
Impact of Prenatal Exposure to Maternal Diabetes and High-Fat Diet on Postnatal Myocardial Ketone Body Metabolism in Rats
 , and
, and 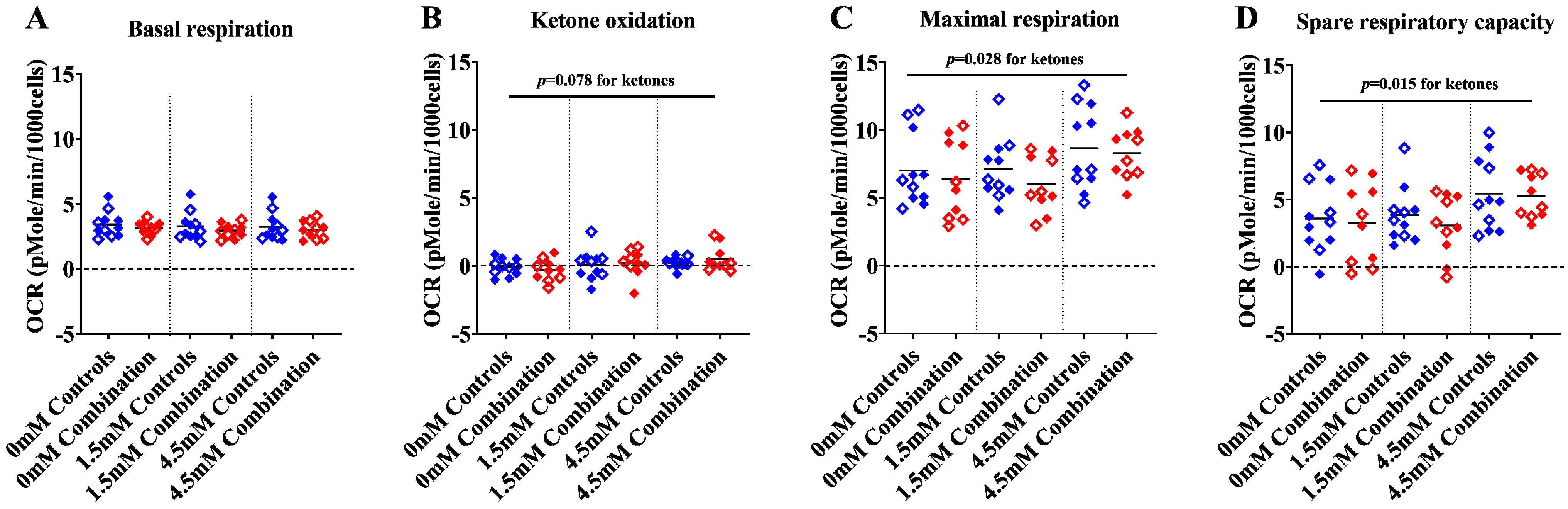{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
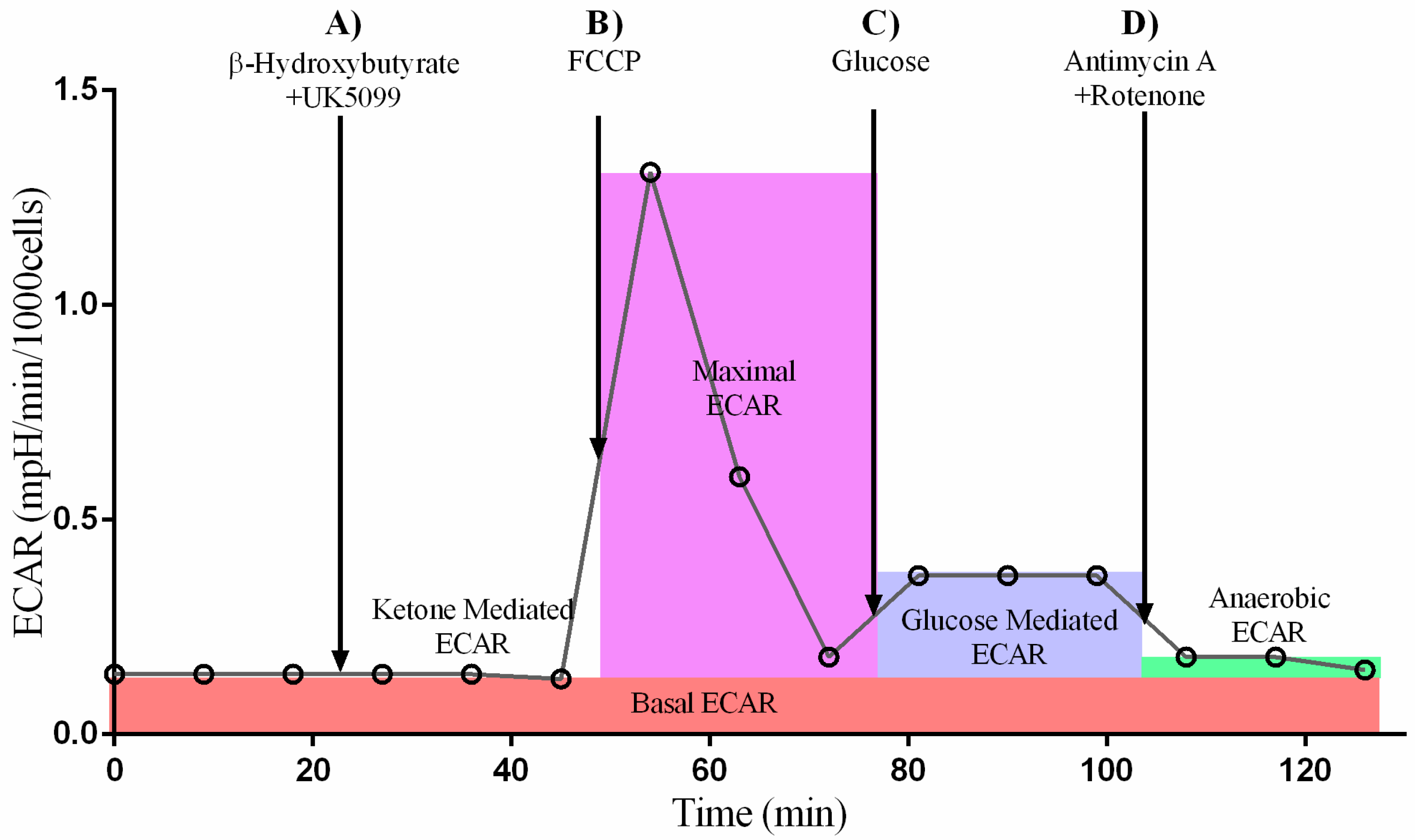
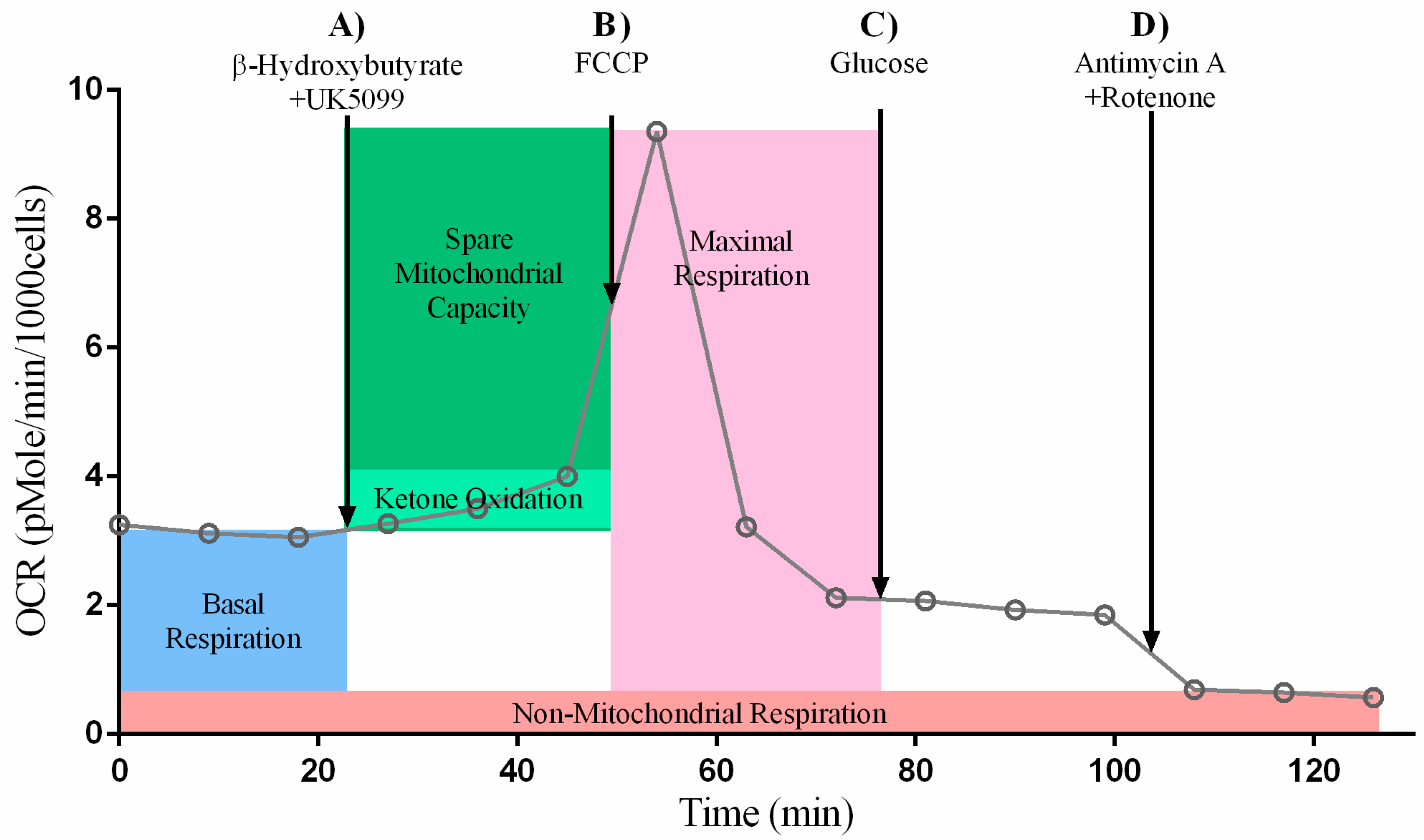
2.1. Ketone Stress Test (KST) in Primary Isolated Cardiomyocytes
2.1.1. Maximal and Spare Respiratory Capacity Increase Directly with Increasing Concentrations of Ketone
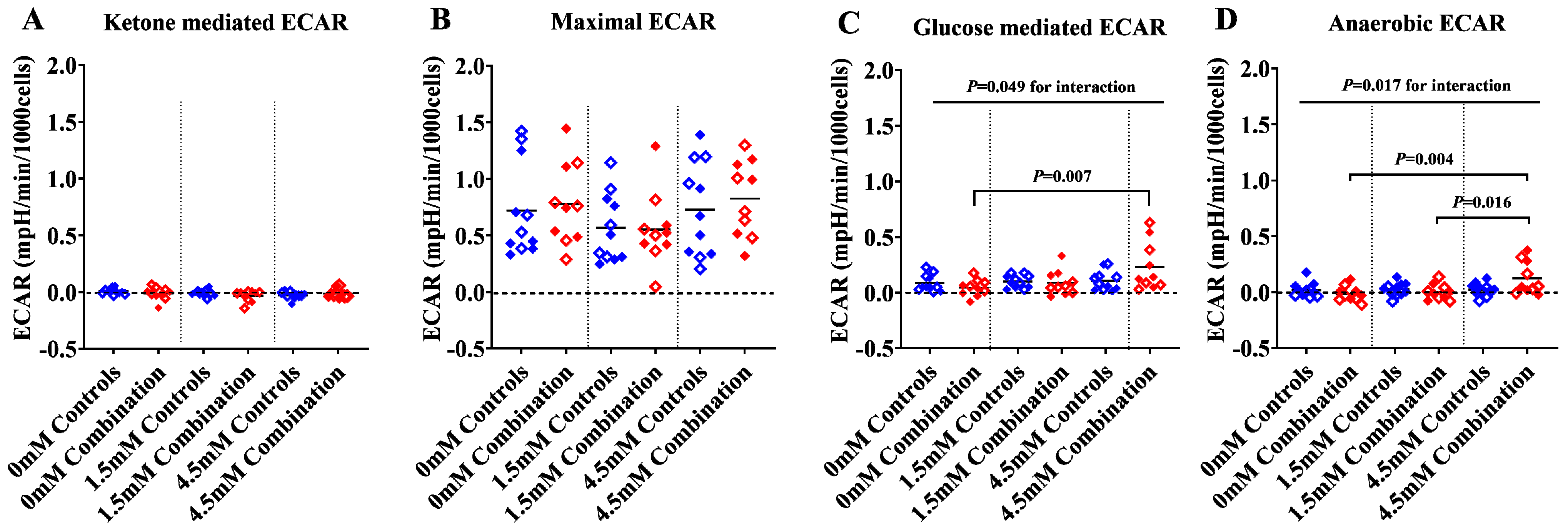
2.1.2. The Presence of Ketones Improves Glycolysis in Diabetes + HFD-Exposed NRCM
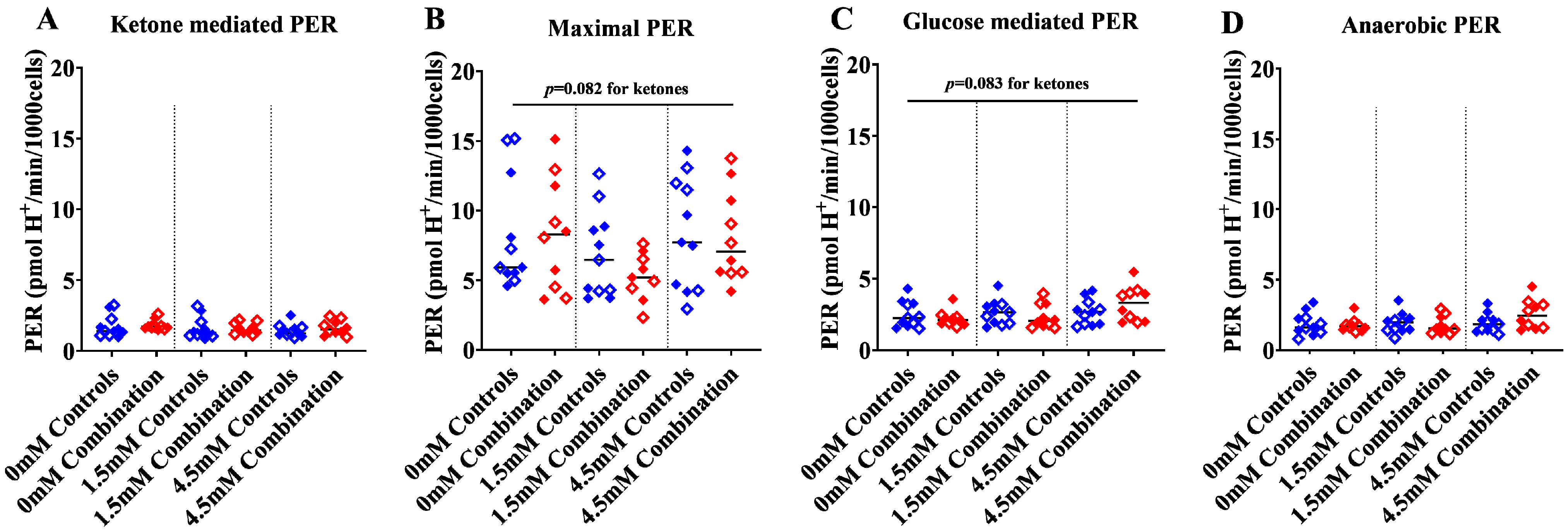
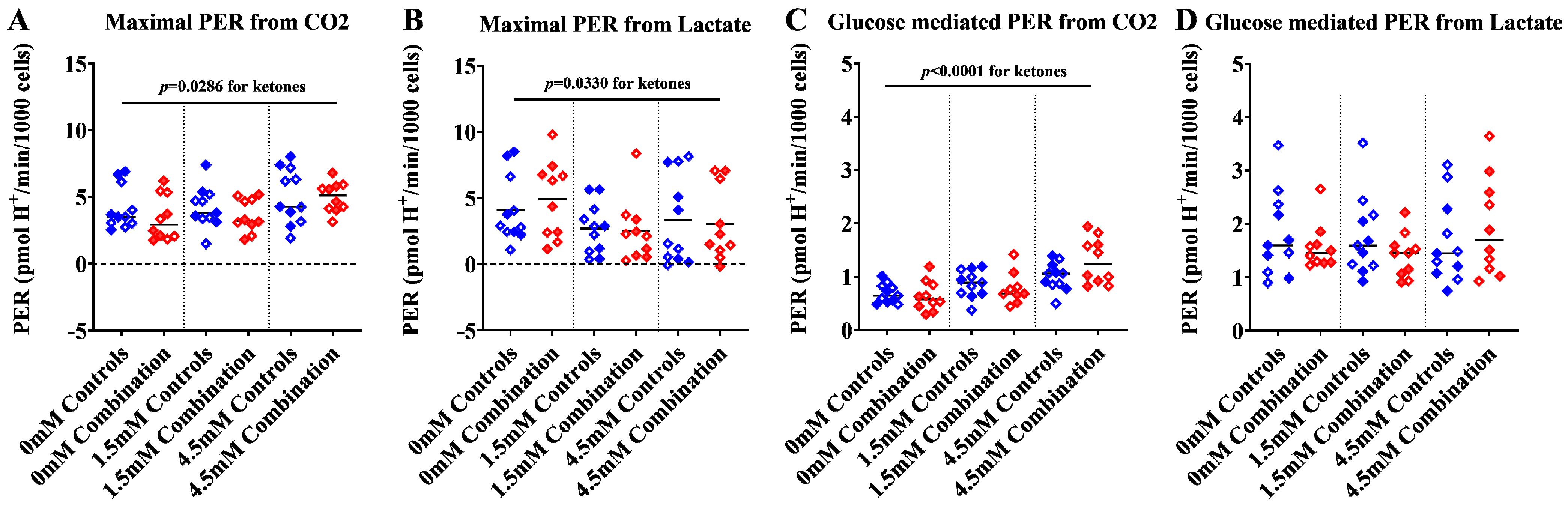
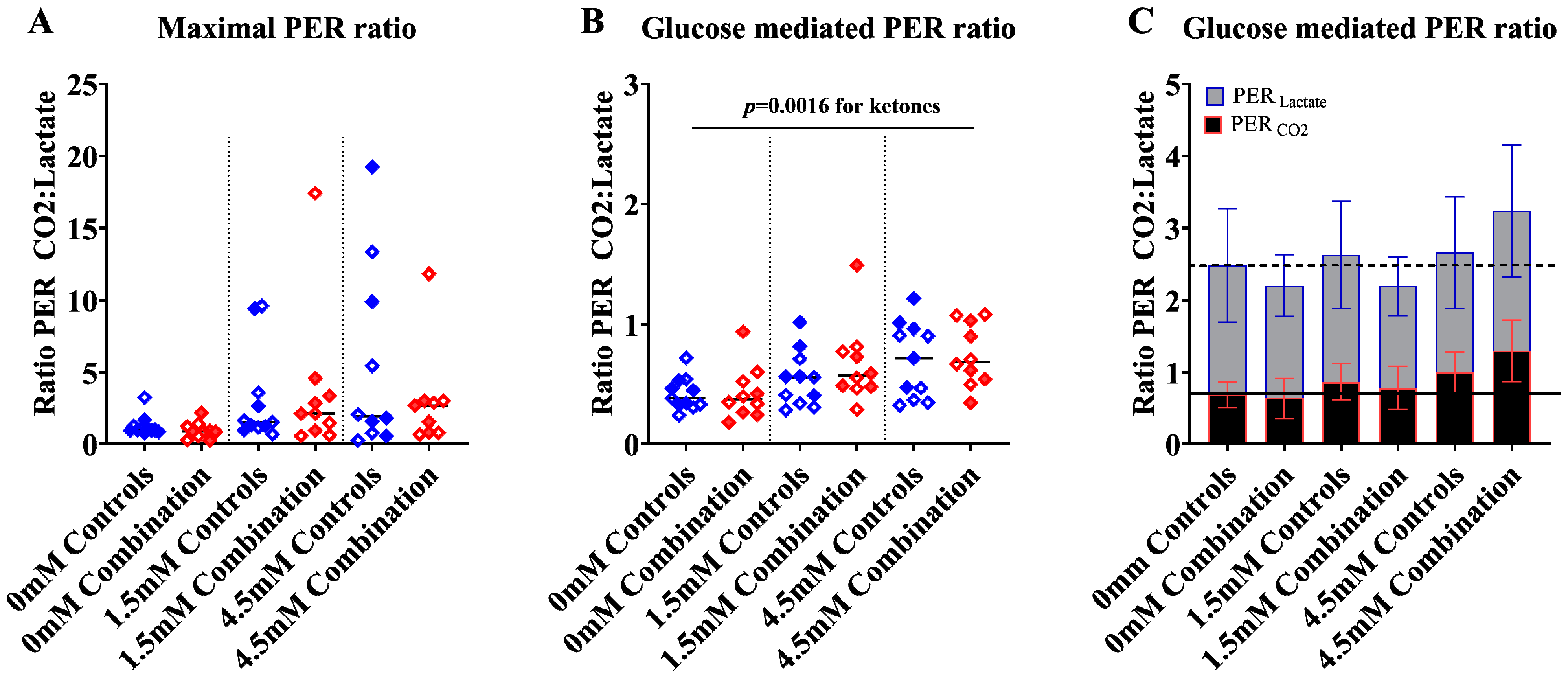
2.1.3. Ketones Improve Proton Efflux Rate (PER) in Diabetes + HFD-Exposed NRCM
2.1.4. Ketones may Enhance Fuel Flexibility More Robustly in Male NRCM
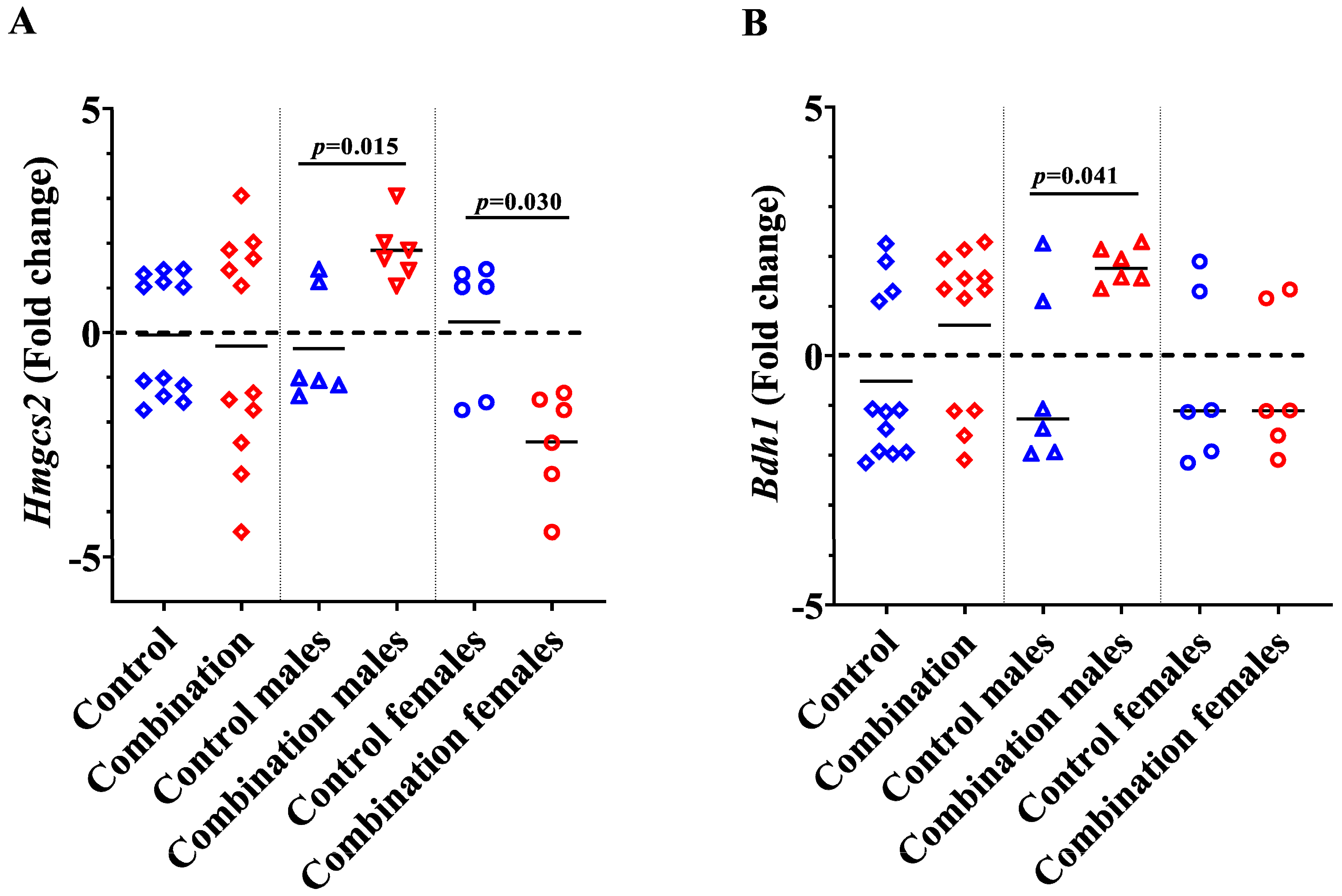
2.2. Maternal Diabetes + HFD Dysregulates Genes Involved in Myocardial Ketone Body Metabolism in a Sex-Divergent Manner
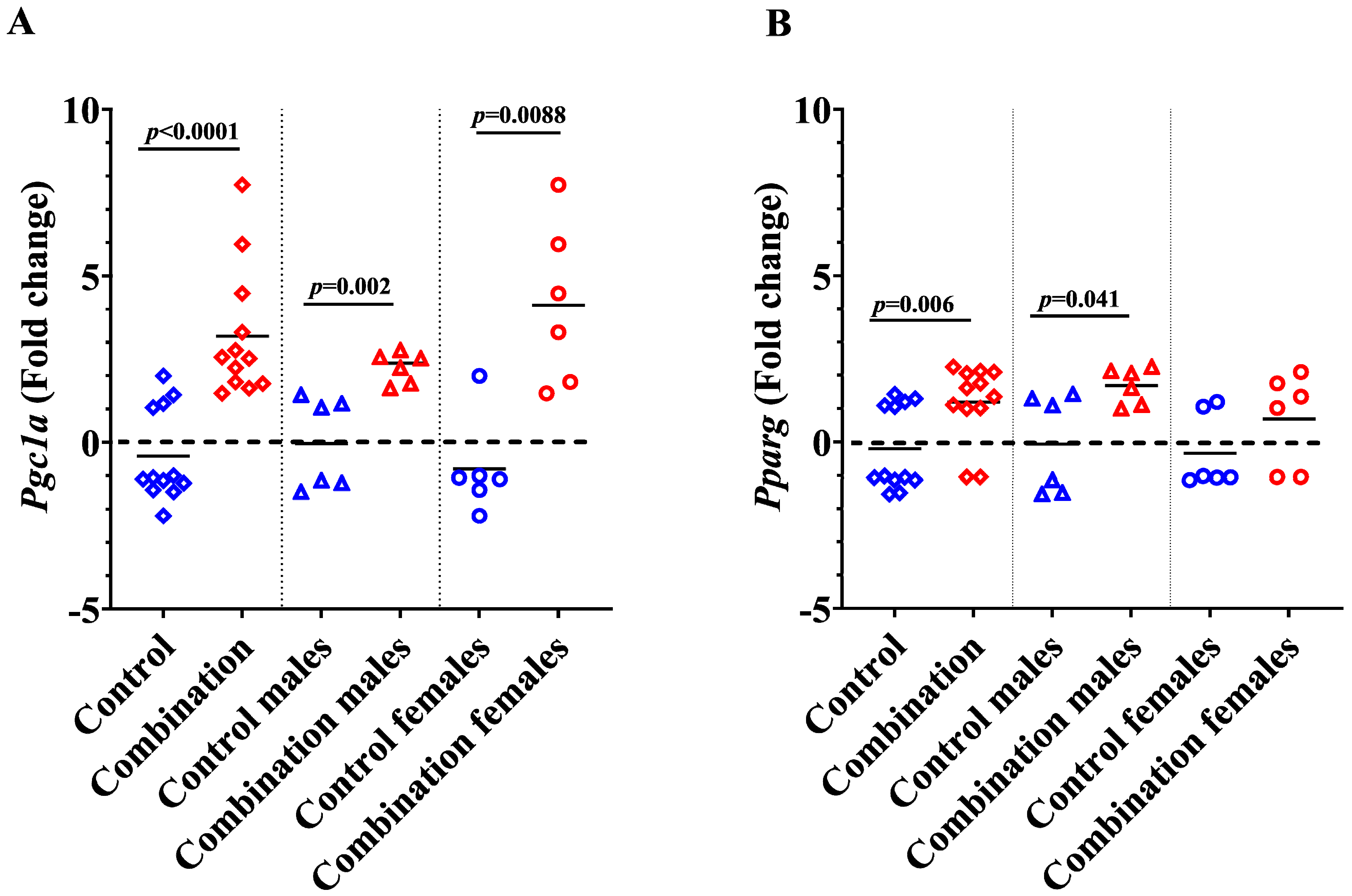
2.3. Maternal Diabetes + HFD Increases Pparg and Pgc1a Expression in Newborn Offspring Hearts
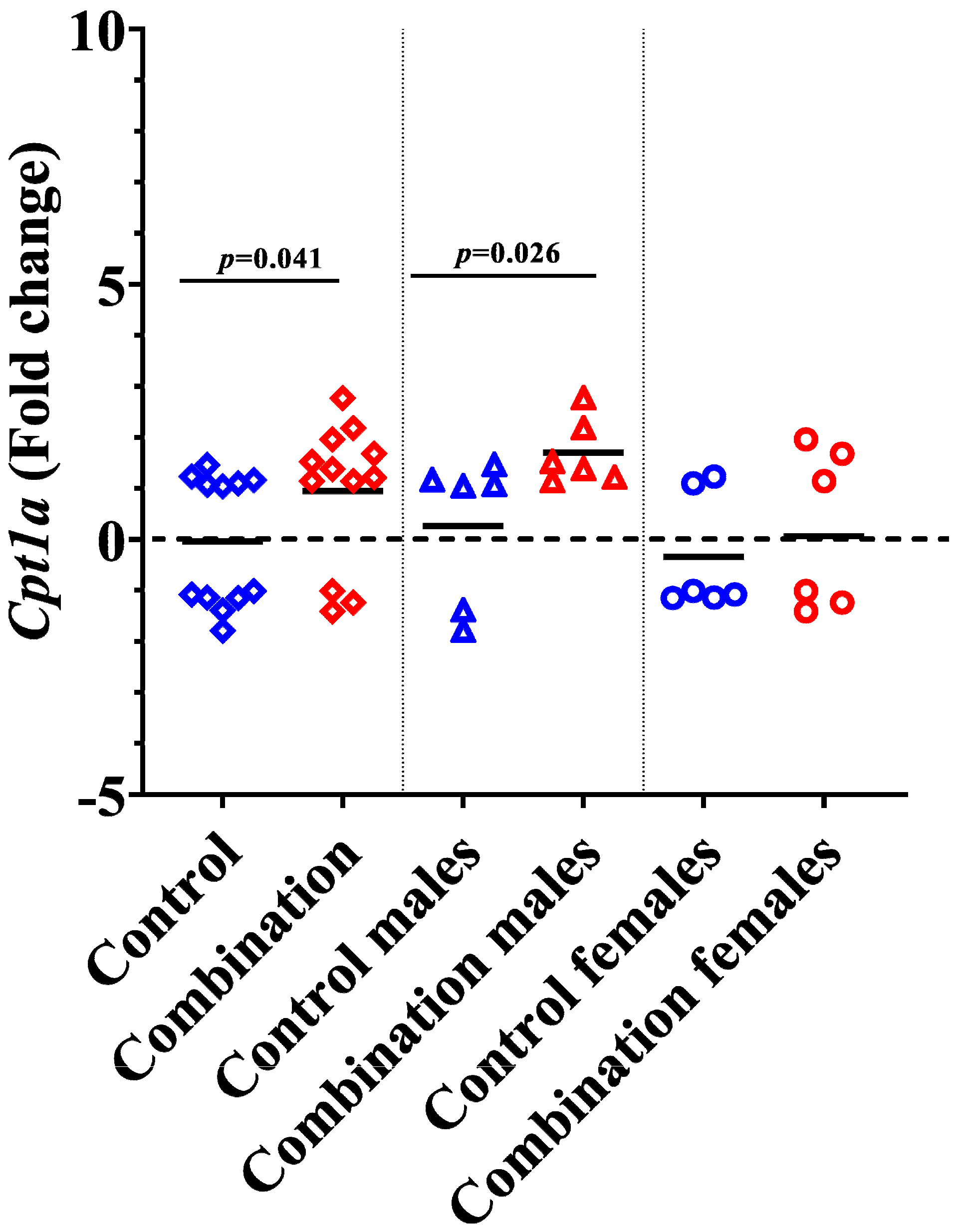
2.4. Diabetes + HFD Increases Expression of Cpt1a in the Neonatal Heart
3. Discussion
4. Materials and Methods
4.1. Animal Care
4.2. Isolation of Neonatal Rat Cardiomyocytes (NRCM)
4.3. Ketone Stress Test (KST) in Isolated NRCM
4.4. Quantitative Real Time PCR
4.5. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Freinkel, N. Banting Lecture 1980: Of pregnancy and progeny. Diabetes 1980, 29, 1023–1035. [Google Scholar] [CrossRef] [PubMed]
- Srinivasan, M.; Katewa, S.D.; Palaniyappan, A.; Pandya, J.D.; Patel, M.S. Maternal high-fat diet consumption results in fetal malprogramming predisposing to the onset of metabolic syndrome-like phenotype in adulthood. Am. J. Physiol. Endocrinol. Metab. 2006, 291, E792–E799. [Google Scholar] [CrossRef]
