

Starch and Sucrose Metabolism and Plant Hormone Signaling Pathways Play Crucial Roles in Aquilegia Salt Stress Adaption
Abstract
:1. Introduction
2. Results
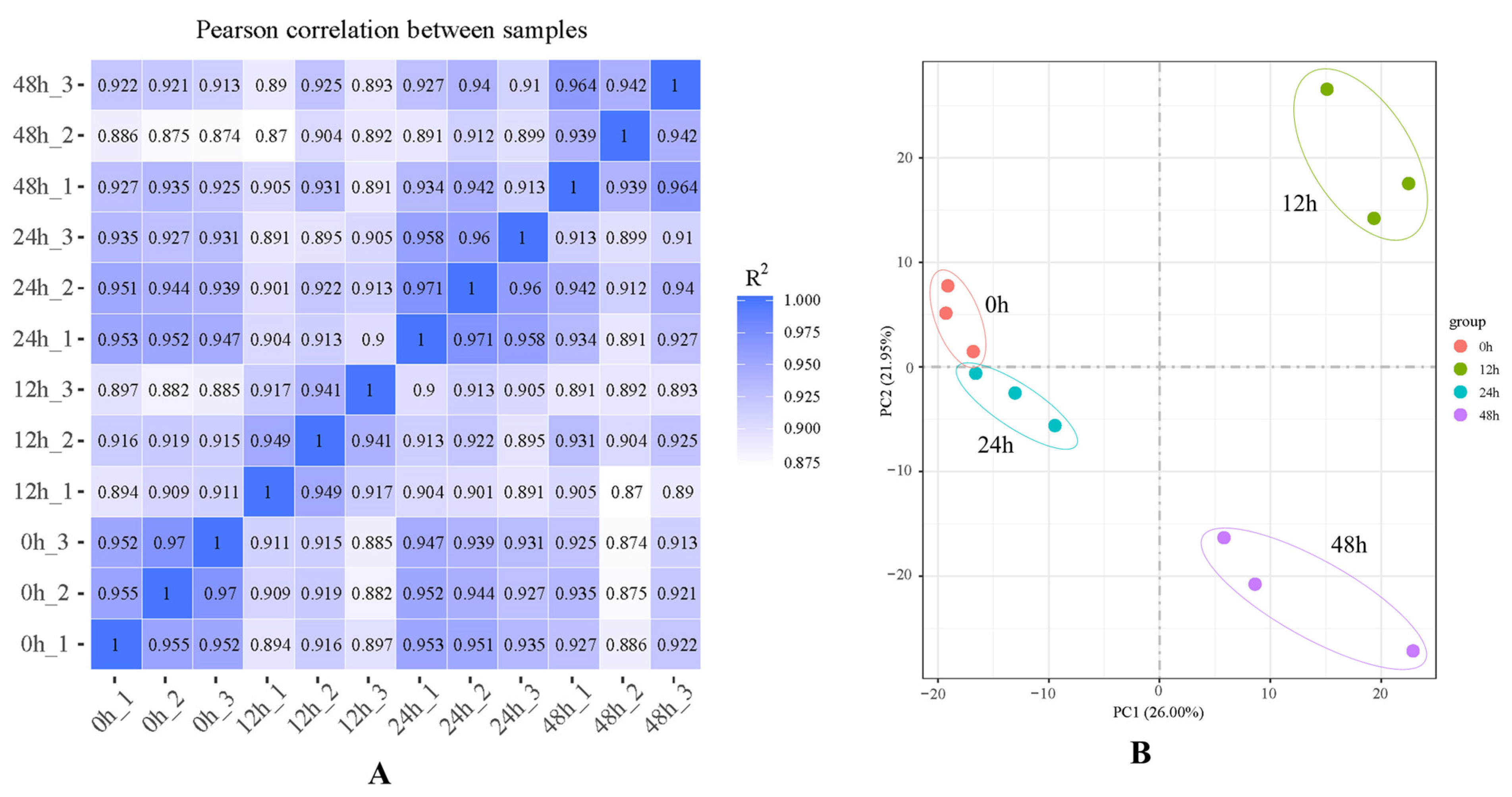
2.1. RNA-Sequencing Quality
2.2. Differentially Expressed Genes (DEGs)
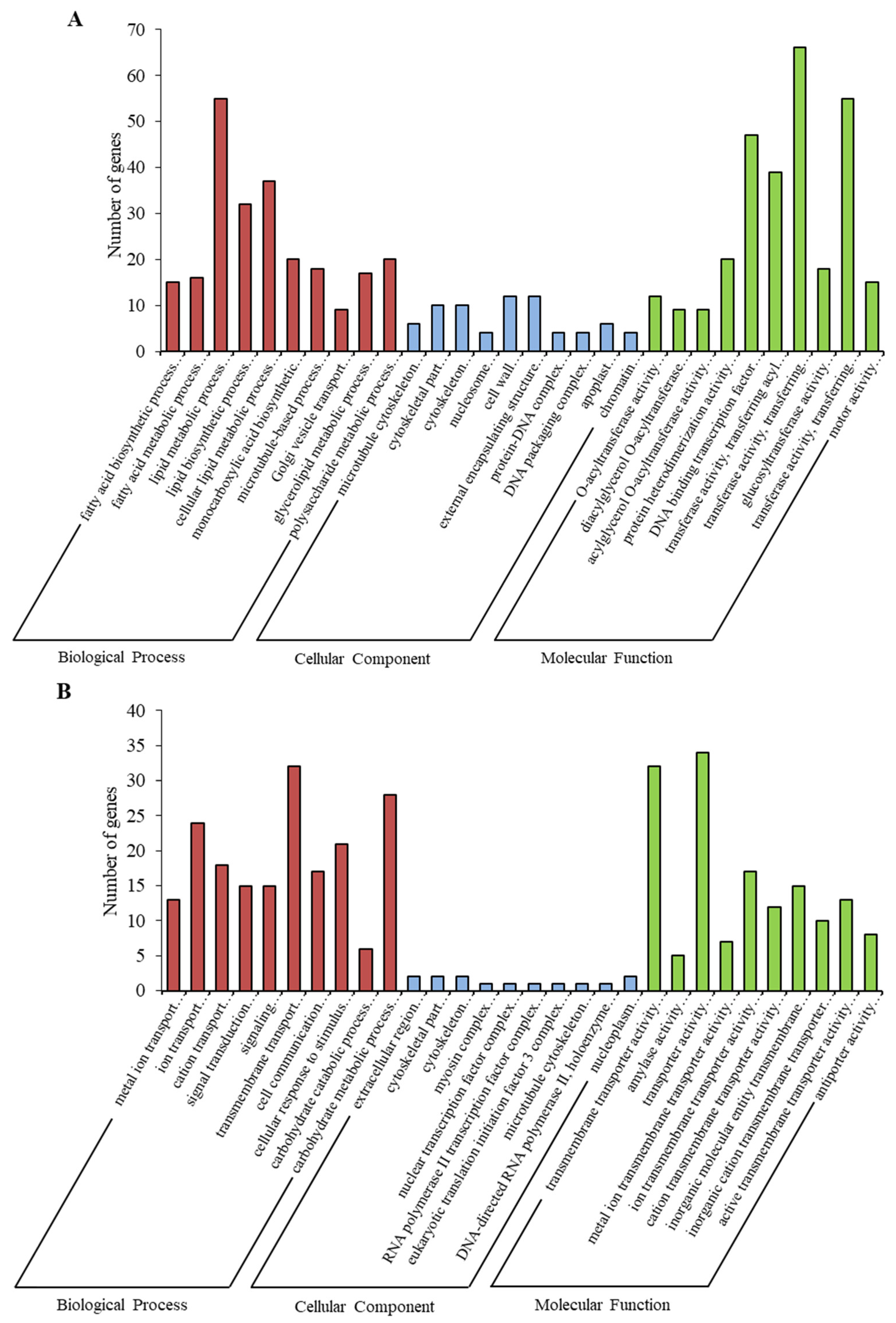
2.3. Gene Ontology (GO) Enrichment Analysis
2.4. Kyoto Encyclopedia of Genes and Genomes (KEGG) Enrichment Analysis
2.5. Gene Expression Analysis in Starch and Sucrose Metabolism Pathway
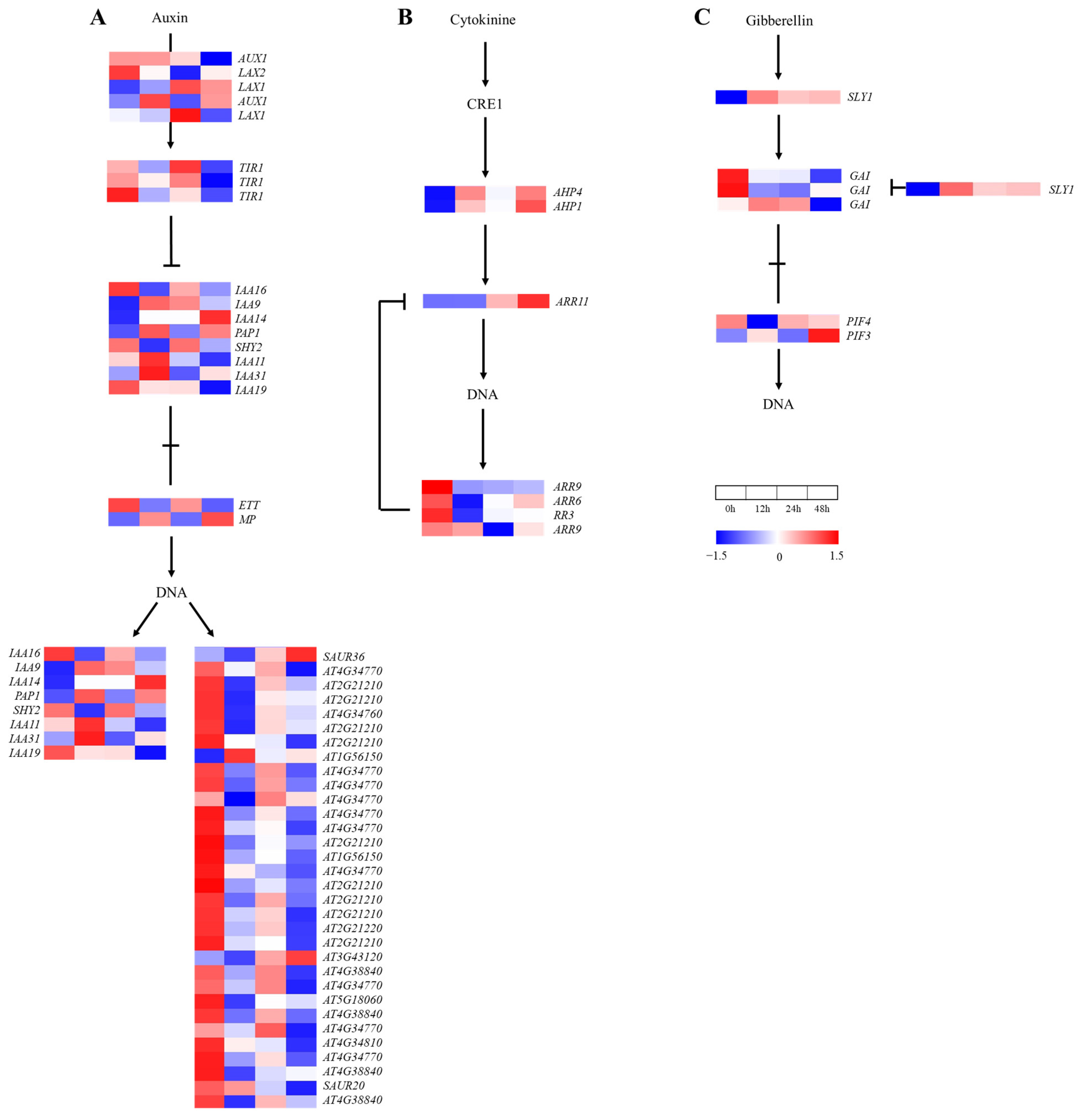
2.6. Gene Expression in Plant Hormone Signal Transduction
2.7. Interaction Network Analysis
2.8. Data Reliability Analysis with Quantitative Real-Time PCR
3. Discussion
4. Materials and Methods
4.1. Plant Materials and Salt Treatment
4.2. RNA Extraction and cDNA Library Construction
4.3. Reference Genome Alignment
4.4. Sample Correlation and Gene Expression Analysis
4.5. Enrichment and Interaction Analyses
4.6. Quantitative Real-Time PCR (qRT-PCR)
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Yu, Z.; Duan, X. How plant hormones mediate salt stress responses. Trends Plant Sci. 2020, 25, 1117–1130. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Yan, G. Elucidating the molecular mechanisms mediating plant salt-stress responses. New Phytol. 2017, 217, 523–539. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nold, R. Columbines: Aquilegia, Paraaquilegia, and Semiaquilegia; Timber Press: Portland, OR, USA, 2003; Volume 3, pp. 124–130. [Google Scholar]
- Kramer, E.M. Aquilegia: A new model for plant development, ecology, and evolution. Annu. Rev. Plant Biol. 2009, 60, 261–277. [Google Scholar] [CrossRef] [PubMed]
- Filiault, D.L.; Ballerini, E.S. The Aquilegia genome provides insight into adaptive radiation and reveals an extraordinarily polymorphic chromosome with a unique history. eLIFE 2018, 7, e36426. [Google Scholar] [CrossRef]
- Han, M.; Zhu, Q. Petal ontogeny, structure, and pollination system of four Aquilegia species in Midwest China. Flora 2022, 286, 151987. [Google Scholar] [CrossRef]
- Min, Y.; Kramer, E.M. Transcriptome profiling and weighted gene co-expression network analysis of early floral development in Aquilegia coerulea. Sci. Rep. 2020, 10, 19637. [Google Scholar] [CrossRef]
