

Autophagy and the Insulin-like Growth Factor (IGF) System in Colonic Cells: Implications for Colorectal Neoplasia

Abstract
:1. Introduction
2. Autophagy in Colorectal Cancer—Clinical Implications
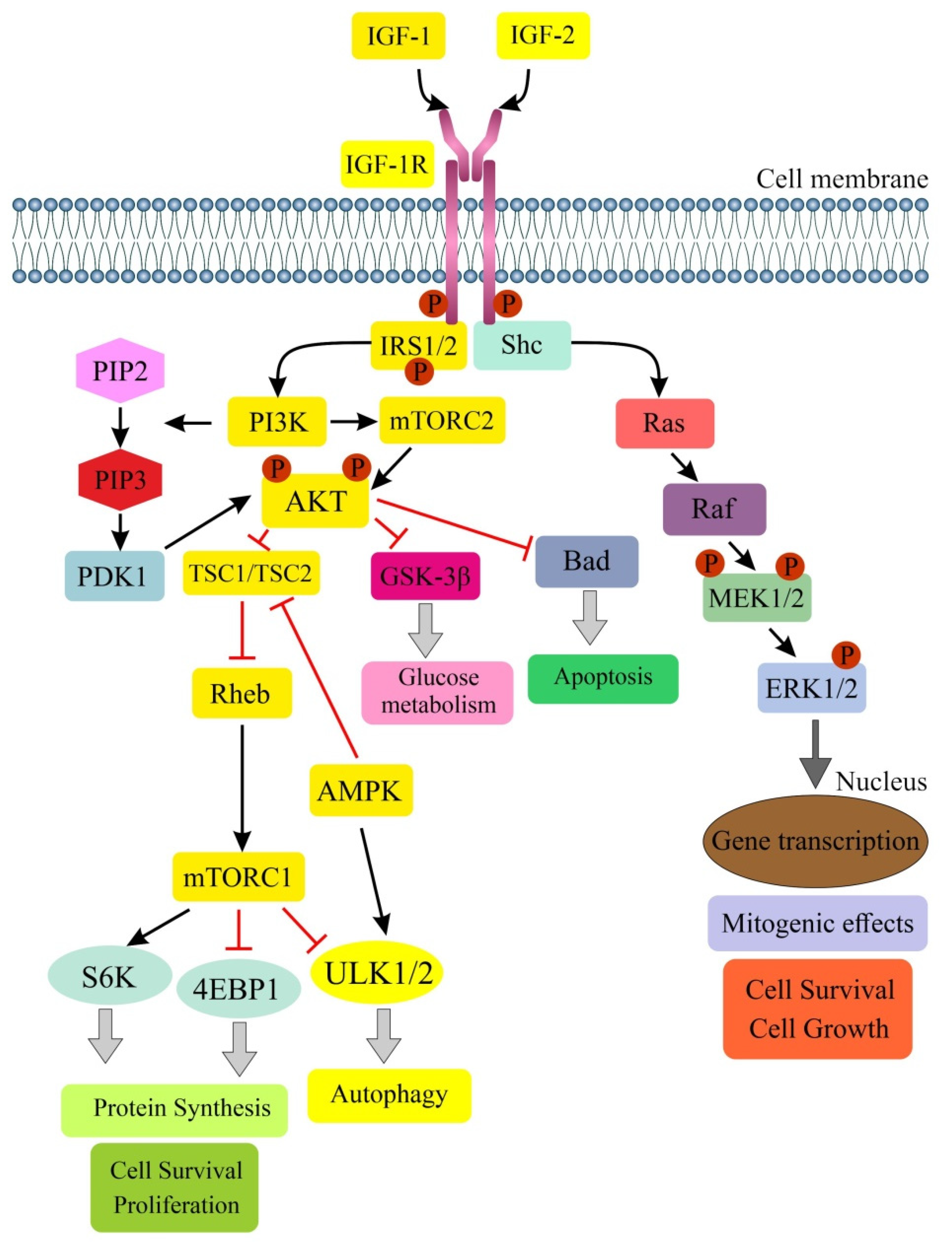
3. IGF Signaling and Colorectal Cancer—A Brief Summary
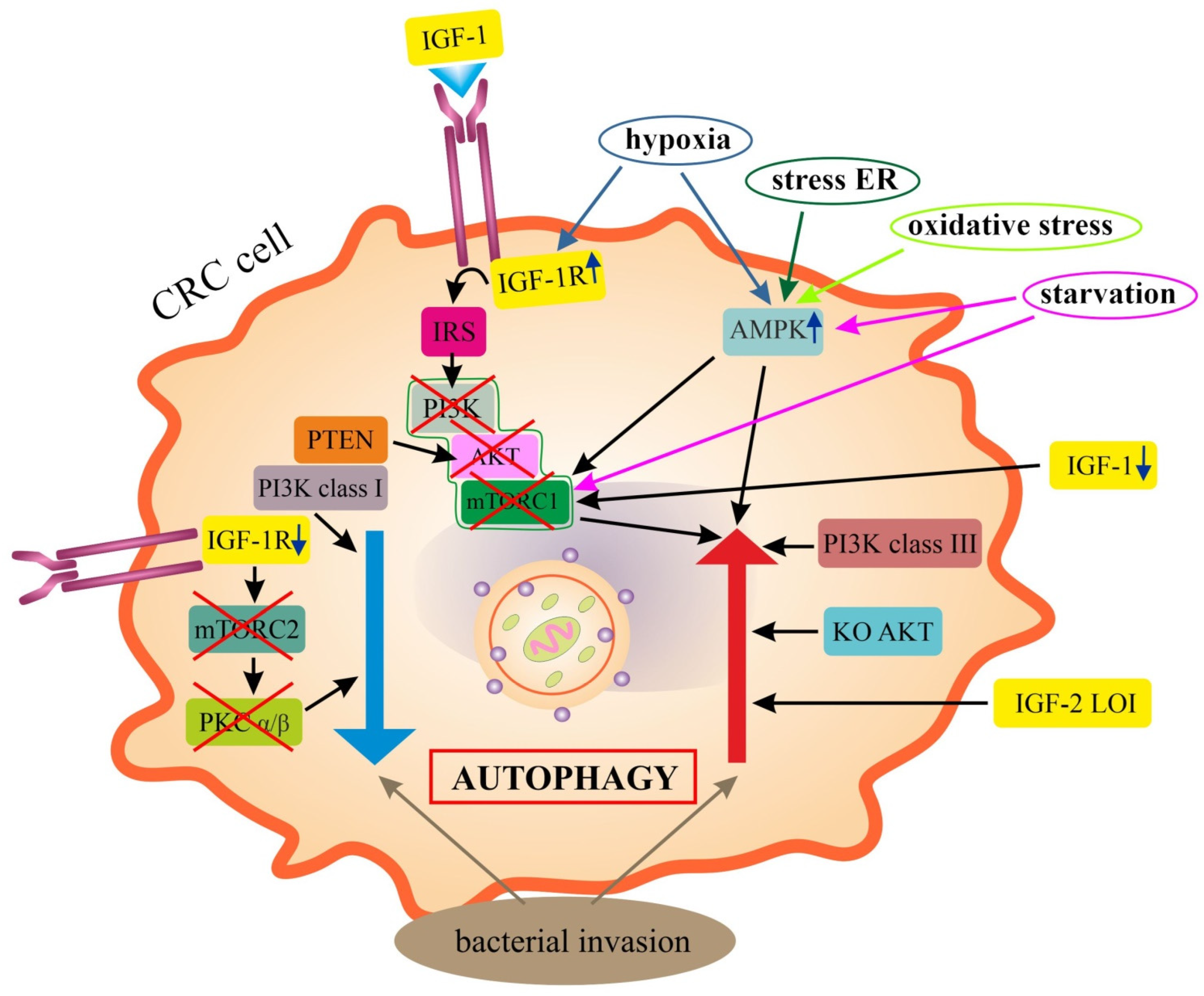
4. Role of IGF Signaling in Regulation of Autophagy
5. Autophagy in Normal Large Intestine Epithelial Cells—Implication for Colorectal Neoplasia
5.1. Colonocytes
5.1.1. Autophagy in Colonocytes
5.1.2. Colonocytes and IGF-1 System
5.2. Goblet Cells
5.2.1. Autophagy in Goblet Cells
5.2.2. Goblet Cells and IGF-1 System
5.3. Intestinal Stem Cells (ISCs)
5.3.1. Autophagy in Intestinal Stem Cells
5.3.2. Intestinal Stem Cells and IGF System
5.4. Paneth Cells and Paneth-like Cells
5.4.1. Autophagy in Paneth Cells
5.4.2. Paneth-like Cells and IGF System
6. Cells of the Tumor Microenvironment
6.1. Epithelial Colorectal Cancer Cells (Epithelial CRC Cells)
IGF Signaling in Autophagy of CRC Cells
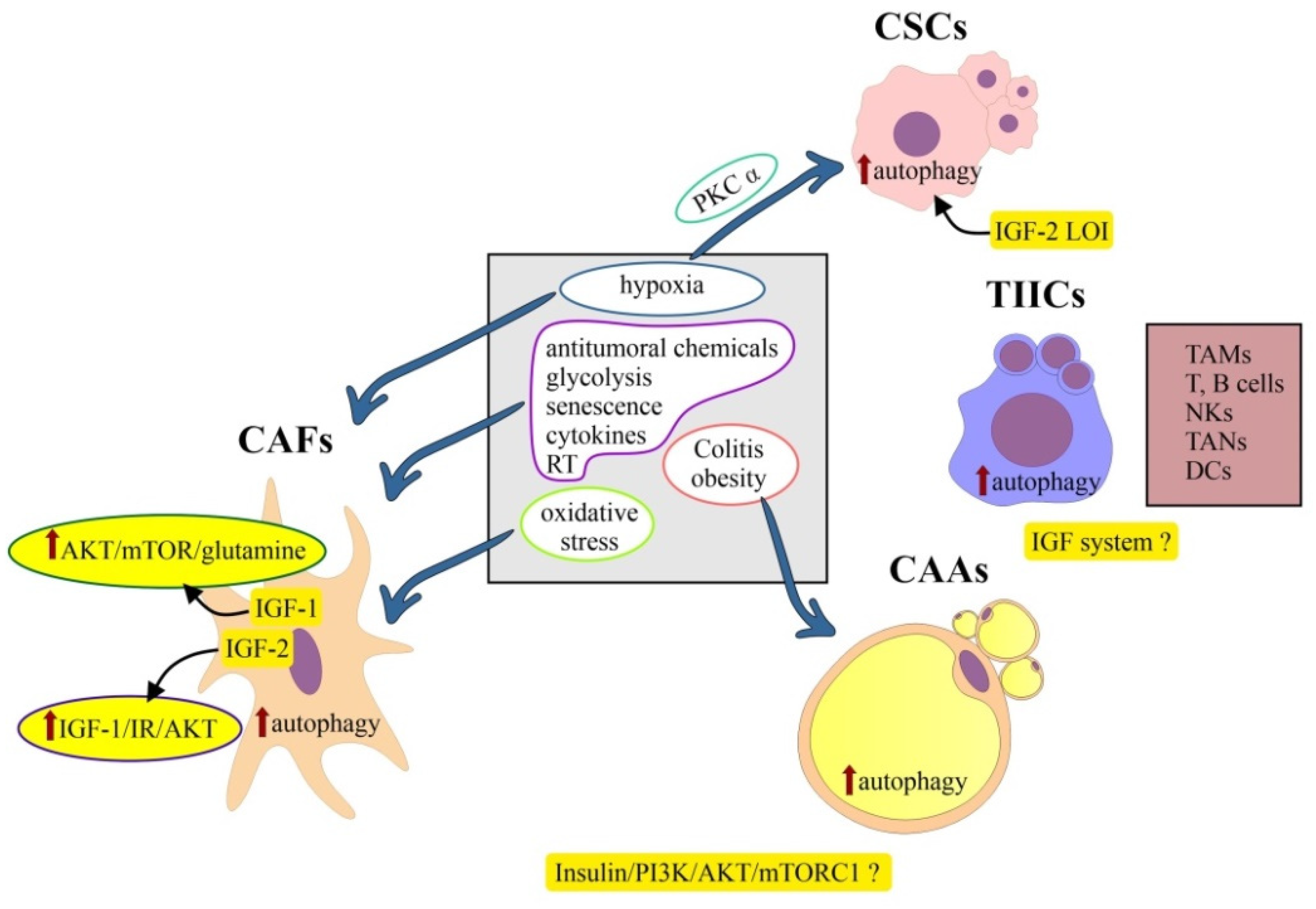
6.2. Colorectal Cancer Stem Cells
6.2.1. Colorectal Cancer Stem Cells and Autophagy
6.2.2. Colorectal Cancer Stem Cells and IGF System
6.3. Cancer-Associated Adipocytes (CAAs)
6.4. Tumor-Infiltrating Immune Cells (TIICs)
6.5. Cancer-Associated Fibroblasts (CAFs)
7. Autophagy and IGF System: Implications for Therapy in CRC
8. Concluding Remarks and Future Directions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| AKT/PKB | serine/threonine kinase/protein kinase B |
| AMBRA1 | Autophagy and beclin 1 regulator 1 |
| AMPK | 5′ Adenosine-monophosphate-activated protein kinase |
| APC | Adenomatous polyposis coli |
| ATG | Autophagy-related genes or proteins |
| BAX | Bcl-2-associated X protein |
| BECN1 | Beclin1, a mammalian ortholog of the yeast Atg6 |
| BEST4 | Bestrophin 4 |
| BNIP3 | Bcl-2-interacting protein 3 |
| BNIP3L | Bcl-2/adenovirus E1B 19kDa protein-interacting protein 3-like |
| BRAF | Proto-oncogene B-Raf; encodes protein called B-Raf |
| CCL2/MCP-1 | Chemokine (C-C Motif) ligand 2/monocyte chemoattractant protein 1 |
| CCL8/MCP-2 | Chemokine (C-C Motif) ligand 8/monocyte chemoattractant protein 2 |
| C/E/I/M(SCs) | Cancer/embryonic/intestinal/mesenchymal stem cells |
| CD | Crohn’s disease |
| CI | Confidence interval |
| CIMP | CpG island methylator phenotype |
| CIN | Chromosomal instability |
| c-MET | Tyrosine-protein kinase Met/hepatocyte growth factor receptor (HGFR) |
| c-Myc | Proto-oncogene protein; transcription factor |
| CQ | Chloroquine |
| CRC | Colorectal cancer |
| CSCs | Cancer stem cells |
| c-Src | Src proto-oncogene, non-receptor tyrosine kinase |
| DECP1 | Double FYVE-containing protein 1 or ZFYVE1 protein |
| EGF/EGFR | Epidermal growth factor/EGF receptor (HER1 in humans) |
| eIF4A3 | Eukaryotic initiation factor 4A-III |
| ERK1/2 | Extracellular signal-regulated kinase 1/2 |
| FAK | Focal adhesion kinase or protein tyrosine kinase 2 |
| FOXO | Forkhead box protein class O |
| GAPs | Goblet cell-associated antigen passage |
| GH/GH-R | Growth hormone/GH-receptor |
| GLP-2 | Glucagon-like peptide 2 |
| GNPTAB | N-acetylglucosamine-1-phosphotransferase subunits alpha/beta |
| GSK-3β | Glycogen synthase kinase-3β |
| HIF-1 | Hypoxia-inducible factor 1 |
| IBD | Inflammatory bowel diseases |
| IECs | Intestinal epithelial cells |
| IGF-1, -2 | Insulin growth factor 1, -2 |
| IGF-1R, -2R | IGF receptor type I, type II |
| IGF-BPs | IGF-binding proteins |
| IR-A | Isoform A of the insulin receptor |
| IRS-1/2 | Insulin receptor substrate 1/2 |
| JAK | Janus kinase |
| KLF4 | Gut-enriched Kruppel-like factor 4; zinc-finger transcription factor |
| KRAS | Kirsten rat sarcoma virus; encodes protein called K-Ras |
| LAMP2 | Lysosome-associated membrane protein 2 |
| (M)LC3B/MAP2 | Microtubule-associated protein 1A/1B light chain 3B/protein 2 |
| LKB1 | Liver kinase B1; serine/threonine protein kinase (STK11) |
| lncRNAs | Long non-coding RNAs |
| LOI | Loss of imprinting |
| MAPK | Mitogen-activated protein kinase |
| MEFs | Mouse embryonic fibroblasts |
| MEK1/2 | Mitogen-activated protein kinase (MAP2K, MEK, MAPKK) |
| MetS | Metabolic syndrome |
| MOI | Maintenance of imprinting |
| MSI-H | High microsatellite instability |
| MSS | Microsatellite stable |
| mTORC1/2 | Mammalian target of rapamycin complex 1/complex 2 |
| NADPH | Nicotinamide adenine dinucleotide phosphate oxidase |
| NANOG | Homebox protein, transcription factor |
| NF-κB | Nuclear factor-kappa B |
| NRF-1/2 | Nuclear respiratory factor 1/2 |
| OCT-4 | Octamer-binding transcription factor 4 |
| OS | Overall survival |
| PARP1 | Poly [ADP-ribose] polymerase 1 |
| PGDF | Platelet-derived growth factor |
| PI3K | Phosphatidylinositol-3-kinase |
| PIK3CA | Gene encoding phosphatidylinositol-4,5-bisphosphate 3-kinase catalytic subunit alpha |
| PINK1 | PTEN-induced kinase 1 |
| PLCs | Paneth-like cells |
| PKCα/PRKCA | Protein kinase Cα |
| PPAR | Peroxisome proliferator-activated receptor |
| PSCs | Pluripotent stem cells |
| PTEN | Phosphatase and tensin homolog deleted on chromosome ten |
| Raf-1 | Proto-oncogene serine/threonine protein kinase |
| Raptor | Regulatory-associated protein of mTOR |
| RAS | Oncogene “Rat sarcoma virus” from three Ras genes: HRAS, KRAS and NRAS |
| ROS | Reactive oxygen species |
| SATB2 | SATB homeobox 2; DNA-binding protein |
| SGK-1 | Serine/threonine-protein kinase/serum and glucocorticoid-regulated kinase 1 |
| SHC | SHC-transforming protein 1 encoded by the SHC gene |
| SIRT1 | NAD-dependent deacetylase sirtuin 1, a mammalian homologue of silent information regulator 2 of the yeast Saccharomyces cerevisiae |
| SLITs/ROBOs | SLIT glycoproteins and their roundabout receptors |
| SNP | Single nucleotide polymorphism |
| SNX9, -18,-33 | Sorting nexin9, -18, -33 |
| SOD3 | Superoxide dismutase 3 |
| SOX2 | Transcription factor 2, known also as sex-determining region Y (SRY)-box 2 |
| STAT3 | Signal transducer and activator of transcription 3 |
| TCGA | The cancer genome atlas |
| TGF-α, β | Tumor growth factor α, β |
| TLRs | Toll-like receptors |
| TME | Tumor microenvironment |
| TP53 | Tumor gene/protein 53 |
| UC | Ulcerative colitis |
| ULK1/2 | Unc-51 like autophagy activating kinase |
| uPA/uPAR | Urokinase-type plasminogen activator/receptor |
| Wnt | Gene wingless + integrated or int-1 |
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, R.; Singh, J.K.; Wunnava, A.; Al-Obeed, O.; Abdulla, M.; Srivastava, S.K. Emerging trends in colorectal cancer: Dysregulated signaling pathways (Review). Int. J. Mol. Med. 2021, 47, 14. [Google Scholar] [CrossRef] [PubMed]
- Stefani, C.; Miricescu, D.; Stanescu-Spinu, I.-I.; Nica, R.I.; Greabu, M.; Totan, A.R.; Jing, M. Growth Factors, PI3K/AKT/mTOR and MAPK Signaling Pathways in Colorectal Cancer Pathogenesis: Where Are We Now? Int. J. Mol. Sci. 2021, 22, 10260. [Google Scholar] [CrossRef]