- Cerf, M.E.; Louw, J. High fat programming induces glucose intolerance in weanling Wistar rats. Horm. Metab. Res. 2010, 42, 307–310. [Google Scholar] [CrossRef] [PubMed]
- Ullmo, S.; Vial, Y.; Di Bernardo, S.; Roth-Kleiner, M.; Mivelaz, Y.; Sekarski, N.; Ruiz, J.; Meijboom, E.J. Pathologic ventricular hypertrophy in the offspring of diabetic mothers: A retrospective study. Eur. Heart J. 2007, 28, 1319–1325. [Google Scholar] [CrossRef]
- Zablah, J.E.; Gruber, D.; Stoffels, G.; Cabezas, E.G.; Hayes, D.A. Subclinical Decrease in Myocardial Function in Asymptomatic Infants of Diabetic Mothers: A Tissue Doppler Study. Pediatr. Cardiol. 2017, 38, 801–806. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.; Arah, O.A.; Liew, Z.; Cnattingius, S.; Olsen, J.; Sorensen, H.T.; Qin, G.; Li, J. Maternal diabetes during pregnancy and early onset of cardiovascular disease in offspring: Population based cohort study with 40 years of follow-up. BMJ 2019, 367, l6398. [Google Scholar] [CrossRef] [PubMed]
- Eriksson, J.G.; Sandboge, S.; Salonen, M.K.; Kajantie, E.; Osmond, C. Long-term consequences of maternal overweight in pregnancy on offspring later health: Findings from the Helsinki Birth Cohort Study. Ann. Med. 2014, 46, 434–438. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.K.; Raja, E.A.; Lee, A.J.; Bhattacharya, S.; Bhattacharya, S.; Norman, J.E.; Reynolds, R.M. Maternal Obesity During Pregnancy Associates With Premature Mortality and Major Cardiovascular Events in Later Life. Hypertension 2015, 66, 938–944. [Google Scholar] [CrossRef]
- Mdaki, K.S.; Larsen, T.D.; Wachal, A.L.; Schimelpfenig, M.D.; Weaver, L.J.; Dooyema, S.D.; Louwagie, E.J.; Baack, M.L. Maternal high-fat diet impairs cardiac function in offspring of diabetic pregnancy through metabolic stress and mitochondrial dysfunction. Am. J. Physiol. Heart Circ. Physiol. 2016, 310, H681–H692. [Google Scholar] [CrossRef]
- Louwagie, E.J.; Larsen, T.D.; Wachal, A.L.; Gandy, T.C.T.; Baack, M.L. Mitochondrial Transfer Improves Cardiomyocyte Bioenergetics and Viability in Male Rats Exposed to Pregestational Diabetes. Int. J. Mol. Sci. 2021, 22, 2382. [Google Scholar] [CrossRef]
