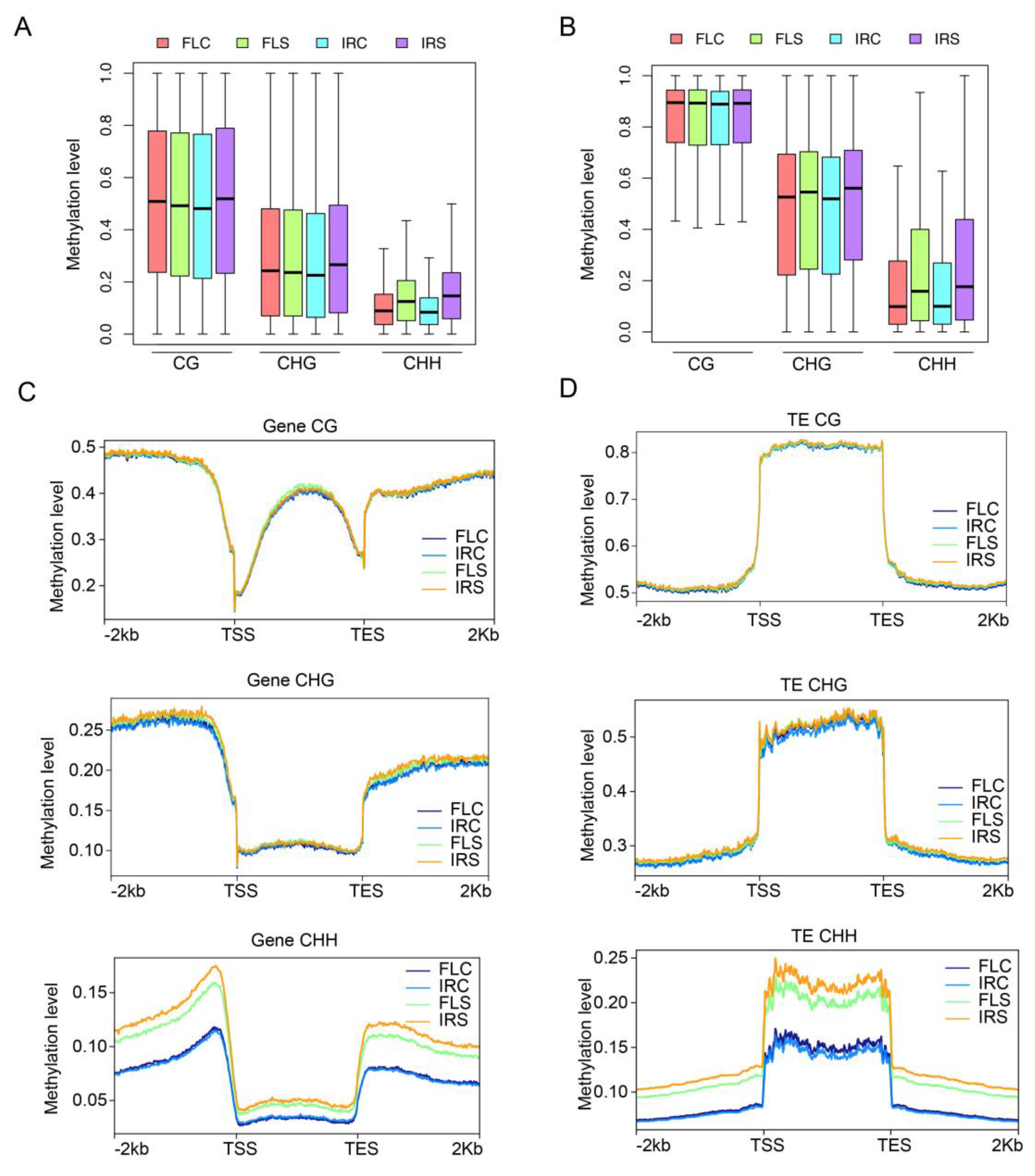
2.7. High CHH Methylation Level in Gene and TE under Salinity Stress
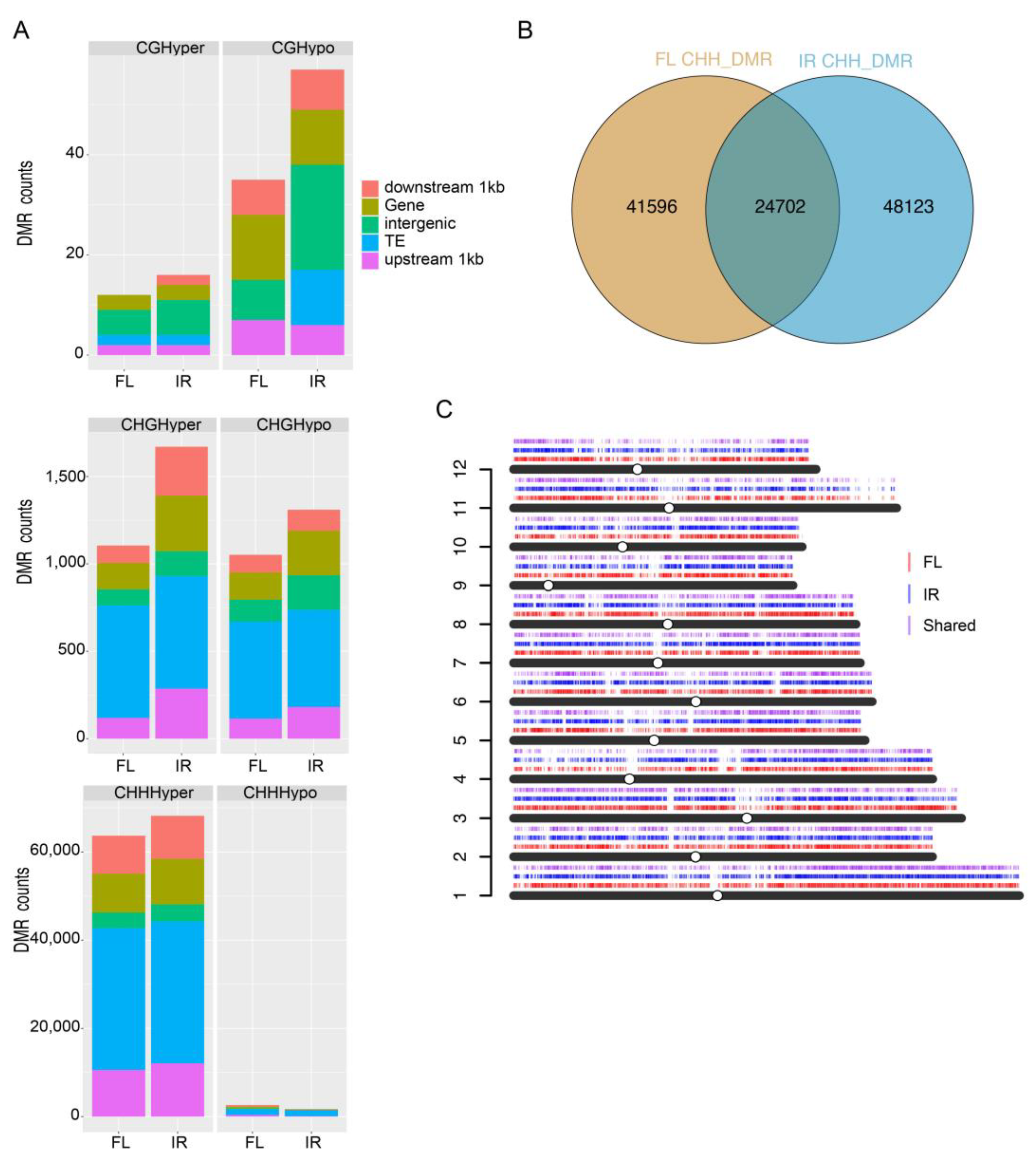
2.8. Differentially Methylated Regions under Salt Stress
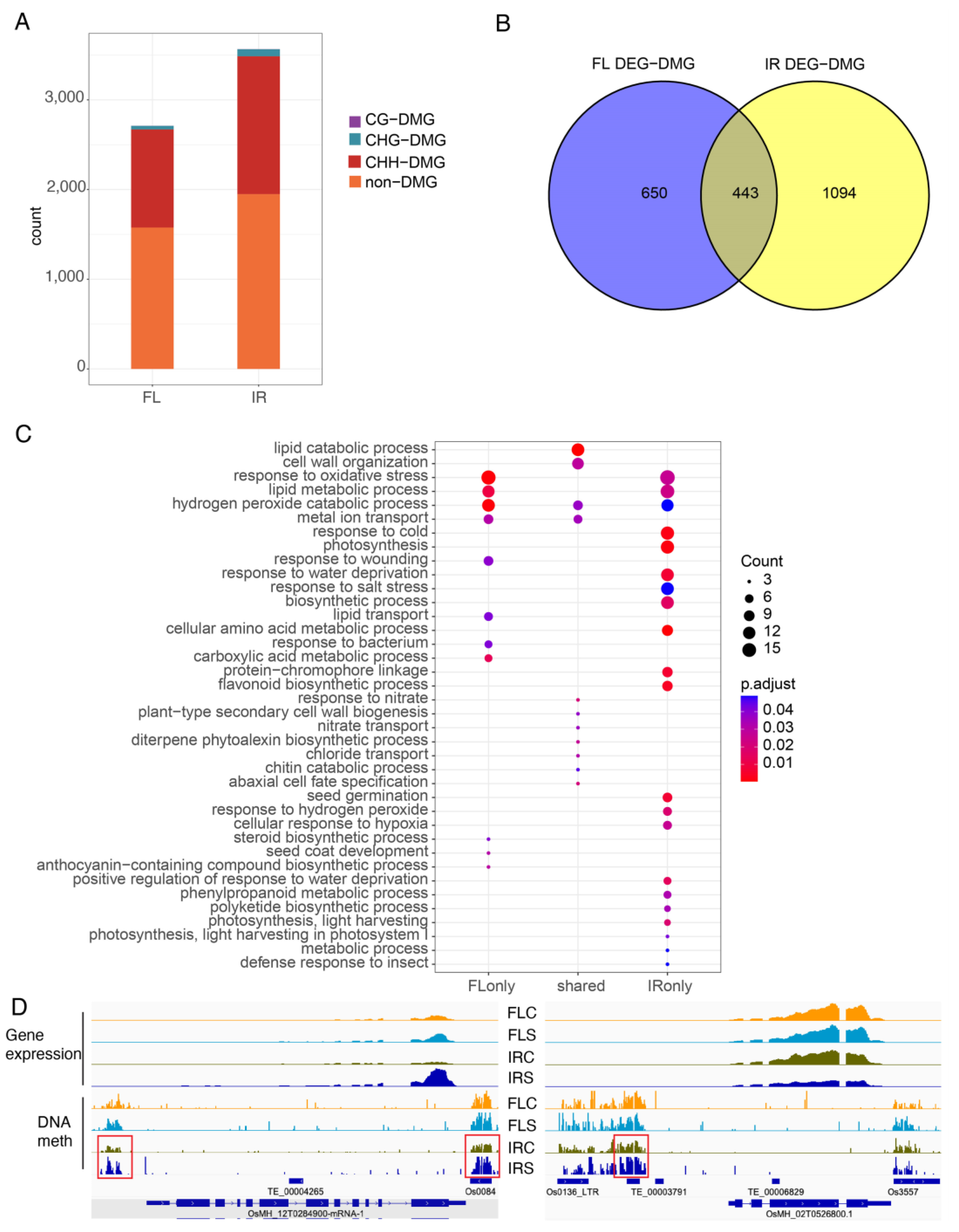
2.9. Differentially Expressed Genes with DMRs
3. Discussion
4. Materials and Methods
4.1. Plant Materials and Salt Treatment
4.2. RNA-Seq and Data Analysis
4.3. Bisulfite-Seq and Data Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Dametto, A.; Sperotto, R.A.; Adamski, J.M.; Blasi, E.A.R.; Cargnelutti, D.; de Oliveira, L.F.V.; Ricachenevsky, F.K.; Fregonezi, J.N.; Mariath, J.E.A.; da Cruz, R.P.; et al. Cold tolerance in rice germinating seeds revealed by deep RNAseq analysis of contrasting indica genotypes. Plant Sci. 2015, 238, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Omuto, C.; Vargas, R.; El Mobarak, A.; Mohamed, N.; Viatkin, K.; Yigini, Y. Mapping of Salt-Affected Soils–Technical Manual; Food and Agriculture Organization of the United Nations: Rome, Italy, 2020. [Google Scholar]
- Wang, W.; Zhao, X.; Pan, Y.; Zhu, L.; Fu, B.; Li, Z. DNA methylation changes detected by methylation-sensitive amplified polymorphism in two contrasting rice genotypes under salt stress. J. Genet. Genom. 2011, 38, 419–424. [Google Scholar] [CrossRef]
- Mardani, Z.; Rabiei, B.; Sabouri, H.; Sabouri, A. Identification of molecular markers linked to salt-tolerant genes at germination stage of rice. Plant Breed. 2014, 133, 196–202. [Google Scholar] [CrossRef]
- Wang, Z.; Wang, J.; Bao, Y.; Wu, Y.; Zhang, H. Quantitative trait loci controlling rice seed germination under salt stress. Euphytica 2011, 178, 297–307. [Google Scholar] [CrossRef]
- Liu, A.; Xiao, Z.; Li, M.-W.; Wong, F.-L.; Yung, W.-S.; Ku, Y.-S.; Wang, Q.; Wang, X.; Xie, M.; Yim, A.K.-Y.; et al. Transcriptomic reprogramming in soybean seedlings under salt stress. Plant Cell Environ. 2019, 42, 98–114. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Li, D.; Zhou, R.; Wang, X.; Dossa, K.; Wang, L.; Zhang, Y.; Yu, J.; Gong, H.; Zhang, X.; et al. Transcriptome and metabolome analyses of two contrasting sesame genotypes reveal the crucial biological pathways involved in rapid adaptive response to salt stress. BMC Plant Biol. 2019, 19, 66. [Google Scholar] [CrossRef] [Green Version]