- Vigneri, P.G.; Tirrò, E.; Pennisi, M.S.; Massimino, M.; Stella, S.; Romano, C.; Manzella, L. The Insulin/IGF System in Colorectal Cancer Development and Resistance to Therapy. Front. Oncol. 2015, 5, 230. [Google Scholar] [CrossRef]
- Kasprzak, A.; Kwasniewski, W.; Adamek, A.; Gozdzicka-Jozefiak, A. Insulin-like growth factor (IGF) axis in cancerogenesis. Mutat. Res. Rev. Mutat. Res. 2017, 772, 78–104. [Google Scholar] [CrossRef] [PubMed]
- Osher, E.; Macaulay, V.M. Therapeutic Targeting of the IGF Axis. Cells 2019, 8, 895. [Google Scholar] [CrossRef]
- Chen, Z.; Gao, S.; Wang, D.; Song, D.; Feng, Y. Colorectal cancer cells are resistant to anti-EGFR monoclonal antibody through adapted autophagy. Am. J. Transl. Res. 2016, 8, 1190–1196. [Google Scholar]
- Koustas, E.; Karamouzis, M.V.; Mihailidou, C.; Schizas, D.; Papavassiliou, A.G. Co-targeting of EGFR and autophagy signaling is an emerging treatment strategy in metastatic colorectal cancer. Cancer Lett. 2017, 396, 94–102. [Google Scholar] [CrossRef]
- Zhou, J.; Ji, Q.; Li, Q. Resistance to anti-EGFR therapies in metastatic colorectal cancer: Underlying mechanisms and reversal strategies. J. Exp. Clin. Cancer Res. 2021, 40, 328. [Google Scholar] [CrossRef]
- Mizushima, N.; Levine, B.; Cuervo, A.M.; Klionsky, D.J. Autophagy fights disease through cellular self-digestion. Nature 2008, 451, 1069–1075. [Google Scholar] [CrossRef]
- Kroemer, G.; Mariño, G.; Levine, B. Autophagy and the integrated stress response. Mol. Cell. 2010, 40, 280–293. [Google Scholar] [CrossRef] [Green Version]
- Li, W.; He, P.; Huang, Y.; Li, Y.F.; Lu, J.; Li, M.; Kurihara, H.; Luo, Z.; Meng, T.; Onishi, M.; et al. Selective autophagy of intracellular organelles: Recent research advances. Theranostics 2021, 11, 222–256. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.J.; Chee, C.E.; Huang, S.; Sinicrope, F.A. The role of autophagy in cancer: Therapeutic implications. Mol. Cancer Ther. 2011, 10, 1533–1541. [Google Scholar] [CrossRef]
- Burada, F.; Nicoli, E.R.; Ciurea, M.E.; Uscatu, D.C.; Ioana, M.; Gheonea, D.I. Autophagy in colorectal cancer: An important switch from physiology to pathology. World J. Gastrointest. Oncol. 2015, 7, 271–284. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; He, S.; Ma, B. Autophagy and autophagy-related proteins in cancer. Mol. Cancer 2020, 19, 12. [Google Scholar] [CrossRef] [PubMed]
- Russell, R.C.; Guan, K.L. The multifaceted role of autophagy in cancer. EMBO J. 2022, 41, e110031. [Google Scholar] [CrossRef]
- Cho, D.-H.; Jo, Y.K.; Kim, S.C.; Park, I.J.; Kim, J.C. Down-regulated expression of ATG5 in colorectal cancer. Anticancer Res. 2012, 32, 4091–4096. [Google Scholar]
- Suares, A.; Medina, M.V.; Coso, O. Autophagy in Viral Development and Progression of Cancer. Front. Oncol. 2021, 11, 603224. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Chen, Y.; Zhou, L.; Chen, H.; Zhou, Z. From Intestinal Epithelial Homeostasis to Colorectal Cancer: Autophagy Regulation in Cellular Stress. Antioxidants 2022, 11, 1308. [Google Scholar] [CrossRef]
- Mahgoub, E.; Taneera, J.; Sulaiman, N.; Saber-Ayad, M. The role of autophagy in colorectal cancer: Impact on pathogenesis and implications in therapy. Front. Med. 2022, 9, 959348. [Google Scholar] [CrossRef]
- Yu, L.; Zhang, M.M.; Hou, J.G. Molecular and cellular pathways in colorectal cancer: Apoptosis, autophagy and inflammation as key players. Scand. J. Gastroenterol. 2022, 57, 1279–1290. [Google Scholar] [CrossRef]
- Koustas, E.; Sarantis, P.; Kyriakopoulou, G.; Papavassiliou, A.G.; Karamouzis, M.V. The Interplay of Autophagy and Tumor Microenvironment in Colorectal Cancer-Ways of Enhancing Immunotherapy Action. Cancers 2019, 11, 533. [Google Scholar] [CrossRef] [Green Version]
- Roshani-Asl, E.; Mansori, B.; Mohammadi, A.; Najafi, S.; Danesh-Pouya, F.; Rasmi, Y. Interaction between DNA damage response and autophagy in colorectal cancer. Gene 2020, 730, 144323. [Google Scholar] [CrossRef] [PubMed]
- Bian-Fang, Y.; Dong-Ning, W.; Dan, T.; Jian-Yu, S.; Shi-Yi, W.; Ben-Jun, W.; Xin, D.; Wen-Wen, Z.; Qing-Feng, W.; Yan, Z.; et al. The Role of Autophagy in Tumor Immune Infiltration in Colorectal Cancer. Anal. Cell Pathol. 2022, 2022, 2055676. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Liu, L. Autophagy is a double-edged sword in the therapy of colorectal cancer. Oncol. Lett. 2021, 21, 378. [Google Scholar] [CrossRef]
- Sipos, F.; Székely, H.; Kis, I.D.; Tulassay, Z.; Műzes, G. Relation of the IGF/IGF1R system to autophagy in colitis and colorectal cancer. World J. Gastroenterol. 2017, 23, 8109–8119. [Google Scholar] [CrossRef] [PubMed]
- Sipos, F.; Bohusné Barta, B.; Simon, Á.; Nagy, L.; Dankó, T.; Raffay, R.E.; Petővári, G.; Zsiros, V.; Wichmann, B.; Sebestyén, A.; et al. Survival of HT29 Cancer Cells Is Affected by IGF1R Inhibition via Modulation of Self-DNA-Triggered TLR9 Signaling and the Autophagy Response. Pathol. Oncol. Res. 2022, 28, 1610322. [Google Scholar] [CrossRef]
- Guo, J.Y.; Chen, H.-Y.; Mathew, R.; Fan, J.; Strohecker, A.M.; Karsli-Uzunbas, G.; Kamphorst, J.J.; Chen, G.; Lemons, J.M.; Karantza, V.; et al. Activated Ras requires autophagy to maintain oxidative metabolism and tumorigenesis. Genes Dev. 2011, 25, 460–470. [Google Scholar] [CrossRef]
- Groulx, J.F.; Khalfaoui, T.; Benoit, Y.D.; Bernatchez, G.; Carrier, J.C.; Basora, N.; Beaulieu, J.F. Autophagy is active in normal colon mucosa. Autophagy 2012, 8, 893–902. [Google Scholar] [CrossRef]
- Sato, K.; Tsuchihara, K.; Fujii, S.; Sugiyama, M.; Goya, T.; Atomi, Y.; Ueno, T.; Ochiai, A.; Esumi, H. Autophagy is activated in colorectal cancer cells and contributes to the tolerance to nutrient deprivation. Cancer Res. 2007, 67, 9677–9684. [Google Scholar] [CrossRef]
- Li, B.X.; Li, C.Y.; Peng, R.Q.; Wu, X.J.; Wang, H.Y.; Wan, D.S.; Zhu, X.F.; Zhang, X.S. The expression of beclin 1 is associated with favorable prognosis in stage IIIB colon cancers. Autophagy 2009, 5, 303–306. [Google Scholar] [CrossRef] [PubMed]
- Mokarram, P.; Albokashy, M.; Zarghooni, M.; Moosavi, M.A.; Sepehri, Z.; Chen, Q.M.; Hudecki, A.; Sargazi, A.; Alizadeh, J.; Moghadam, A.R.; et al. New frontiers in the treatment of colorectal cancer: Autophagy and the unfolded protein response as promising targets. Autophagy 2017, 13, 781–819. [Google Scholar] [CrossRef]
- Bednarczyk, M.; Muc-Wierzgoń, M.; Waniczek, D.; Fatyga, E.; Klakla, K.; Mazurek, U.; Wierzgoń, J. Autophagy-related gene expression in colorectal cancer patients. J. Biol. Regul. Homeost. Agents 2017, 31, 923–927. [Google Scholar]
- Liu, H.; Lou, J.; Liu, Y.; Liu, Z.; Xie, J.; Sun, J.; Pan, H.; Han, W. Intestinal epithelial cell autophagy deficiency suppresses inflammation-associated colon tumorigenesis. Mol. Ther. Nucleic Acids 2022, 28, 35–46. [Google Scholar] [CrossRef] [PubMed]
- Lévy, J.; Romagnolo, B. Autophagy, microbiota and intestinal oncogenesis. Oncotarget 2015, 6, 34067–34068. [Google Scholar] [CrossRef] [PubMed]