- Ballerini, E.S.; Min, Y. POPOVICH, encoding a C2H2 zinc-finger transcription factor, plays a central role in the development of a key innovation, floral nectar spurs, in Aquilegia. Proc. Natl. Acad. Sci. USA 2020, 117, 202006912. [Google Scholar] [CrossRef]
- Shi, T.; Chen, J. A reappraisal of the phylogenetic placement of the Aquilegia whole-genome duplication. Genom. Biol. 2020, 21, 295. [Google Scholar] [CrossRef]
- Mushtaq, S.; Aga, M.A. Isolation, characterization and HPLC quantification of compounds from Aquilegia fragrans Benth: Their in vitro antibacterial activities against bovine mastitis pathogens. J. Ethnopharmacol. 2016, 178, 9–12. [Google Scholar] [CrossRef]
- Chen, L.; Yuan, M. Physio-biochemical responses of three Aquilegia species seedlings to salt stress. Agronomy 2022, 12, 2841. [Google Scholar] [CrossRef]
- El Moukhtari, A.; Cabassa-Hourton, C. How does proline treatment promote salt stress tolerance during crop plant development? Front. Plant Sci. 2020, 11, 1127. [Google Scholar] [CrossRef]
- Chen, Y.; Jiang, Y. Uncovering candidate genes responsive to salt stress in Salix matsudana (Koidz) by transcriptomic analysis. PLoS ONE 2020, 15, e0236129. [Google Scholar] [CrossRef]
- Liu, Z.; Hua, Y. Analysis of the Prunellae spica transcriptome under salt stress. Plant Physiol. Biochem. 2020, 156, 314–322. [Google Scholar] [CrossRef]
- Deng, C.; Zhang, Z. Salt-responsive transcriptome analysis of triticale reveals candidate genes involved in the key metabolic pathway in response to salt stress. Sci. Rep. 2020, 10, 20669. [Google Scholar] [CrossRef]
- Yang, J.; Ma, L. Comprehensive identification and characterization of abiotic stress and hormone responsive glycosyl hydrolase family 1 genes in Medicago truncatula. Plant Physiol. Biochem. 2021, 158, 21–33. [Google Scholar] [CrossRef]
- Samadi, S.; Habibi, G. Exogenous trehalose alleviates the inhibitory effects of salt stress in strawberry plants. Acta Physiol. Plant 2019, 41, 112. [Google Scholar] [CrossRef]