- Cotsaftis, O.; Plett, D.; Johnson, A.A.T.; Walia, H.; Wilson, C.; Ismail, A.M.; Close, T.J.; Tester, M.; Baumann, U. Root-Specific Transcript Profiling of Contrasting Rice Genotypes in Response to Salinity Stress. Mol. Plant 2011, 4, 25–41. [Google Scholar] [CrossRef] [Green Version]
- Chen, M.-X.; Lu, C.-C.; Sun, P.-C.; Nie, Y.-X.; Tian, Y.; Hu, Q.-J.; Das, D.; Hou, X.-X.; Gao, B.; Chen, X.; et al. Comprehensive transcriptome and proteome analyses reveal a novel sodium chloride responsive gene network in maize seed tissues during germination. Plant Cell Environ. 2021, 44, 88–101. [Google Scholar] [CrossRef]
- Wu, T.-Y.; Goh, H.; Azodi, C.B.; Krishnamoorthi, S.; Liu, M.-J.; Urano, D. Evolutionarily conserved hierarchical gene regulatory networks for plant salt stress response. Nat. Plants 2021, 7, 787–799. [Google Scholar] [CrossRef]
- Kucera, B.; Cohn, M.A.; Leubner-Metzger, G. Plant hormone interactions during seed dormancy release and germination. Seed Sci. Res. 2005, 15, 281–307. [Google Scholar] [CrossRef]
- Yang, J.; Su, L.; Li, D.; Luo, L.; Sun, K.; Yang, M.; Gu, F.; Xia, A.; Liu, Y.; Wang, H.; et al. Dynamic transcriptome and metabolome analyses of two types of rice during the seed germination and young seedling growth stages. BMC Genom. 2020, 21, 603. [Google Scholar] [CrossRef] [PubMed]
- Zhang, R.; Hussain, S.; Wang, Y.; Liu, Y.; Li, Q.; Chen, Y.; Wei, H.; Gao, P.; Dai, Q. Comprehensive Evaluation of Salt Tolerance in Rice (Oryza sativa L.) Germplasm at the Germination Stage. Agronomy 2021, 11, 1569. [Google Scholar] [CrossRef]
- Liu, L.; Xia, W.; Li, H.; Zeng, H.; Wei, B.; Han, S.; Yin, C. Salinity Inhibits Rice Seed Germination by Reducing α-Amylase Activity via Decreased Bioactive Gibberellin Content. Front. Plant Sci. 2018, 9, 275. [Google Scholar] [CrossRef]