- Devenport, S.N.; Singhal, R.; Radyk, M.D.; Taranto, J.G.; Kerk, S.A.; Chen, B.; Goyert, J.W.; Jain, C.; Das, N.K.; Oravecz-Wilson, K.; et al. Colorectal cancer cells utilize autophagy to maintain mitochondrial metabolism for cell proliferation under nutrient stress. JCI Insight 2021, 6, e138835. [Google Scholar] [CrossRef] [PubMed]
- Fritz, T.; Niederreiter, L.; Adolph, T.; Blumberg, R.S.; Kaser, A. Crohn’s disease: NOD2, autophagy and ER stress converge. Gut 2011, 60, 1580–1588. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Eun, H.S.; Jo, E.K. Roles of Autophagy-Related Genes in the Pathogenesis of Inflammatory Bowel Disease. Cells 2019, 8, 77. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.; Holowatyj, A.N.; Roy, T.; Pronovost, S.M.; Marchetti, M.; Liu, H.; Ulrich, C.M.; Edgar, B.A. An SH3PX1-Dependent Endocytosis-Autophagy Network Restrains Intestinal Stem Cell Proliferation by Counteracting EGFR-ERK Signaling. Dev. Cell 2019, 49, 574–589.e5. [Google Scholar] [CrossRef]
- Kang, M.R.; Kim, M.S.; Oh, J.E.; Kim, Y.R.; Song, S.Y.; Kim, S.S.; Ahn, C.H.; Yoo, N.J.; Lee, S.H. Frameshift mutations of autophagy-related genes ATG2B, ATG5, ATG9B and ATG12 in gastric and colorectal cancers with microsatellite instability. J. Pathol. 2009, 217, 702–706. [Google Scholar] [CrossRef] [PubMed]
- Bednarczyk, M.; Fatyga, E.; Dzięgielewska-Gęsiak, S.; Waniczek, D.; Grabarek, B.; Zmarzły, N.; Janikowska, G.; Muc-Wierzgoń, M. The Expression Patterns of BECN1, LAMP2, and PINK1 Genes in Colorectal Cancer Are Potentially Regulated by Micrornas and CpG Islands: An in silico Study. J. Clin. Med. 2020, 9, 4020. [Google Scholar] [CrossRef]
- Grimm, W.A.; Messer, J.S.; Murphy, S.F.; Nero, T.; Lodolce, J.P.; Weber, C.R.; Logsdon, M.F.; Bartulis, S.; Sylvester, B.E.; Springer, A.; et al. The Thr300Ala variant in ATG16L1 is associated with improved survival in human colorectal cancer and enhanced production of type I interferon. Gut 2016, 65, 456–464. [Google Scholar] [CrossRef]
- Yu, T.; Ben, S.; Ma, L.; Jiang, L.; Chen, S.; Lin, Y.; Chen, T.; Li, S.; Zhu, L. Genetic variants in autophagy-related gene ATG2B predict the prognosis of colorectal cancer patients receiving chemotherapy. Front. Oncol. 2022, 12, 876424. [Google Scholar] [CrossRef] [PubMed]
- Koustas, E.; Sarantis, P.; Theoharis, S.; Saetta, A.A.; Chatziandreou, I.; Kyriakopoulou, G.; Giannopoulou, I.; Michelli, M.; Schizas, D.; Papavassiliou, A.G.; et al. Autophagy-related Proteins as a Prognostic Factor of Patients with Colorectal Cancer. Am. J. Clin. Oncol. 2019, 42, 767–776. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Liu, S.; Chen, X.; Xu, H.; Tang, Y. Machine learning identifies two autophagy-related genes as markers of recurrence in colorectal cancer. J. Int. Med. Res. 2020, 48, 300060520958808. [Google Scholar] [CrossRef]
- Wang, X.; Xu, Y.; Li, T.; Chen, B.; Yang, W. Development of prognosis model for colon cancer based on autophagy-related genes. World J. Surg. Oncol. 2020, 18, 285. [Google Scholar] [CrossRef]
- Liu, T.T.; Liu, S.M. Prediction of Prognostic Biomarkers and Construction of an Autophagy Prognostic Model for Colorectal Cancer Using Bioinformatics. Technol. Cancer Res. Treat. 2020, 19, 1533033820984177. [Google Scholar] [CrossRef]
- He, Q.; Li, Z.; Yin, J.; Li, Y.; Yin, Y.; Lei, X.; Zhu, W. Prognostic Significance of Autophagy-Relevant Gene Markers in Colorectal Cancer. Front. Oncol. 2021, 11, 566539. [Google Scholar] [CrossRef] [PubMed]
- Haruki, K.; Kosumi, K.; Hamada, T.; Twombly, T.S.; Väyrynen, J.P.; Kim, S.A.; Masugi, Y.; Qian, Z.R.; Mima, K.; Baba, Y.; et al. Association of autophagy status with amount of Fusobacterium nucleatum in colorectal cancer. J. Pathol. 2020, 250, 397–408. [Google Scholar] [CrossRef]
- Chen, Y.; Chen, Y.; Zhang, J.; Cao, P.; Su, W.; Deng, Y.; Zhan, N.; Fu, X.; Huang, Y.; Dong, W.; et al. Fusobacterium nucleatum Promotes Metastasis in Colorectal Cancer by Activating Autophagy Signaling via the Upregulation of CARD3 Expression. Theranostics 2020, 10, 323–339. [Google Scholar] [CrossRef]
- Yu, T.; Guo, F.; Yu, Y.; Sun, T.; Ma, D.; Han, J.; Qian, Y.; Kryczek, I.; Sun, D.; Nagarsheth, N.; et al. Fusobacterium nucleatum Promotes Chemoresistance to Colorectal Cancer by Modulating Autophagy. Cell 2017, 170, 548–563.e16. [Google Scholar] [CrossRef]
- Sun, C.H.; Li, B.B.; Wang, B.; Zhao, J.; Zhang, X.Y.; Li, T.T.; Li, W.B.; Tang, D.; Qiu, M.J.; Wang, X.C.; et al. The role of Fusobacterium nucleatum in colorectal cancer: From carcinogenesis to clinical management. Chronic Dis. Transl. Med. 2019, 5, 178–187. [Google Scholar] [CrossRef]
- Lucas, C.; Salesse, L.; Hoang, M.H.T.; Bonnet, M.; Sauvanet, P.; Larabi, A.; Godfraind, C.; Gagnière, J.; Pezet, D.; Rosenstiel, P.; et al. Autophagy of Intestinal Epithelial Cells Inhibits Colorectal Carcinogenesis Induced by Colibactin-Producing Escherichia coli in ApcMin/+ Mice. Gastroenterology 2020, 158, 1373–1388. [Google Scholar] [CrossRef] [Green Version]
- Salesse, L.; Lucas, C.; Hoang, M.H.T.; Sauvanet, P.; Rezard, A.; Rosenstiel, P.; Damon-Soubeyrand, C.; Barnich, N.; Godfraind, C.; Dalmasso, G.; et al. Colibactin-Producing Escherichia coli Induce the Formation of Invasive Carcinomas in a Chronic Inflammation-Associated Mouse Model. Cancers 2021, 13, 2060. [Google Scholar] [CrossRef] [PubMed]
- Siddle, K. Signalling by insulin and IGF receptors: Supporting acts and new players. J. Mol. Endocrinol. 2011, 47, R1–R10. [Google Scholar] [CrossRef] [PubMed]
- LeRoith, D.; Roberts, C.T., Jr. The insulin-like growth factor system and cancer. Cancer Lett. 2003, 195, 127–137. [Google Scholar] [CrossRef]
- Baserga, R.; Peruzzi, F.; Reiss, K. The IGF-1 receptor in cancer biology. Int. J. Cancer 2003, 107, 873–877. [Google Scholar] [CrossRef]
- Lelbach, A.; Muzes, G.; Feher, J. The insulin-like growth factor system: IGFs, IGF-binding proteins and IGFBP-proteases. Acta Physiol. Hung. 2005, 92, 97–107. [Google Scholar] [CrossRef]
- Abajo, A.; Bitarte, N.; Zarate, R.; Boni, V.; Lopez, I.; Gonzalez-Huarriz, M.; Rodriguez, J.; Bandres, E.; Garcia-Foncillas, J. Identification of colorectal cancer metastasis markers by an angiogenesis-related cytokine-antibody array. World J. Gastroenterol. 2012, 18, 637–645. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.S.; Cui, W. Proliferation, survival and metabolism: The role of PI3K/AKT/mTOR signalling in pluripotency and cell fate determination. Development 2016, 143, 3050–3060. [Google Scholar] [CrossRef]
- Oshima, T.; Akaike, M.; Yoshihara, K.; Shiozawa, M.; Yamamoto, N.; Sato, T.; Yamada, R.; Fujii, S.; Rino, Y.; Kunisaki, C.; et al. Clinicopathological significance of the gene expression of matrix metalloproteinase-7, insulin-like growth factor-1, insulin-like growth factor-2 and insulin-like growth factor-1 receptor in patients with colorectal cancer: Insulin-like growth factor-1 receptor gene expression is a useful predictor of liver metastasis from colorectal cancer. Oncol. Rep. 2008, 20, 359–364. [Google Scholar]