- Louwagie, E.J.; Larsen, T.D.; Wachal, A.L.; Baack, M.L. Placental lipid processing in response to a maternal high-fat diet and diabetes in rats. Pediatr. Res. 2017, 83, 712–722. [Google Scholar] [CrossRef] [PubMed]
- Larsen, T.D.; Sabey, K.H.; Knutson, A.J.; Gandy, T.C.T.; Louwagie, E.J.; Lauterboeck, L.; Mdaki, K.S.; Baack, M.L. Diabetic Pregnancy and Maternal High-Fat Diet Impair Mitochondrial Dynamism in the Developing Fetal Rat Heart by Sex-Specific Mechanisms. Int. J. Mol. Sci. 2019, 20, 3090. [Google Scholar] [CrossRef] [PubMed]
- Louwagie, E.J.; Larsen, T.D.; Wachal, A.L.; Gandy, T.C.T.; Eclov, J.A.; Rideout, T.C.; Kern, K.A.; Cain, J.T.; Anderson, R.H.; Mdaki, K.S.; et al. Age and Sex Influence Mitochondria and Cardiac Health in Offspring Exposed to Maternal Glucolipotoxicity. iScience 2020, 23, 101746. [Google Scholar] [CrossRef] [PubMed]
- Mdaki, K.S.; Larsen, T.D.; Weaver, L.J.; Baack, M.L. Age Related Bioenergetics Profiles in Isolated Rat Cardiomyocytes Using Extracellular Flux Analyses. PLoS ONE 2016, 11, e0149002. [Google Scholar] [CrossRef] [PubMed]
- Qian, M.; Wu, N.; Li, L.; Yu, W.; Ouyang, H.; Liu, X.; He, Y.; Al-Mureish, A. Effect of Elevated Ketone Body on Maternal and Infant Outcome of Pregnant Women with Abnormal Glucose Metabolism During Pregnancy. Diabetes Metab. Syndr. Obes. 2020, 13, 4581–4588. [Google Scholar] [CrossRef]
- Aubert, G.; Martin, O.J.; Horton, J.L.; Lai, L.; Vega, R.B.; Leone, T.C.; Koves, T.; Gardell, S.J.; Krüger, M.; Hoppel, C.L.; et al. The Failing Heart Relies on Ketone Bodies as a Fuel. Circulation 2016, 133, 698–705. [Google Scholar] [CrossRef]
- Cotter, D.G.; Schugar, R.C.; Crawford, P.A. Ketone body metabolism and cardiovascular disease. Am. J. Physiol. Heart Circ. Physiol. 2013, 304, H1060–H1076. [Google Scholar] [CrossRef]
- Bayeva, M.; Sawicki, K.T.; Ardehali, H. Taking diabetes to heart--deregulation of myocardial lipid metabolism in diabetic cardiomyopathy. J. Am. Heart Assoc. 2013, 2, e000433. [Google Scholar] [CrossRef]