- Law, J.A.; Jacobsen, S.E. Establishing, maintaining and modifying DNA methylation patterns in plants and animals. Nat. Rev. Genet. 2010, 11, 204–220. [Google Scholar] [CrossRef] [PubMed]
- Erdmann, R.M.; Picard, C.L. RNA-directed DNA Methylation. PLoS Genet. 2020, 16, e1009034. [Google Scholar] [CrossRef]
- Zhu, J.-K. Active DNA Demethylation Mediated by DNA Glycosylases. Annu. Rev. Genet. 2009, 43, 143–166. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dowen, R.H.; Pelizzola, M.; Schmitz, R.J.; Lister, R.; Dowen, J.M.; Nery, J.R.; Dixon, J.E.; Ecker, J.R. Widespread dynamic DNA methylation in response to biotic stress. Proc. Natl. Acad. Sci. USA 2012, 109, 2183–2191. [Google Scholar] [CrossRef] [Green Version]
- Secco, D.; Wang, C.; Shou, H.X.; Schultz, M.D.; Chiarenza, S.; Nussaume, L.; Ecker, J.R.; Whelan, J.; Lister, R. Stress induced gene expression drives transient DNA methylation changes at adjacent repetitive elements. eLife 2015, 4, e09343. [Google Scholar] [CrossRef] [Green Version]
- Karan, R.; DeLeon, T.; Biradar, H.; Subudhi, P.K. Salt Stress Induced Variation in DNA Methylation Pattern and Its Influence on Gene Expression in Contrasting Rice Genotypes. PLoS ONE 2012, 7, e40203. [Google Scholar] [CrossRef]
- Munns, R.; Tester, M. Mechanisms of salinity tolerance. Annu. Rev. Plant Biol. 2008, 59, 651–681. [Google Scholar] [CrossRef] [Green Version]
- Baek, D.; Jiang, J.; Chung, J.-S.; Wang, B.; Chen, J.; Xin, Z.; Shi, H. Regulated AtHKT1 Gene Expression by a Distal Enhancer Element and DNA Methylation in the Promoter Plays an Important Role in Salt Tolerance. Plant Cell Physiol. 2011, 52, 149–161. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, S.; Beena, A.S.; Awana, M.; Singh, A. Salt-Induced Tissue-Specific Cytosine Methylation Downregulates Expression of HKT Genes in Contrasting Wheat (Triticum aestivum L.) Genotypes. DNA Cell Biol. 2017, 36, 283–294. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferreira, L.J.; Azevedo, V.; Maroco, J.; Oliveira, M.M.; Santos, A.P. Salt Tolerant and Sensitive Rice Varieties Display Differential Methylome Flexibility under Salt Stress. PLoS ONE 2015, 10, e0124060. [Google Scholar] [CrossRef] [Green Version]