- Zhang, R.; Xu, G.L.; Li, Y.; He, L.J.; Chen, L.M.; Wang, G.B.; Lin, S.Y.; Luo, G.Y.; Gao, X.Y.; Shan, H.B.; et al. The role of insulin-like growth factor 1 and its receptor in the formation and development of colorectal carcinoma. J. Int. Med. Res. 2013, 41, 1228–1235. [Google Scholar] [CrossRef] [PubMed]
- Shiratsuchi, I.; Akagi, Y.; Kawahara, A.; Kinugasa, T.; Romeo, K.; Yoshida, T.; Ryu, Y.; Gotanda, Y.; Kage, M.; Shirouzu, K.; et al. Expression of IGF-1 and IGF-1R and their relation to clinicopathological factors in colorectal cancer. Anticancer Res. 2011, 31, 2541–2545. [Google Scholar] [PubMed]
- Nakamura, M.; Miyamoto, S.; Maeda, H.; Zhang, S.C.; Sangai, T.; Ishii, G.; Hasebe, T.; Endoh, Y.; Saito, N.; Asaka, M.; et al. Low levels of insulin-like growth factor type 1 receptor expression at cancer cell membrane predict liver metastasis in Dukes’ C human colorectal cancers. Clin. Cancer Res. 2004, 10, 8434–8441. [Google Scholar] [CrossRef] [Green Version]
- Li, Z.J.; Ying, X.J.; Chen, H.L.; Ye, P.J.; Chen, Z.L.; Li, G.; Jiang, H.F.; Liu, J.; Zhou, S.Z. Insulin-like growth factor-1 induces lymphangiogenesis and facilitates lymphatic metastasis in colorectal cancer. World J. Gastroenterol. 2013, 19, 7788–7794. [Google Scholar] [CrossRef]
- Wu, X.Y.; Wu, Z.F.; Cao, Q.H.; Chen, C.; Chen, Z.W.; Xu, Z.; Li, W.S.; Liu, F.K.; Yao, X.Q.; Li, G.; et al. Insulin-like growth factor receptor-1 overexpression is associated with poor response of rectal cancers to radiotherapy. World J. Gastroenterol. 2014, 20, 16268–16274. [Google Scholar] [CrossRef]
- Codony-Servat, J.; Cuatrecasas, M.; Asensio, E.; Montironi, C.; Martínez-Cardús, A.; Marín-Aguilera, M.; Horndler, C.; Martínez-Balibrea, E.; Rubini, M.; Jares, P.; et al. Nuclear IGF-1R predicts chemotherapy and targeted therapy resistance in metastatic colorectal cancer. Br. J. Cancer 2017, 117, 1777–1786. [Google Scholar] [CrossRef] [PubMed]
- Sekharam, M.; Nasir, A.; Kaiser, H.E.; Coppola, D. Insulin-like growth factor 1 receptor activates c-SRC and modifies transformation and motility of colon cancer in vitro. Anticancer Res. 2003, 23, 1517–1524. [Google Scholar]
- Bauer, T.W.; Fan, F.; Liu, W.; Johnson, M.; Parikh, N.U.; Parry, G.C.; Callahan, J.; Mazar, A.P.; Gallick, G.E.; Ellis, L.M.; et al. Insulinlike growth factor-I-mediated migration and invasion of human colon carcinoma cells requires activation of c-Met and urokinase plasminogen activator receptor. Ann. Surg. 2005, 241, 748–756. [Google Scholar] [CrossRef]
- Bauer, T.W.; Fan, F.; Liu, W.; Camp, E.R.; Yang, A.; Somcio, R.J.; Bucana, C.D.; Singh, R.; Ellis, L.M. Targeting of insulin-like growth factor-I receptor with a monoclonal antibody inhibits growth of hepatic metastases from human colon carcinoma in mice. Ann. Surg. Oncol. 2007, 14, 2838–2846. [Google Scholar] [CrossRef]
- Guijarro, L.G.; Cano-Martínez, D.; Toledo-Lobo, M.V.; Salinas, P.S.; Chaparro, M.; Gómez-Lahoz, A.M.; Zoullas, S.; Rodríguez-Torres, R.; Román, I.D.; Monasor, L.S.; et al. Relationship between IGF-1 and body weight in inflammatory bowel diseases: Cellular and molecular mechanisms involved. Biomed. Pharmacother. 2021, 144, 112239. [Google Scholar] [CrossRef] [PubMed]
- Kasprzak, A. Insulin-like Growth Factor 1 (IGF-1) Signaling in Glucose Metabolism in Colorectal Cancer. Int. J. Mol. Sci. 2021, 22, 6434. [Google Scholar] [CrossRef] [PubMed]
- Mukherjee, A.; Alzhanov, D.; Rotwein, P. Defining human insulin-like growth factor I gene regulation. Am. J. Physiol. Endocrinol. Metab. 2016, 311, E519–E529. [Google Scholar] [CrossRef]
- Rotwein, P. Regulation of gene expression by growth hormone. Mol. Cell Endocrinol. 2020, 507, 110788. [Google Scholar] [CrossRef] [PubMed]
- Koveitypour, Z.; Panahi, F.; Vakilian, M.; Peymani, M.; Seyed Forootan, F.; Nasr Esfahani, M.H.; Ghaedi, K. Signaling pathways involved in colorectal cancer progression. Cell Biosci. 2019, 9, 97. [Google Scholar] [CrossRef] [Green Version]
- Chen, P.C.; Kuo, Y.C.; Chuong, C.M.; Huang, Y.H. Niche Modulation of IGF-1R Signaling: Its Role in Stem Cell Pluripotency, Cancer Reprogramming, and Therapeutic Applications. Front. Cell Dev. Biol. 2021, 8, 625943. [Google Scholar] [CrossRef]
- Tiwari, A.; Saraf, S.; Verma, A.; Panda, P.K.; Jain, S.K. Novel targeting approaches and signaling pathways of colorectal cancer: An insight. World J. Gastroenterol. 2018, 24, 4428–4435. [Google Scholar] [CrossRef]
- Laplante, M.; Sabatini, D.M. mTOR signaling in growth control and disease. Cell 2012, 149, 274–293. [Google Scholar] [CrossRef]
- Yu, J.S.; Ramasamy, T.S.; Murphy, N.; Holt, M.K.; Czapiewski, R.; Wei, S.K.; Cui, W. PI3K/mTORC2 regulates TGF-β/Activin signalling by modulating Smad2/3 activity via linker phosphorylation. Nat. Commun. 2015, 6, 7212. [Google Scholar] [CrossRef]
- Yao, C.; Su, L.; Shan, J.; Zhu, C.; Liu, L.; Liu, C.; Xu, Y.; Yang, Z.; Bian, X.; Shao, J.; et al. IGF/STAT3/NANOG/Slug Signaling Axis Simultaneously Controls Epithelial-Mesenchymal Transition and Stemness Maintenance in Colorectal Cancer. Stem Cells 2016, 34, 820–831. [Google Scholar] [CrossRef]
- Ding, J.; Li, C.; Tang, J.; Yi, C.; Liu, J.Y.; Qiu, M. Higher Expression of Proteins in IGF/IR Axes in Colorectal Cancer is Associated with Type 2 Diabetes Mellitus. Pathol. Oncol. Res. 2016, 22, 773–779. [Google Scholar] [CrossRef]
- Kasprzak, A.; Adamek, A. Insulin-like Growth Factor 2 (IGF2) Signaling in Colorectal Cancer-From Basic Research to Potential Clinical Applications. Int. J. Mol. Sci. 2019, 20, 4915. [Google Scholar] [CrossRef]
- Klionsky, D.J.; Abdel-Aziz, A.K.; Abdelfatah, S.; Abdellatif, M.; Abdoli, A.; Abel, S.; Abeliovich, H.; Abildgaard, M.H.; Abudu, Y.P.; Acevedo-Arozena, A.; et al. Guidelines for the use and interpretation of assays for monitoring autophagy (4th edition). Autophagy 2021, 17, 1–382. [Google Scholar] [CrossRef]
- Levine, B.; Kroemer, G. Biological Functions of Autophagy Genes: A Disease Perspective. Cell 2019, 176, 11–42. [Google Scholar] [CrossRef] [PubMed]
- Ktistakis, N.T.; Tooze, S.A. Digesting the Expanding Mechanisms of Autophagy. Trends Cell Biol. 2016, 26, 624–635. [Google Scholar] [CrossRef]
- Dupont, N.; Nascimbeni, A.C.; Morel, E.; Codogno, P. Molecular Mechanisms of Noncanonical Autophagy. Int. Rev. Cell Mol. Biol. 2017, 328, 1–23. [Google Scholar] [CrossRef] [PubMed]
- Neufeld, T.P. TOR-dependent control of autophagy: Biting the hand that feeds. Curr. Opin. Cell Biol. 2010, 22, 157–168. [Google Scholar] [CrossRef]
- El-Kott, A.F.; Al-Kahtani, M.A.; Shati, A.A. Calycosin induces apoptosis in adenocarcinoma HT29 cells by inducing cytotoxic autophagy mediated by SIRT1/AMPK-induced inhibition of Akt/mTOR. Clin. Exp. Pharmacol. Physiol. 2019, 46, 944–954. [Google Scholar] [CrossRef] [PubMed]
- Liang, J.; Shao, S.H.; Xu, Z.X.; Hennessy, B.; Ding, Z.; Larrea, M.; Kondo, S.; Dumont, D.J.; Gutterman, J.U.; Walker, C.L.; et al. The energy sensing LKB1-AMPK pathway regulates p27kip1 phosphorylation mediating the decision to enter autophagy or apoptosis. Nat. Cell Biol. 2007, 9, 218–224. [Google Scholar] [CrossRef]