- Thalmann, M.; Pazmino, D. Regulation of leaf starch degradation by abscisic acid is important for osmotic stress tolerance in plants. Plant Cell 2016, 28, 1860–1878. [Google Scholar] [CrossRef] [Green Version]
- Dar, N.A.; Amin, I. Abscisic acid: A key regulator of abiotic stress tolerance in plants. Plant Gene 2017, 11, 106–111. [Google Scholar] [CrossRef]
- Muchate, N.S.; Nikalje, G.C. Plant salt stress: Adaptive responses, tolerance mechanism and bioengineering for salt tolerance. Bot. Rev. 2016, 82, 1–36. [Google Scholar] [CrossRef]
- Pavlović, I.; Pěnčík, A. Short-term salt stress in Brassica rapa seedlings causes alterations in auxin metabolism. Plant Physiol. Biochem. 2018, 125, 74–87. [Google Scholar] [CrossRef]
- Van den Ende, W.; El-Esawe, S.K. Sucrose signaling pathways leading to fructan and anthocyanin accumulation: A dual function in abiotic and biotic stress responses? Environ. Exp. Bot. 2014, 108, 4–13. [Google Scholar] [CrossRef]
- Li, L.; Sheen, J. Dynamic and diverse sugar signaling. Curr. Opin. Plant Biol. 2016, 33, 116–125. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ceusters, N.; Van den Ende, W. Exploration of Sweet Immunity to Enhance Abiotic Stress Tolerance in Plants: Lessons from CAM; Springer: Cham, Switzerland, 2016; Volume 78, pp. 145–166. [Google Scholar] [CrossRef]
- Martínez-Noël, G.M.A.; Tognetti, J.A. Chapter 22—Sugar Signaling Under Abiotic Stress in Plants. In Plant Metabolites and Regulation under Environmental Stress; Academic Press: San Diego, CA, USA, 2018; pp. 397–406. [Google Scholar] [CrossRef]
- Saddhe, A.; Manuka, R. Plant sugars: Homeostasis and transport under abiotic stress in plants. Physiol. Plant. 2020, 171, 739–755. [Google Scholar] [CrossRef] [PubMed]
- Sakr, S.; Wang, M. The sugar-signaling hub: Overview of regulators and interaction with the hormonal and metabolic network. Int. J. Mol. Sci. 2018, 19, 2506. [Google Scholar] [CrossRef] [Green Version]
- Kushwah, S.; Laxmi, A. The interaction between glucose and cytokinin signal transduction pathway in Arabidopsis thaliana. Plant Cell Environ. 2014, 37, 235–253. [Google Scholar] [CrossRef]
- Paparelli, E.; Parlanti, S. Nighttime sugar starvation orchestrates gibberellin biosynthesis and plant growth in Arabidopsis. Plant Cell 2013, 25, 3760–3769. [Google Scholar] [CrossRef] [Green Version]