- Fukushima, A.; Lopaschuk, G.D. Cardiac fatty acid oxidation in heart failure associated with obesity and diabetes. Biochim. Biophys. Acta 2016, 1861, 1525–1534. [Google Scholar] [CrossRef]
- Bugger, H.; Abel, E.D. Molecular mechanisms of diabetic cardiomyopathy. Diabetologia 2014, 57, 660–671. [Google Scholar] [CrossRef] [Green Version]
- Bedi, K.C., Jr.; Snyder, N.W.; Brandimarto, J.; Aziz, M.; Mesaros, C.; Worth, A.J.; Wang, L.L.; Javaheri, A.; Blair, I.A.; Margulies, K.B.; et al. Evidence for Intramyocardial Disruption of Lipid Metabolism and Increased Myocardial Ketone Utilization in Advanced Human Heart Failure. Circulation 2016, 133, 706–716. [Google Scholar] [CrossRef] [PubMed]
- Brahma, M.K.; Ha, C.M.; Pepin, M.E.; Mia, S.; Sun, Z.; Chatham, J.C.; Habegger, K.M.; Abel, E.D.; Paterson, A.J.; Young, M.E.; et al. Increased Glucose Availability Attenuates Myocardial Ketone Body Utilization. J. Am. Heart Assoc. 2020, 9, e013039. [Google Scholar] [CrossRef] [PubMed]
- Preston, C.C.; Larsen, T.D.; Eclov, J.A.; Louwagie, E.J.; Gandy, T.C.T.; Faustino, R.S.; Baack, M.L. Maternal High Fat Diet and Diabetes Disrupts Transcriptomic Pathways That Regulate Cardiac Metabolism and Cell Fate in Newborn Rat Hearts. Front. Endocrinol. 2020, 11, 570846. [Google Scholar] [CrossRef]
- Lin, J.; Handschin, C.; Spiegelman, B.M. Metabolic control through the PGC-1 family of transcription coactivators. Cell Metab. 2005, 1, 361–370. [Google Scholar] [CrossRef] [PubMed]
- Di, W.; Lv, J.; Jiang, S.; Lu, C.; Yang, Z.; Ma, Z.; Hu, W.; Yang, Y.; Xu, B. PGC-1: The Energetic Regulator in Cardiac Metabolism. Curr. Issues Mol. Biol. 2018, 28, 29–46. [Google Scholar] [CrossRef] [PubMed]
- Wagner, N.; Wagner, K.D. The Role of PPARs in Disease. Cells 2020, 9, 2367. [Google Scholar] [CrossRef] [PubMed]
- Sikder, K.; Shukla, S.K.; Patel, N.; Singh, H.; Rafiq, K. High Fat Diet Upregulates Fatty Acid Oxidation and Ketogenesis via Intervention of PPAR-γ. Cell. Physiol. Biochem. 2018, 48, 1317–1331. [Google Scholar] [CrossRef]