- Efeyan, A.; Sabatini, D.M. mTOR and cancer: Many loops in one pathway. Curr. Opin. Cell Biol. 2010, 22, 169–176. [Google Scholar] [CrossRef]
- Vellai, T.; Bicsák, B.; Tóth, M.L.; Takács-Vellai, K.; Kovács, A.L. Regulation of cell growth by autophagy. Autophagy 2008, 4, 507–509. [Google Scholar] [CrossRef] [PubMed]
- Renna, M.; Bento, C.F.; Fleming, A.; Menzies, F.M.; Siddiqi, F.H.; Ravikumar, B.; Puri, C.; Garcia-Arencibia, M.; Sadiq, O.; Corrochano, S.; et al. IGF-1 receptor antagonism inhibits autophagy. Hum. Mol. Genet. 2013, 22, 4528–4544. [Google Scholar] [CrossRef] [PubMed]
- Takasaka, N.; Araya, J.; Hara, H.; Ito, S.; Kobayashi, K.; Kurita, Y.; Wakui, H.; Yoshii, Y.; Yumino, Y.; Fujii, S.; et al. Autophagy induction by SIRT6 through attenuation of insulin-like growth factor signaling is involved in the regulation of human bronchial epithelial cell senescence. J. Immunol. 2014, 192, 958–968. [Google Scholar] [CrossRef] [PubMed]
- Bitto, A.; Lerner, C.; Torres, C.; Roell, M.; Malaguti, M.; Perez, V.; Lorenzini, A.; Hrelia, S.; Ikeno, Y.; Matzko, M.E.; et al. Long-term IGF-I exposure decreases autophagy and cell viability. PLoS ONE 2010, 5, e12592. [Google Scholar] [CrossRef]
- Gu, Y.; Wang, C.; Cohen, A. Effect of IGF-1 on the balance between autophagy of dysfunctional mitochondria and apoptosis. FEBS Lett. 2004, 577, 357–360. [Google Scholar] [CrossRef] [Green Version]
- Lyons, A.; Coleman, M.; Riis, S.; Favre, C.; O’Flanagan, C.H.; Zhdanov, A.V.; Papkovsky, D.B.; Hursting, S.D.; O’Connor, R. Insulin-like growth factor 1 signaling is essential for mitochondrial biogenesis and mitophagy in cancer cells. J. Biol. Chem. 2017, 292, 16983–16998. [Google Scholar] [CrossRef]
- Riis, S.; Murray, J.B.; O’Connor, R. IGF-1 Signalling Regulates Mitochondria Dynamics and Turnover through a Conserved GSK-3β-Nrf2-BNIP3 Pathway. Cells 2020, 9, 147. [Google Scholar] [CrossRef]
- Liu, Q.; Guan, J.Z.; Sun, Y.; Le, Z.; Zhang, P.; Yu, D.; Liu, Y. Insulin-like growth factor 1 receptor-mediated cell survival in hypoxia depends on the promotion of autophagy via suppression of the PI3K/Akt/mTOR signaling pathway. Mol. Med. Rep. 2017, 15, 2136–2142. [Google Scholar] [CrossRef]
- Ricci-Vitiani, L.; Fabrizi, E.; Palio, E.; de Maria, R. Colon cancer stem cells. J. Mol. Med. 2009, 87, 1097–1104. [Google Scholar] [CrossRef] [PubMed]
- Pott, J.; Hornef, M. Innate immune signalling at the intestinal epithelium in homeostasis and disease. EMBO Rep. 2012, 13, 684–698. [Google Scholar] [CrossRef]
- Wang, Y.; Song, W.; Yu, S.; Liu, Y.; Chen, Y.G. Intestinal cellular heterogeneity and disease development revealed by single-cell technology. Cell Regen. 2022, 11, 26. [Google Scholar] [CrossRef]
- Wang, Y.; Song, W.; Wang, J.; Wang, T.; Xiong, X.; Qi, Z.; Fu, W.; Yang, X.; Chen, Y.G. Single-cell transcriptome analysis reveals differential nutrient absorption functions in human intestine. J. Exp. Med. 2020, 217, e20191130. [Google Scholar] [CrossRef]
- Parikh, K.; Antanaviciute, A.; Fawkner-Corbett, D.; Jagielowicz, M.; Aulicino, A.; Lagerholm, C.; Davis, S.; Kinchen, J.; Chen, H.H.; Alham, N.K.; et al. Colonic epithelial cell diversity in health and inflammatory bowel disease. Nature 2019, 567, 49–55. [Google Scholar] [CrossRef]
- Smillie, C.S.; Biton, M.; Ordovas-Montanes, J.; Sullivan, K.M.; Burgin, G.; Graham, D.B.; Herbst, R.H.; Rogel, N.; Slyper, M.; Waldman, J.; et al. Intra- and Inter-cellular Rewiring of the Human Colon during Ulcerative Colitis. Cell 2019, 178, 714–730.e22. [Google Scholar] [CrossRef] [PubMed]
- Barker, N.; van Es, J.H.; Kuipers, J.; Kujala, P.; van den Born, M.; Cozijnsen, M.; Haegebarth, A.; Korving, J.; Begthel, H.; Peters, P.J.; et al. Identification of stem cells in small intestine and colon by marker gene Lgr5. Nature 2007, 449, 1003–1007. [Google Scholar] [CrossRef] [PubMed]
- Van der Wath, R.C.; Gardiner, B.S.; Burgess, A.W.; Smith, D.W. Cell organisation in the colonic crypt: A theoretical comparison of the pedigree and niche concepts. PLoS ONE 2013, 8, e73204. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.S.; Ho, S.B. Intestinal goblet cells and mucins in health and disease: Recent insights and progress. Curr. Gastroenterol. Rep. 2010, 12, 319–330. [Google Scholar] [CrossRef]
- Johansson, M.E.; Gustafsson, J.K.; Holmén-Larsson, J.; Jabbar, K.S.; Xia, L.; Xu, H.; Ghishan, F.K.; Carvalho, F.A.; Gewirtz, A.T.; Sjövall, H.; et al. Bacteria penetrate the normally impenetrable inner colon mucus layer in both murine colitis models and patients with ulcerative colitis. Gut 2014, 63, 281–291. [Google Scholar] [CrossRef]
- Paone, P.; Cani, P.D. Mucus barrier, mucins and gut microbiota: The expected slimy partners? Gut 2020, 69, 2232–2243. [Google Scholar] [CrossRef]
- Kabat, A.M.; Pott, J.; Maloy, K.J. The Mucosal Immune System and Its Regulation by Autophagy. Front. Immunol. 2016, 7, 240. [Google Scholar] [CrossRef]
- Ke, P.; Shao, B.Z.; Xu, Z.Q.; Chen, X.W.; Liu, C. Intestinal Autophagy and Its Pharmacological Control in Inflammatory Bowel Disease. Front. Immunol. 2017, 7, 695. [Google Scholar] [CrossRef] [PubMed]
- Foerster, E.G.; Mukherjee, T.; Cabral-Fernandes, L.; Rocha, J.D.B.; Girardin, S.E.; Philpott, D.J. How autophagy controls the intestinal epithelial barrier. Autophagy 2022, 18, 86–103. [Google Scholar] [CrossRef] [PubMed]
- Lévy, J.; Cacheux, W.; Bara, M.A.; L’Hermitte, A.; Lepage, P.; Fraudeau, M.; Trentesaux, C.; Lemarchand, J.; Durand, A.; Crain, A.M.; et al. Intestinal inhibition of Atg7 prevents tumour initiation through a microbiome-influenced immune response and suppresses tumour growth. Nat. Cell Biol. 2015, 17, 1062–1073. [Google Scholar] [CrossRef]
- Tunçer, S.; Sade-Memişoğlu, A.; Keşküş, A.G.; Sheraj, I.; Güner, G.; Akyol, A.; Banerjee, S. Enhanced expression of HNF4α during intestinal epithelial differentiation is involved in the activation of ER stress. FEBS J. 2020, 287, 2504–2523. [Google Scholar] [CrossRef] [PubMed]
- Tsuboi, K.; Nishitani, M.; Takakura, A.; Imai, Y.; Komatsu, M.; Kawashima, H. Autophagy Protects against Colitis by the Maintenance of Normal Gut Microflora and Secretion of Mucus. J. Biol. Chem. 2015, 290, 20511–20526. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nishiumi, S.; Fujishima, Y.; Inoue, J.; Masuda, A.; Azuma, T.; Yoshida, M. Autophagy in the intestinal epithelium is not involved in the pathogenesis of intestinal tumors. Biochem. Biophys. Res. Commun. 2012, 421, 768–772. [Google Scholar] [CrossRef]
- Robert, M.E.; Singh, S.K.; Ikuma, M.; Jain, D.; Ardito, T.; Binder, H.J. Morphology of isolated colonic crypts. Cells Tissues Organs 2001, 168, 246–251. [Google Scholar] [CrossRef]
- Potten, C.S.; Kellett, M.; Roberts, S.A.; Rew, D.A.; Wilson, G.D. Measurement of in vivo proliferation in human colorectal mucosa using bromodeoxyuridine. Gut 1992, 33, 71–78. [Google Scholar] [CrossRef]