- Wang, W.; Liang, J. Overexpression of PpSnRK1α in tomato enhanced salt tolerance by regulating ABA signaling pathway and reactive oxygen metabolism. BMC Plant Biol. 2020, 20, 128. [Google Scholar] [CrossRef]
- Han, C.; Kim, S. Cross-talk between ABA and sugar signaling is mediated by the ACGT core and CE1 element reciprocally in OsTIP3;1 promoter. J. Plant Physiol. 2018, 224–225, 103–111. [Google Scholar] [CrossRef]
- Ren, R.; Li, D. Specific roles of Os4BGlu10, Os6BGlu24, and Os9BGlu33 in seed germination, root elongation, and drought tolerance in rice. Planta 2019, 249, 1851–1861. [Google Scholar] [CrossRef]
- Wei, J.; Chen, Q. Genome-wide identification and expression analysis of tomato glycoside hydrolase family 1 β-glucosidase genes in response to abiotic stresses. Biotechnol. Biotechnol. Equip. 2022, 36, 268–280. [Google Scholar] [CrossRef]
- Baba, S.A.; Vishwakarma, R.A. Functional characterization of CsBGlu12, a β-Glucosidase from Crocus sativus, provides insights into its role in abiotic stress through accumulation of antioxidant flavonols. J. Biol. Chem. 2017, 292, 4700–4713. [Google Scholar] [CrossRef] [Green Version]
- Cao, Y.; Yang, J. A Phylogenetically informed comparison of GH1 hydrolases between Arabidopsis and rice response to stressors. Front. Plant Sci. 2017, 8, 350. [Google Scholar] [CrossRef] [Green Version]
- Wani, S.H.; Kumar, V. Phytohormones and their metabolic engineering for abiotic stress tolerance in crop plants. Crop J. 2016, 4, 162–176. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Liu, P. Comparative transcriptome analyses of maize seedling root responses to salt stress. PeerJ 2020, 9, e10765. [Google Scholar] [CrossRef]
- Zhang, M.; Hong, L. Transcriptome analyses revealed molecular responses of Cynanchum auriculatum leaves to saline stress. Sci. Rep. 2020, 10, 449. [Google Scholar] [CrossRef] [Green Version]
- Bao, Y.; Chen, C. Comparative transcriptome analysis of Rosa chinensis ‘Old Blush’ provides insights into the crucial and signaling pathways in salt stress response. Agron. J. 2021, 113, 3031–3050. [Google Scholar] [CrossRef]