- Casals, N.; Zammit, V.; Herrero, L.; Fadó, R.; Rodríguez-Rodríguez, R.; Serra, D. Carnitine palmitoyltransferase 1C: From cognition to cancer. Prog. Lipid Res. 2016, 61, 134–148. [Google Scholar] [CrossRef] [PubMed]
- Schreurs, M.; Kuipers, F.; van der Leij, F.R. Regulatory enzymes of mitochondrial beta-oxidation as targets for treatment of the metabolic syndrome. Obes. Rev. Off. J. Int. Assoc. Study Obes. 2010, 11, 380–388. [Google Scholar] [CrossRef]
- Lopaschuk, G.D.; Karwi, Q.G.; Tian, R.; Wende, A.R.; Abel, E.D. Cardiac Energy Metabolism in Heart Failure. Circ. Res. 2021, 128, 1487–1513. [Google Scholar] [CrossRef]
- Chu, Y.; Zhang, C.; Xie, M. Beta-Hydroxybutyrate, Friend or Foe for Stressed Hearts. Front. Aging 2021, 2, 681513. [Google Scholar] [CrossRef] [PubMed]
- Lopaschuk, G.D.; Ussher, J.R.; Folmes, C.D.; Jaswal, J.S.; Stanley, W.C. Myocardial fatty acid metabolism in health and disease. Physiol. Rev. 2010, 90, 207–258. [Google Scholar] [CrossRef] [PubMed]
- Persad, K.L.; Lopaschuk, G.D. Energy Metabolism on Mitochondrial Maturation and Its Effects on Cardiomyocyte Cell Fate. Front. Cell Dev. Biol. 2022, 10, 886393. [Google Scholar] [CrossRef]
- Mishra, P.K. Why the diabetic heart is energy inefficient: A ketogenesis and ketolysis perspective. Am. J. Physiology. Heart Circ. Physiol. 2021, 321, H751–H755. [Google Scholar] [CrossRef] [PubMed]
- Kolwicz, S.C., Jr. Ketone Body Metabolism in the Ischemic Heart. Front. Cardiovasc. Med. 2021, 8, 789458. [Google Scholar] [CrossRef]
- Vijay, V.; Han, T.; Moland, C.L.; Kwekel, J.C.; Fuscoe, J.C.; Desai, V.G. Sexual dimorphism in the expression of mitochondria-related genes in rat heart at different ages. PloS ONE 2015, 10, e0117047. [Google Scholar] [CrossRef]
- Rattanasopa, C.; Phungphong, S.; Wattanapermpool, J.; Bupha-Intr, T. Significant role of estrogen in maintaining cardiac mitochondrial functions. J. Steroid Biochem. Mol. Biol. 2015, 147, 1–9. [Google Scholar] [CrossRef]