- Raskov, H.; Pommergaard, H.C.; Burcharth, J.; Rosenberg, J. Colorectal carcinogenesis--update and perspectives. World J. Gastroenterol. 2014, 20, 18151–18164. [Google Scholar] [CrossRef]
- Van Lieshout, E.M.; van Doesburg, W.; van der Meer, R. Real-time PCR of host DNA in feces to study differential exfoliation of colonocytes between rats and humans. Scand. J. Gastroenterol. 2004, 39, 852–857. [Google Scholar] [CrossRef]
- Barkla, D.H.; Gibson, P.R. The fate of epithelial cells in the human large intestine. Pathology 1999, 31, 230–238. [Google Scholar] [CrossRef]
- Sandle, G.I. Salt and water absorption in the human colon: A modern appraisal. Gut 1998, 43, 294–299. [Google Scholar] [CrossRef] [PubMed]
- Cosme, D.; Estevinho, M.M.; Rieder, F.; Magro, F. Potassium channels in intestinal epithelial cells and their pharmacological modulation: A systematic review. Am. J. Physiol. Cell Physiol. 2021, 320, C520–C546. [Google Scholar] [CrossRef] [PubMed]
- Al-Hazza, A.; Linley, J.E.; Aziz, Q.; Maclennan, K.A.; Hunter, M.; Sandle, G.I. Potential role of reduced basolateral potassium (IKCa3.1) channel expression in the pathogenesis of diarrhoea in ulcerative colitis. J. Pathol. 2012, 226, 463–470. [Google Scholar] [CrossRef]
- Antico, S.; Lionetto, M.G.; Giordano, M.E.; Caricato, R.; Schettino, T. Cell volume regulation and apoptotic volume decrease in rat distal colon superficial enterocytes. Cell. Physiol. Biochem. 2013, 32, 1551–1565. [Google Scholar] [CrossRef] [PubMed]
- Sandle, G.I.; Perry, M.D.; Mathialahan, T.; Linley, J.E.; Robinson, P.; Hunter, M.; MacLennan, K.A. Altered cryptal expression of luminal potassium (BK) channels in ulcerative colitis. J. Pathol. 2007, 212, 66–73. [Google Scholar] [CrossRef] [PubMed]
- Negussie, A.B.; Dell, A.C.; Davis, B.A.; Geibel, J.P. Colonic Fluid and Electrolyte Transport 2022: An Update. Cells 2022, 11, 1712. [Google Scholar] [CrossRef]
- Donohoe, D.R.; Garge, N.; Zhang, X.; Sun, W.; O’Connell, T.M.; Bunger, M.K.; Bultman, S.J. The microbiome and butyrate regulate energy metabolism and autophagy in the mammalian colon. Cell Metab. 2011, 13, 517–526. [Google Scholar] [CrossRef]
- Litvak, Y.; Byndloss, M.X.; Bäumler, A.J. Colonocyte metabolism shapes the gut microbiota. Science 2018, 362, eaat9076. [Google Scholar] [CrossRef]
- Park, C.H.; Eun, C.S.; Han, D.S. Intestinal microbiota, chronic inflammation, and colorectal cancer. Intest. Res. 2018, 16, 338–345. [Google Scholar] [CrossRef]
- Li, S.; Liu, J.; Zheng, X.; Ren, L.; Yang, Y.; Li, W.; Fu, W.; Wang, J.; Du, G. Tumorigenic bacteria in colorectal cancer: Mechanisms and treatments. Cancer Biol. Med. 2021, 19, 147–162. [Google Scholar] [CrossRef] [PubMed]
- Yu, L.C.; Wei, S.C.; Li, Y.H.; Lin, P.Y.; Chang, X.Y.; Weng, J.P.; Shue, Y.W.; Lai, L.C.; Wang, J.T.; Jeng, Y.M.; et al. Invasive Pathobionts Contribute to Colon Cancer Initiation by Counterbalancing Epithelial Antimicrobial Responses. Cell. Mol. Gastroenterol. Hepatol. 2022, 13, 57–79. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Thornton, W.H.; MacDonald, R.S. Insulin-like growth factor-I and II receptor expression in rat colon mucosa are affected by dietary lipid intake. J. Nutr. 1998, 128, 158–165. [Google Scholar] [CrossRef]
- Breininger, S.P.; Malcomson, F.C.; Afshar, S.; Turnbull, D.M.; Greaves, L.; Mathers, J.C. Effects of obesity and weight loss on mitochondrial structure and function and implications for colorectal cancer risk. Proc. Nutr. Soc. 2019, 78, 426–437. [Google Scholar] [CrossRef]
- Chesnokova, V.; Zonis, S.; Barrett, R.J.; Gleeson, J.P.; Melmed, S. Growth Hormone Induces Colon DNA Damage Independent of IGF-1. Endocrinology 2019, 160, 1439–1447. [Google Scholar] [CrossRef]
- Chen, T.; Zheng, F.; Tao, J.; Tan, S.; Zeng, L.; Peng, X.; Wu, B. Insulin-like Growth Factor-1 Contributes to Mucosal Repair by β-Arrestin2-Mediated Extracellular Signal-Related Kinase Signaling in Experimental Colitis. Am. J. Pathol. 2015, 185, 2441–2453. [Google Scholar] [CrossRef]
- Xu, J.; Wang, X.; Chen, J.; Chen, S.; Li, Z.; Liu, H.; Bai, Y.; Zhi, F. Embryonic stem cell-derived mesenchymal stem cells promote colon epithelial integrity and regeneration by elevating circulating IGF-1 in colitis mice. Theranostics 2020, 10, 12204–12222. [Google Scholar] [CrossRef]
- Zheng, Y.; Song, Y.; Han, Q.; Liu, W.; Xu, J.; Yu, Z.; Zhang, R.; Li, N. Intestinal epithelial cell-specific IGF-1 promotes the expansion of intestinal stem cells during epithelial regeneration and functions on the intestinal immune homeostasis. Am. J. Physiol. Endocrinol. Metab. 2018, 315, E638–E649. [Google Scholar] [CrossRef]
- Chen, G.Y.; Stappenbeck, T.S. Mucus, it is not just a static barrier. Sci. Signal. 2014, 7, pe11. [Google Scholar] [CrossRef] [PubMed]
- Wlodarska, M.; Thaiss, C.A.; Nowarski, R.; Henao-Mejia, J.; Zhang, J.P.; Brown, E.M.; Frankel, G.; Levy, M.; Katz, M.N.; Philbrick, W.M.; et al. NLRP6 inflammasome orchestrates the colonic host-microbial interface by regulating goblet cell mucus secretion. Cell 2014, 156, 1045–1059. [Google Scholar] [CrossRef]
- Knoop, K.A.; McDonald, K.G.; McCrate, S.; McDole, J.R.; Newberry, R.D. Microbial sensing by goblet cells controls immune surveillance of luminal antigens in the colon. Mucosal Immunol. 2015, 8, 198–210. [Google Scholar] [CrossRef] [PubMed]
- Knoop, K.A.; McDonald, K.G.; Kulkarni, D.H.; Newberry, R.D. Antibiotics promote inflammation through the translocation of native commensal colonic bacteria. Gut 2016, 65, 1100–1109. [Google Scholar] [CrossRef]
- Zhang, M.; Wu, C. The relationship between intestinal goblet cells and the immune response. Biosci. Rep. 2020, 40, BSR20201471. [Google Scholar] [CrossRef] [PubMed]
- Gustafsson, J.K.; Johansson, M.E.V. The role of goblet cells and mucus in intestinal homeostasis. Nat. Rev. Gastroenterol. Hepatol. 2022, 19, 785–803. [Google Scholar] [CrossRef]
- Loktionov, A. Colon mucus in colorectal neoplasia and beyond. World J. Gastroenterol. 2022, 28, 4475–4492. [Google Scholar] [CrossRef]
- Van der Sluis, M.; de Koning, B.A.; de Bruijn, A.C.; Velcich, A.; Meijerink, J.P.; van Goudoever, J.B.; Büller, H.A.; Dekker, J.; van Seuningen, I.; Renes, I.B.; et al. Muc2-deficient mice spontaneously develop colitis, indicating that MUC2 is critical for colonic protection. Gastroenterology 2006, 131, 117–129. [Google Scholar] [CrossRef] [PubMed]
- Johansson, M.E.; Hansson, G.C. Is the intestinal goblet cell a major immune cell? Cell Host Microbe 2014, 15, 251–252. [Google Scholar] [CrossRef]
- McGuckin, M.A.; Hasnain, S.Z. Goblet cells as mucosal sentinels for immunity. Mucosal Immunol. 2017, 10, 1118–1121. [Google Scholar] [CrossRef]
- Dilly, A.K.; Honick, B.D.; Lee, Y.J.; Bartlett, D.L.; Choudry, H.A. Synergistic apoptosis following endoplasmic reticulum stress aggravation in mucinous colon cancer. Orphanet J. Rare Dis. 2020, 15, 211. [Google Scholar] [CrossRef]