- Ribba, T.; Garrido, F. Auxin-mediated responses under salt stress from developmental regulation to biotechnological applications. J. Exp. Bot. 2020, 71, 3843–3853. [Google Scholar] [CrossRef]
- Lei, P.; Liu, Z. Transcriptome analysis of salt stress responsiveness in the seedlings of wild and cultivated Ricinus communis L. J. Biotechnol. 2021, 327, 106–116. [Google Scholar] [CrossRef]
- Cadavid, I.C.; Guzman, F. Transcriptional analyses of two soybean cultivars under salt stress. Mol. Biol. Rep. 2020, 47, 2871–2888. [Google Scholar] [CrossRef]
- Strader, L.C.; Zhao, Y. Auxin perception and downstream events. Curr. Opin. Plant Biol. 2016, 33, 8–14. [Google Scholar] [CrossRef] [Green Version]
- Ren, H.; Gray, W.M. SAUR proteins as effectors of hormonal and environmental signals in plant growth. Mol. Plant 2015, 8, 1153–1164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stortenbeker, N.; Bemer, M. The SAUR gene family: The plant’s toolbox for adaptation of growth and development. J. Exp. Bot. 2018, 70, 17–27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Atamian, H.S.; Creux, N.M. Circadian regulation of sunflower heliotropism, floral orientation, and pollinator visits. Science 2016, 353, 587–590. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luo, J.; Zhou, J. Aux/IAA gene family in plants: Molecular structure, regulation, and function. Int. J. Mol. Sci. 2018, 19, 259. [Google Scholar] [CrossRef] [Green Version]
- Purgatto, E.; Lajolo, F.M. Inhibition of β-amylase activity, starch degradation and sucrose formation by indole-3-acetic acid during banana ripening. Planta 2001, 212, 823–828. [Google Scholar] [CrossRef]
- Sami, F.; Yusuf, M. Role of sugars under abiotic stress. Plant Physiol. Biochem. 2016, 109, 54–61. [Google Scholar] [CrossRef]
- Bian, Z.; Wang, D. Analysis of Populus glycosyl hydrolase family I members and their potential role in the ABA treatment and drought stress response. Plant Physiol. Biochem. 2021, 163, 178–788. [Google Scholar] [CrossRef]
- Davière, J.M.; Achard, P. Gibberellin signaling in plants. Development 2013, 140, 1147–1151. [Google Scholar] [CrossRef] [Green Version]