- Jiang, S.; Teague, A.M.; Tryggestad, J.B.; Aston, C.E.; Lyons, T.; Chernausek, S.D. Effects of maternal diabetes and fetal sex on human placenta mitochondrial biogenesis. Placenta 2017, 57, 26–32. [Google Scholar] [CrossRef]
- Gyllenhammer, L.E.; Entringer, S.; Buss, C.; Wadhwa, P.D. Developmental programming of mitochondrial biology: A conceptual framework and review. Proc. Biol. Sci. 2020, 287, 20192713. [Google Scholar] [CrossRef]
- Groban, L.; Tran, Q.K.; Ferrario, C.M.; Sun, X.; Cheng, C.P.; Kitzman, D.W.; Wang, H.; Lindsey, S.H. Female Heart Health: Is GPER the Missing Link? Front. Endocrinol. 2019, 10, 919. [Google Scholar] [CrossRef] [Green Version]
- Montaigne, D.; Butruille, L.; Staels, B. PPAR control of metabolism and cardiovascular functions. Nat. Rev. Cardiol. 2021, 18, 809–823. [Google Scholar] [CrossRef] [PubMed]
- Costantino, S.; Akhmedov, A.; Melina, G.; Mohammed, S.A.; Othman, A.; Ambrosini, S.; Wijnen, W.J.; Sada, L.; Ciavarella, G.M.; Liberale, L.; et al. Obesity-induced activation of JunD promotes myocardial lipid accumulation and metabolic cardiomyopathy. Eur. Heart J. 2019, 40, 97–1008. [Google Scholar] [CrossRef] [PubMed]
- Son, N.H.; Park, T.S.; Yamashita, H.; Yokoyama, M.; Huggins, L.A.; Okajima, K.; Homma, S.; Szabolcs, M.J.; Huang, L.S.; Goldberg, I.J. Cardiomyocyte expression of PPARgamma leads to cardiac dysfunction in mice. J. Clin. Investig. 2007, 117, 2791–2801. [Google Scholar] [CrossRef] [PubMed]
- Oka, S.I.; Sabry, A.D.; Cawley, K.M.; Warren, J.S. Multiple Levels of PGC-1α Dysregulation in Heart Failure. Front. Cardiovasc. Med. 2020, 7, 2. [Google Scholar] [CrossRef]
- Jeong, S.; Yoon, M. 17β-Estradiol inhibition of PPARγ-induced adipogenesis and adipocyte-specific gene expression. Acta Pharmacol. Sin 2011, 32, 230–238. [Google Scholar] [CrossRef]