- Tiwari, S.; Begum, S.; Moreau, F.; Gorman, H.; Chadee, K. Autophagy is required during high MUC2 mucin biosynthesis in colonic goblet cells to contend metabolic stress. Am. J. Physiol. Gastrointest. Liver Physiol. 2021, 321, G489–G499. [Google Scholar] [CrossRef]
- Papandreou, M.E.; Tavernarakis, N. Autophagy and the endo/exosomal pathways in health and disease. Biotechnol. J. 2017, 12, 1600175. [Google Scholar] [CrossRef]
- Patel, K.K.; Miyoshi, H.; Beatty, W.L.; Head, R.D.; Malvin, N.P.; Cadwell, K.; Guan, J.L.; Saitoh, T.; Akira, S.; Seglen, P.O.; et al. Autophagy proteins control goblet cell function by potentiating reactive oxygen species production. EMBO J. 2013, 32, 3130–3144. [Google Scholar] [CrossRef] [PubMed]
- Ghimire, L.; Paudel, S.; Jin, L.; Jeyaseelan, S. The NLRP6 inflammasome in health and disease. Mucosal Immunol. 2020, 13, 388–398. [Google Scholar] [CrossRef]
- Volk, J.K.; Nyström, E.E.L.; van der Post, S.; Abad, B.M.; Schroeder, B.O.; Johansson, Å.; Svensson, F.; Jäverfelt, S.; Johansson, M.E.V.; Hansson, G.C.; et al. The Nlrp6 inflammasome is not required for baseline colonic inner mucus layer formation or function. J. Exp. Med. 2019, 216, 2602–2618. [Google Scholar] [CrossRef] [PubMed]
- Birchenough, G.M.; Nyström, E.E.; Johansson, M.E.; Hansson, G.C. A sentinel goblet cell guards the colonic crypt by triggering Nlrp6-dependent Muc2 secretion. Science 2016, 352, 1535–1542. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nyström, E.E.L.; Martinez-Abad, B.; Arike, L.; Birchenough, G.M.H.; Nonnecke, E.B.; Castillo, P.A.; Svensson, F.; Bevins, C.L.; Hansson, G.C.; Johansson, M.E.V. An intercrypt subpopulation of goblet cells is essential for colonic mucus barrier function. Science 2021, 372, abb1590. [Google Scholar] [CrossRef]
- Howarth, G.S.; Xian, C.J.; Read, L.C. Insulin-like growth factor-I partially attenuates colonic damage in rats with experimental colitis induced by oral dextran sulphate sodium. Scand. J. Gastroenterol. 1998, 33, 180–190. [Google Scholar] [CrossRef]
- Bach, S.P.; Renehan, A.G.; Potten, C.S. Stem cells: The intestinal stem cell as a paradigm. Carcinogenesis 2000, 21, 469–476. [Google Scholar] [CrossRef]
- Huels, D.J.; Sansom, O.J. Stem vs. non-stem cell origin of colorectal cancer. Br. J. Cancer. 2015, 113, 1–5. [Google Scholar] [CrossRef]
- McKernan, D.P.; Egan, L.J. The intestinal epithelial cell cycle: Uncovering its ‘cryptic’ nature. Curr. Opin. Gastroenterol. 2015, 31, 124–129. [Google Scholar] [CrossRef]
- Shih, I.M.; Wang, T.L.; Traverso, G.; Romans, K.; Hamilton, S.R.; Ben-Sasson, S.; Kinzler, K.W.; Vogelstein, B. Top-down morphogenesis of colorectal tumors. Proc. Natl. Acad. Sci. USA 2001, 98, 2640–2645. [Google Scholar] [CrossRef] [PubMed]
- Preston, S.L.; Wong, W.M.; Chan, A.O.; Poulsom, R.; Jeffery, R.; Goodlad, R.A.; Mandir, N.; Elia, G.; Novelli, M.; Bodmer, W.F.; et al. Bottom-up histogenesis of colorectal adenomas: Origin in the monocryptal adenoma and initial expansion by crypt fission. Cancer Res. 2003, 63, 3819–3825. [Google Scholar] [PubMed]
- Guan, J.L.; Simon, A.K.; Prescott, M.; Menendez, J.A.; Liu, F.; Wang, F.; Wang, C.; Wolvetang, E.; Vazquez-Martin, A.; Zhang, J.; et al. Autophagy in stem cells. Autophagy 2013, 9, 830–849. [Google Scholar] [CrossRef]
- Asano, J.; Sato, T.; Ichinose, S.; Kajita, M.; Onai, N.; Shimizu, S.; Ohteki, T. Intrinsic Autophagy Is Required for the Maintenance of Intestinal Stem Cells and for Irradiation-Induced Intestinal Regeneration. Cell Rep. 2017, 20, 1050–1060. [Google Scholar] [CrossRef] [PubMed]
- Baulies, A.; Angelis, N.; Li, V.S.W. Hallmarks of intestinal stem cells. Development 2020, 147, dev182675. [Google Scholar] [CrossRef]
- Wang, S.; Xia, P.; Rehm, M.; Fan, Z. Autophagy and cell reprogramming. Cell Mol. Life Sci. 2015, 72, 1699–1713. [Google Scholar] [CrossRef]
- Zhang, P.; Holowatyj, A.N.; Ulrich, C.M.; Edgar, B.A. Tumor suppressive autophagy in intestinal stem cells controls gut homeostasis. Autophagy 2019, 15, 1668–1670. [Google Scholar] [CrossRef]
- Buszczak, M.; Krämer, H. Autophagy Keeps the Balance in Tissue Homeostasis. Dev. Cell 2019, 49, 499–500. [Google Scholar] [CrossRef] [PubMed]
- Xie, J.; Li, L.; Deng, S.; Chen, J.; Gu, Q.; Su, H.; Wen, L.; Wang, S.; Lin, C.; Qi, C.; et al. Slit2/Robo1 Mitigates DSS-induced Ulcerative Colitis by Activating Autophagy in Intestinal Stem Cell. Int. J. Biol Sci. 2020, 16, 1876–1887. [Google Scholar] [CrossRef]
- Nigro, G.; Rossi, R.; Commere, P.H.; Jay, P.; Sansonetti, P.J. The cytosolic bacterial peptidoglycan sensor Nod2 affords stem cell protection and links microbes to gut epithelial regeneration. Cell Host Microbe 2014, 15, 792–798. [Google Scholar] [CrossRef]
- Levy, A.; Stedman, A.; Deutsch, E.; Donnadieu, F.; Virgin, H.W.; Sansonetti, P.J.; Nigro, G. Innate immune receptor NOD2 mediates LGR5+ intestinal stem cell protection against ROS cytotoxicity via mitophagy stimulation. Proc. Natl. Acad. Sci. USA 2020, 117, 1994–2003. [Google Scholar] [CrossRef] [PubMed]
- Bohin, N.; McGowan, K.P.; Keeley, T.M.; Carlson, E.A.; Yan, K.S.; Samuelson, L.C. Insulin-like Growth Factor-1 and mTORC1 Signaling Promote the Intestinal Regenerative Response After Irradiation Injury. Cell. Mol. Gastroenterol. Hepatol. 2020, 10, 797–810. [Google Scholar] [CrossRef]
- Chen, M.E.; Naeini, S.M.; Srikrishnaraj, A.; Drucker, D.J.; Fesler, Z.; Brubaker, P.L. Glucagon-like Peptide-2 Stimulates S-Phase Entry of Intestinal Lgr5+ Stem Cells. Cell. Mol. Gastroenterol. Hepatol. 2022, 13, 1829–1842. [Google Scholar] [CrossRef]
- Sato, T.; van Es, J.H.; Snippert, H.J.; Stange, D.E.; Vries, R.G.; van den Born, M.; Barker, N.; Shroyer, N.F.; van de Wetering, M.; Clevers, H. Paneth cells constitute the niche for Lgr5 stem cells in intestinal crypts. Nature 2011, 469, 415–418. [Google Scholar] [CrossRef] [PubMed]
- Bel, S.; Hooper, L.V. Secretory autophagy of lysozyme in Paneth cells. Autophagy 2018, 14, 719–721. [Google Scholar] [CrossRef] [Green Version]
- Bel, S.; Pendse, M.; Wang, Y.; Li, Y.; Ruhn, K.A.; Hassell, B.; Leal, T.; Winter, S.E.; Xavier, R.J.; Hooper, L.V.; et al. Paneth cells secrete lysozyme via secretory autophagy during bacterial infection of the intestine. Science 2017, 357, 1047–1052. [Google Scholar] [CrossRef] [PubMed]
- Bjerknes, M.; Cheng, H. The stem-cell zone of the small intestinal epithelium. I. Evidence from Paneth cells in the adult mouse. Am. J. Anat. 1981, 160, 51–63. [Google Scholar] [CrossRef]
- Igarashi, M.; Guarente, L. mTORC1 and SIRT1 Cooperate to Foster Expansion of Gut Adult Stem Cells during Calorie Restriction. Cell 2016, 166, 436–450. [Google Scholar] [CrossRef]
- Van Es, J.H.; Wiebrands, K.; López-Iglesias, C.; van de Wetering, M.; Zeinstra, L.; van den Born, M.; Korving, J.; Sasaki, N.; Peters, P.J.; van Oudenaarden, A.; et al. Enteroendocrine and tuft cells support Lgr5 stem cells on Paneth cell depletion. Proc. Natl. Acad. Sci. USA 2019, 116, 26599–26605. [Google Scholar] [CrossRef]