- Xu, R.; Wang, Y.; Zheng, H.; Lu, W.; Wu, C.; Huang, J.; Yan, K.; Yang, G.; Zheng, C. Salt-induced transcription factor MYB74 is regulated by the RNA-directed DNA methylation pathway in Arabidopsis. J. Exp. Bot. 2015, 66, 5997–6008. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, W.; Wang, N.; Yang, J.; Guo, H.; Liu, Z.; Zheng, X.; Li, S.; Xiang, F. The salt-induced transcription factor GmMYB84 confers salinity tolerance in soybean. Plant Sci. 2020, 291, 110326. [Google Scholar] [CrossRef]
- Fang, Y.; Li, J.; Jiang, J.; Geng, Y.; Wang, J.; Wang, Y. Physiological and epigenetic analyses of Brassica napus seed germination in response to salt stress. Acta Physiol. Plant. 2017, 39, 128. [Google Scholar] [CrossRef]
- Guo, X.; Hou, X.; Fang, J.; Wei, P.; Xu, B.; Chen, M.; Feng, Y.; Chu, C. The rice GERMINATION DEFECTIVE 1, encoding a B3 domain transcriptional repressor, regulates seed germination and seedling development by integrating GA and carbohydrate metabolism. Plant J. 2013, 75, 403–416. [Google Scholar] [CrossRef] [Green Version]
- Li, W.; Niu, Y.; Zheng, Y.; Wang, Z. Advances in the Understanding of Reactive Oxygen Species-Dependent Regulation on Seed Dormancy, Germination, and Deterioration in Crops. Front. Plant Sci. 2022, 13, 826809. [Google Scholar] [CrossRef]
- Stroud, H.; Greenberg, M.V.C.; Feng, S.; Bernatavichute, Y.V.; Jacobsen, S.E. Comprehensive Analysis of Silencing Mutants Reveals Complex Regulation of the Arabidopsis Methylome. Cell 2013, 152, 352–364. [Google Scholar] [CrossRef] [Green Version]
- Khayatnezhad, M.; Gholamin, R. Effects of salt stress levels on five maize (Zea mays L.) cultivars at germination stage. Afr. J. Biotechnol. 2011, 10, 12909–12915. [Google Scholar] [CrossRef] [Green Version]