- Yepuru, M.; Eswaraka, J.; Kearbey, J.D.; Barrett, C.M.; Raghow, S.; Veverka, K.A.; Miller, D.D.; Dalton, J.T.; Narayanan, R. Estrogen receptor-{beta}-selective ligands alleviate high-fat diet- and ovariectomy-induced obesity in mice. J. Biol. Chem. 2010, 285, 31292–31303. [Google Scholar] [CrossRef]
- Seeland, U.; Regitz-Zagrosek, V. Genes and hormones: Sex differences in myocardial hypertrophy. Clin. Res. Cardiol. Suppl. 2013, 8, 6–13. [Google Scholar] [CrossRef]
- Cook, G.A.; Edwards, T.L.; Jansen, M.S.; Bahouth, S.W.; Wilcox, H.G.; Park, E.A. Differential regulation of carnitine palmitoyltransferase-I gene isoforms (CPT-I alpha and CPT-I beta) in the rat heart. J. Mol. Cell. Cardiol. 2001, 33, 317–329. [Google Scholar] [CrossRef]
- Song, S.; Zhang, Y.; Ma, K.; Jackson-Hayes, L.; Lavrentyev, E.N.; Cook, G.A.; Elam, M.B.; Park, E.A. Peroxisomal proliferator activated receptor gamma coactivator (PGC-1alpha) stimulates carnitine palmitoyltransferase I (CPT-Ialpha) through the first intron. Biochim. Biophys. Acta 2004, 1679, 164–173. [Google Scholar] [CrossRef]
- Baack, M.L.; Wang, C.; Hu, S.; Segar, J.L.; Norris, A.W. Hyperglycemia induces embryopathy, even in the absence of systemic maternal diabetes: An in vivo test of the fuel mediated teratogenesis hypothesis. Reprod. Toxicol. 2014, 46, 129–136. [Google Scholar] [CrossRef] [Green Version]
- Sussman, D.; van Eede, M.; Wong, M.D.; Adamson, S.L.; Henkelman, M. Effects of a ketogenic diet during pregnancy on embryonic growth in the mouse. BMC Pregnancy Childbirth 2013, 13, 109. [Google Scholar] [CrossRef]
- Nielsen, R.; Møller, N.; Gormsen, L.C.; Tolbod, L.P.; Hansson, N.H.; Sorensen, J.; Harms, H.J.; Frøkiær, J.; Eiskjaer, H.; Jespersen, N.R.; et al. Cardiovascular Effects of Treatment With the Ketone Body 3-Hydroxybutyrate in Chronic Heart Failure Patients. Circulation 2019, 139, 2129–2141. [Google Scholar] [CrossRef] [PubMed]
- Murashige, D.; Jang, C.; Neinast, M.; Edwards, J.J.; Cowan, A.; Hyman, M.C.; Rabinowitz, J.D.; Frankel, D.S.; Arany, Z. Comprehensive quantification of fuel use by the failing and nonfailing human heart. Science 2020, 370, 364–368. [Google Scholar] [CrossRef]