- Ito, T.; Okada, K. DELLA-dependent and –independent gibberellin signaling. Plant Signal. Behav. 2018, 13, e1445933. [Google Scholar] [CrossRef] [Green Version]
- Silverstone, A.L.; Tseng, T. Functional analysis of SPINDLY in gibberellin signaling in Arabidopsis. Plant Physiol. 2007, 143, 987–1000. [Google Scholar] [CrossRef] [Green Version]
- Jue, D.; Sang, X. Comprehensive analysis of the longan transcriptome reveals distinct regulatory programs during the floral transition. BMC Genom. 2019, 20, 126. [Google Scholar] [CrossRef]
- Dugasa, M.T.; Feng, X. Comparative transcriptome and tolerance mechanism analysis in the two contrasting wheat (Triticum aestivum L.) cultivars in response to drought and salinity stresses. Plant Growth Regul. 2021, 94, 101–114. [Google Scholar] [CrossRef]
- Kim, D.; Paggi, J.M. Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nat. Biotechnol. 2019, 37, 907–915. [Google Scholar] [CrossRef]
- Liao, Y.; Smyth, G.K. featureCounts: An efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 2014, 30, 923–930. [Google Scholar] [CrossRef] [Green Version]
- Love, M.I.; Huber, W. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [Green Version]
- Benjamini, Y.; Hochberg, Y. On the adaptive control of the false discovery rate in multiple testing with independent statistics. J. Educ. Behav. Stat. 2000, 25, 60–83. [Google Scholar] [CrossRef] [Green Version]
- Yu, G.; Wang, L.-G.; Han, Y.; He, Q.-Y. clusterProfiler: An R package for comparing biological themes among gene clusters. OMICS J. Integr. Biol. 2012, 16, 284–287. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- Schmittgen, T.D.; Livak, K.J. Analyzing real time PCR data by the comparative CT methods. Nat. Protoc. 2008, 3, 1101–1108. [Google Scholar] [CrossRef]













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Raw Reads | Clean Reads | Clean Bases | Error Rate | Q20 | Q30 | GC PCT |
|---|---|---|---|---|---|---|---|
| 0 h_1 | 47,647,814 | 46,506,466 | 6.98 G | 0.02 | 98.37 | 94.88 | 42.31 |
| 0 h_2 | 47,064,990 | 45,853,912 | 6.88 G | 0.02 | 98.41 | 94.96 | 42.58 |
| 0 h_3 | 46,642,716 | 45,494,096 | 6.82 G | 0.02 | 98.48 | 95.14 | 42.72 |