- Funck, D.; Baumgarten, L.; Stift, M.; von Wirén, N.; Schönemann, L. Differential Contribution of P5CS Isoforms to Stress Tolerance in Arabidopsis. Front. Plant Sci. 2020, 11, 565134. [Google Scholar] [CrossRef]
- Huang, S.; Spielmeyer, W.; Lagudah, E.S.; James, R.A.; Platten, J.D.; Dennis, E.S.; Munns, R. A Sodium Transporter (HKT7) Is a Candidate for Nax1, a Gene for Salt Tolerance in Durum Wheat. Plant Physiol. 2006, 142, 1718–1727. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yuan, W.; Zhang, D.; Song, T.; Xu, F.; Lin, S.; Xu, W.; Li, Q.; Zhu, Y.; Liang, J.; Zhang, J. Arabidopsis plasma membrane H+-ATPase genes AHA2 and AHA7 have distinct and overlapping roles in the modulation of root tip H+ efflux in response to low-phosphorus stress. J. Exp. Bot. 2017, 68, 1731–1741. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Naito, K.; Zhang, F.; Tsukiyama, T.; Saito, H.; Hancock, C.N.; Richardson, A.O.; Okumoto, Y.; Tanisaka, T.; Wessler, S.R. Unexpected consequences of a sudden and massive transposon amplification on rice gene expression. Nature 2009, 461, 1130–1134. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Wu, X.; Yao, X.; Yu, R.; Larkin, P.J.; Liu, C.-M. Mutations in the DNA demethylase OsROS1 result in a thickened aleurone and improved nutritional value in rice grains. Proc. Natl. Acad. Sci. USA 2018, 115, 11327–11332. [Google Scholar] [CrossRef] [Green Version]
- Lin, J.-Y.; Le, B.H.; Chen, M.; Henry, K.F.; Hur, J.; Hsieh, T.-F.; Chen, P.-Y.; Pelletier, J.M.; Pellegrini, M.; Fischer, R.L.; et al. Similarity between soybean and Arabidopsis seed methylomes and loss of non-CG methylation does not affect seed development. Proc. Natl. Acad. Sci. USA 2017, 114, 9730–9739. [Google Scholar] [CrossRef] [Green Version]
- Kawakatsu, T.; Nery, J.R.; Castanon, R.; Ecker, J.R. Dynamic DNA methylation reconfiguration during seed development and germination. Genome Biol. 2017, 18, 171. [Google Scholar] [CrossRef] [Green Version]