- Taylor, S.I.; Blau, J.E.; Rother, K.I. SGLT2 Inhibitors May Predispose to Ketoacidosis. J. Clin. Endocrinol. Metab. 2015, 100, 2849–2852. [Google Scholar] [CrossRef]
- Ferrannini, E.; Baldi, S.; Frascerra, S.; Astiarraga, B.; Heise, T.; Bizzotto, R.; Mari, A.; Pieber, T.R.; Muscelli, E. Shift to Fatty Substrate Utilization in Response to Sodium-Glucose Cotransporter 2 Inhibition in Subjects Without Diabetes and Patients With Type 2 Diabetes. Diabetes 2016, 65, 1190–1195. [Google Scholar] [CrossRef] [PubMed]
- Baack, M.L.; Forred, B.J.; Larsen, T.D.; Jensen, D.N.; Wachal, A.L.; Khan, M.A.; Vitiello, P.F. Consequences of a Maternal High-Fat Diet and Late Gestation Diabetes on the Developing Rat Lung. PLoS ONE 2016, 11, e0160818. [Google Scholar] [CrossRef] [PubMed]
- Mookerjee, S.A.; Gerencser, A.A.; Nicholls, D.G.; Brand, M.D. Quantifying intracellular rates of glycolytic and oxidative ATP production and consumption using extracellular flux measurements. J. Biol. Chem. 2017, 292, 7189–7207. [Google Scholar] [CrossRef] [Green Version]










Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ayyappan, P.; Larsen, T.D.; Gandy, T.C.T.; Louwagie, E.J.; Baack, M.L. Impact of Prenatal Exposure to Maternal Diabetes and High-Fat Diet on Postnatal Myocardial Ketone Body Metabolism in Rats. Int. J. Mol. Sci. 2023, 24, 3684. https://doi.org/10.3390/ijms24043684
Ayyappan P, Larsen TD, Gandy TCT, Louwagie EJ, Baack ML. Impact of Prenatal Exposure to Maternal Diabetes and High-Fat Diet on Postnatal Myocardial Ketone Body Metabolism in Rats. International Journal of Molecular Sciences. 2023; 24(4):3684. https://doi.org/10.3390/ijms24043684
Chicago/Turabian StyleAyyappan, Prathapan, Tricia D. Larsen, Tyler C. T. Gandy, Eli J. Louwagie, and Michelle L. Baack. 2023. "Impact of Prenatal Exposure to Maternal Diabetes and High-Fat Diet on Postnatal Myocardial Ketone Body Metabolism in Rats" International Journal of Molecular Sciences 24, no. 4: 3684. https://doi.org/10.3390/ijms24043684