| 12 h_1 | 42,376,174 | 41,521,502 | 6.23 G | 0.02 | 98.34 | 94.87 | 42.23 |
| 12 h_2 | 47,123,344 | 45,962,436 | 6.89 G | 0.02 | 98.34 | 94.91 | 42.05 |
| 12 h_3 | 49,174,170 | 47,628,388 | 7.14 G | 0.02 | 98.48 | 95.19 | 41.85 |
| 24 h_1 | 45,614,350 | 44,336,182 | 6.65 G | 0.02 | 98.54 | 95.31 | 42.25 |
| 24 h_2 | 46,115,956 | 44,862,266 | 6.73 G | 0.02 | 98.5 | 95.13 | 42.31 |
| 24 h_3 | 51,810,052 | 50,770,432 | 7.62 G | 0.02 | 98.55 | 95.26 | 42.29 |
| 48 h_1 | 45,223,922 | 44,376,174 | 6.66 G | 0.02 | 98.43 | 95.14 | 42.09 |
| 48 h_2 | 47,939,292 | 46,686,300 | 7.0 G | 0.02 | 98.3 | 94.81 | 41.51 |
| 48 h_3 | 48,060,240 | 46,931,440 | 7.04 G | 0.02 | 98.41 | 95.1 | 42.2 |
| Sample | Total Reads | Total Map | Multi Map |
|---|---|---|---|
| 0 h_1 | 46,506,466 | 91.08% | 2.20% |
| 0 h_2 | 45,853,912 | 91.21% | 2.24% |
| 0 h_3 | 45,494,096 | 92.07% | 2.26% |
| 12 h_1 | 41,521,502 | 91.56% | 2.42% |
| 12 h_2 | 45,962,436 | 91.05% | 2.36% |
| 12 h_3 | 47,628,388 | 90.82% | 2.54% |
| 24 h_1 | 44,336,182 | 91.62% | 2.37% |
| 24 h_2 | 44,862,266 | 91.86% | 2.32% |
| 24 h_3 | 50,770,432 | 91.77% | 2.42% |
| 48 h_1 | 44,376,174 | 91.15% | 2.50% |
| 48 h_2 | 46,686,300 | 90.59% | 2.52% |
| 48 h_3 | 46,931,440 | 91.46% | 2.53% |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, L.; Meng, Y.; Bai, Y.; Yu, H.; Qian, Y.; Zhang, D.; Zhou, Y. Starch and Sucrose Metabolism and Plant Hormone Signaling Pathways Play Crucial Roles in Aquilegia Salt Stress Adaption. Int. J. Mol. Sci. 2023, 24, 3948. https://doi.org/10.3390/ijms24043948
Chen L, Meng Y, Bai Y, Yu H, Qian Y, Zhang D, Zhou Y. Starch and Sucrose Metabolism and Plant Hormone Signaling Pathways Play Crucial Roles in Aquilegia Salt Stress Adaption. International Journal of Molecular Sciences. 2023; 24(4):3948. https://doi.org/10.3390/ijms24043948
Chicago/Turabian StyleChen, Lifei, Yuan Meng, Yun Bai, Haihang Yu, Ying Qian, Dongyang Zhang, and Yunwei Zhou. 2023. "Starch and Sucrose Metabolism and Plant Hormone Signaling Pathways Play Crucial Roles in Aquilegia Salt Stress Adaption" International Journal of Molecular Sciences 24, no. 4: 3948. https://doi.org/10.3390/ijms24043948