- Lu, B.; Dong, L.; Yi, D.; Zhang, M.; Zhu, C.; Li, X.; Yi, C. Transposase-assisted tagmentation of RNA/DNA hybrid duplexes. eLife 2020, 9, 54919. [Google Scholar] [CrossRef]
- Kim, D.; Paggi, J.M.; Park, C.; Bennett, C.; Salzberg, S.L. Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nat. Biotechnol. 2019, 37, 907–915. [Google Scholar] [CrossRef]
- Liao, Y.; Smyth, G.K.; Shi, W. featureCounts: An efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 2014, 30, 923–930. [Google Scholar] [CrossRef] [Green Version]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krueger, F.; Andrews, S.R. Bismark: A flexible aligner and methylation caller for Bisulfite-Seq applications. Bioinformatics 2011, 27, 1571–1572. [Google Scholar] [CrossRef] [Green Version]
- Catoni, M.; Tsang, J.M.F.; Greco, A.P.; Zabet, N.R. DMRcaller: A versatile R/Bioconductor package for detection and visualization of differentially methylated regions in CpG and non-CpG contexts. Nucleic Acids Res. 2018, 46, e114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krzywinski, M.; Schein, J.; Birol, I.; Connors, J.; Gascoyne, R.; Horsman, D.; Jones, S.J.; Marra, M.A. Circos: An information aesthetic for comparative genomics. Genome Res. 2009, 19, 1639–1645. [Google Scholar] [CrossRef] [PubMed] [Green Version]








Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, Y.; Guo, D. Transcriptome and DNA Methylome Analysis of Two Contrasting Rice Genotypes under Salt Stress during Germination. Int. J. Mol. Sci. 2023, 24, 3978. https://doi.org/10.3390/ijms24043978
Li Y, Guo D. Transcriptome and DNA Methylome Analysis of Two Contrasting Rice Genotypes under Salt Stress during Germination. International Journal of Molecular Sciences. 2023; 24(4):3978. https://doi.org/10.3390/ijms24043978
Chicago/Turabian StyleLi, Yongqiang, and Dianjing Guo. 2023. "Transcriptome and DNA Methylome Analysis of Two Contrasting Rice Genotypes under Salt Stress during Germination" International Journal of Molecular Sciences 24, no. 4: 3978. https://doi.org/10.3390/ijms24043978