- Naik, R.; Galande, S. SATB family chromatin organizers as master regulators of tumor progression. Oncogene 2019, 38, 1989–2004. [Google Scholar] [CrossRef]
- Cadwell, K.; Liu, J.Y.; Brown, S.L.; Miyoshi, H.; Loh, J.; Lennerz, J.K.; Kishi, C.; Kc, W.; Carrero, J.A.; Hunt, S.; et al. A key role for autophagy and the autophagy gene Atg16l1 in mouse and human intestinal Paneth cells. Nature 2008, 456, 259–263. [Google Scholar] [CrossRef] [PubMed]
- Cadwell, K.; Patel, K.K.; Komatsu, M.; Virgin, H.W., 4th; Stappenbeck, T.S. A common role for Atg16L1, Atg5 and Atg7 in small intestinal Paneth cells and Crohn disease. Autophagy 2009, 5, 250–252. [Google Scholar] [CrossRef] [PubMed]
- Inoue, J.; Nishiumi, S.; Fujishima, Y.; Masuda, A.; Shiomi, H.; Yamamoto, K.; Nishida, M.; Azuma, T.; Yoshida, M. Autophagy in the intestinal epithelium regulates Citrobacter rodentium infection. Arch. Biochem. Biophys. 2012, 521, 95–101. [Google Scholar] [CrossRef] [PubMed]
- Delorme-Axford, E.; Klionsky, D.J. Secretory autophagy holds the key to lysozyme secretion during bacterial infection of the intestine. Autophagy 2018, 4, 365–367. [Google Scholar] [CrossRef]
- Deng, G.; Lei, Q.; Gao, X.; Zhang, Y.; Zheng, H.; Bi, J.; Wang, X. Glucagon-like Peptide-2 Modulates Enteric Paneth Cells Immune Response and Alleviates Gut Inflammation During Intravenous Fluid Infusion in Mice with a Central Catheter. Front. Nutr. 2021, 8, 688715. [Google Scholar] [CrossRef]
- Mizuno, R.; Kawada, K.; Itatani, Y.; Ogawa, R.; Kiyasu, Y.; Sakai, Y. The Role of Tumor-Associated Neutrophils in Colorectal Cancer. Int. J. Mol. Sci. 2019, 20, 529. [Google Scholar] [CrossRef]
- Mizuno, R.; Kawada, K.; Sakai, Y. Prostaglandin E2/EP Signaling in the Tumor Microenvironment of Colorectal Cancer. Int. J. Mol. Sci. 2019, 20, 6254. [Google Scholar] [CrossRef]
- Roma-Rodrigues, C.; Mendes, R.; Baptista, P.V.; Fernandes, A.R. Targeting Tumor Microenvironment for Cancer Therapy. Int. J. Mol. Sci. 2019, 20, 840. [Google Scholar] [CrossRef]
- Li, X.L.; Zhou, J.; Xia, C.J.; Min, H.; Lu, Z.K.; Chen, Z.R. PRIMA-1met induces autophagy in colorectal cancer cells through upregulation of the mTOR/AMPK-ULK1-Vps34 signaling cascade. Oncol. Rep. 2021, 45, 86. [Google Scholar] [CrossRef]
- Bellot, G.; Garcia-Medina, R.; Gounon, P.; Chiche, J.; Roux, D.; Pouysségur, J.; Mazure, N.M. Hypoxia-induced autophagy is mediated through hypoxia-inducible factor induction of BNIP3 and BNIP3L via their BH3 domains. Mol. Cell Biol. 2009, 29, 2570–2581. [Google Scholar] [CrossRef]
- Petiot, A.; Ogier-Denis, E.; Blommaart, E.F.; Meijer, A.J.; Codogno, P. Distinct classes of phosphatidylinositol 3′-kinases are involved in signaling pathways that control macroautophagy in HT-29 cells. J. Biol. Chem. 2000, 275, 992–998. [Google Scholar] [CrossRef]
- Arico, S.; Petiot, A.; Bauvy, C.; Dubbelhuis, P.F.; Meijer, A.J.; Codogno, P.; Ogier-Denis, E. The tumor suppressor PTEN positively regulates macroautophagy by inhibiting the phosphatidylinositol 3-kinase/protein kinase B pathway. J. Biol. Chem. 2001, 276, 35243–35246. [Google Scholar] [CrossRef] [PubMed]
- Degtyarev, M.; de Mazière, A.; Orr, C.; Lin, J.; Lee, B.B.; Tien, J.Y.; Prior, W.W.; van Dijk, S.; Wu, H.; Gray, D.C.; et al. Akt inhibition promotes autophagy and sensitizes PTEN-null tumors to lysosomotropic agents. J. Cell Biol. 2008, 183, 101–116. [Google Scholar] [CrossRef]
- Ballesteros-Álvarez, J.; Andersen, J.K. mTORC2: The other mTOR in autophagy regulation. Aging Cell 2021, 20, e13431. [Google Scholar] [CrossRef] [PubMed]
- Lampada, A.; O’Prey, J.; Szabadkai, G.; Ryan, K.M.; Hochhauser, D.; Salomoni, P. mTORC1-independent autophagy regulates receptor tyrosine kinase phosphorylation in colorectal cancer cells via an mTORC2-mediated mechanism. Cell Death Differ. 2017, 24, 1045–1062. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baghdiguian, S.; Verrier, B.; Gerard, C.; Fantini, J. Insulin like growth factor I is an autocrine regulator of human colon cancer cell differentiation and growth. Cancer Lett. 1992, 62, 23–33. [Google Scholar] [CrossRef]
- Donovan, E.A.; Kummar, S. Role of insulin-like growth factor-1R system in colorectal carcinogenesis. Crit. Rev. Oncol. Hematol. 2008, 66, 91–98. [Google Scholar] [CrossRef]
- Fleming-de-Moraes, C.D.; Rocha, M.R.; Tessmann, J.W.; de Araujo, W.M.; Morgado-Diaz, J.A. Crosstalk between PI3K/Akt and Wnt/β-catenin pathways promote colorectal cancer progression regardless of mutational status. Cancer Biol. Ther. 2022, 23, 1–13. [Google Scholar] [CrossRef]
- Sanchez-Lopez, E.; Flashner-Abramson, E.; Shalapour, S.; Zhong, Z.; Taniguchi, K.; Levitzki, A.; Karin, M. Targeting colorectal cancer via its microenvironment by inhibiting IGF-1 receptor-insulin receptor substrate and STAT3 signaling. Oncogene 2016, 35, 2634–2644. [Google Scholar] [CrossRef]
- Habeeb, B.S.; Kitayama, J.; Nagawa, H. Adiponectin supports cell survival in glucose deprivation through enhancement of autophagic response in colorectal cancer cells. Cancer Sci. 2011, 102, 999–1006. [Google Scholar] [CrossRef]
- Ma, Z.; Lou, S.; Jiang, Z. PHLDA2 regulates EMT and autophagy in colorectal cancer via the PI3K/AKT signaling pathway. Aging 2020, 12, 7985–8000. [Google Scholar] [CrossRef]
- Wang, S.; Gu, K. Insulin-like growth factor 1 inhibits autophagy of human colorectal carcinoma drug-resistant cells via the protein kinase B/mammalian target of rapamycin signaling pathway. Mol. Med. Rep. 2018, 17, 2952–2956. [Google Scholar] [CrossRef]
- Shimokawa, M.; Ohta, Y.; Nishikori, S.; Matano, M.; Takano, A.; Fujii, M.; Date, S.; Sugimoto, S.; Kanai, T.; Sato, T.; et al. Visualization and targeting of LGR5+ human colon cancer stem cells. Nature 2017, 545, 187–192. [Google Scholar] [CrossRef] [PubMed]
- Munro, M.J.; Wickremesekera, S.K.; Peng, L.; Tan, S.T.; Itinteang, T. Cancer stem cells in colorectal cancer: A review. J. Clin. Pathol. 2018, 71, 110–116. [Google Scholar] [CrossRef]
- De Sousa e Melo, F.; Kurtova, A.V.; Harnoss, J.M.; Kljavin, N.; Hoeck, J.D.; Hung, J.; Anderson, J.E.; Storm, E.E.; Modrusan, Z.; Koeppen, H.; et al. A distinct role for Lgr5+ stem cells in primary and metastatic colon cancer. Nature 2017, 543, 676–680. [Google Scholar] [CrossRef] [PubMed]
- Heurtier, V.; Owens, N.; Gonzalez, I.; Mueller, F.; Proux, C.; Mornico, D.; Clerc, P.; Dubois, A.; Navarro, P. The molecular logic of Nanog-induced self-renewal in mouse embryonic stem cells. Nat. Commun. 2019, 10, 1109. [Google Scholar] [CrossRef] [PubMed]
- Nazio, F.; Bordi, M.; Cianfanelli, V.; Locatelli, F.; Cecconi, F. Autophagy and cancer stem cells: Molecular mechanisms and therapeutic applications. Cell Death Differ. 2019, 26, 690–702. [Google Scholar] [CrossRef]
- Wu, S.; Wang, X.; Chen, J.; Chen, Y. Autophagy of cancer stem cells is involved with chemoresistance of colon cancer cells. Biochem. Biophys. Res. Commun. 2013, 434, 898–903. [Google Scholar] [CrossRef]
- Qureshi-Baig, K.; Kuhn, D.; Viry, E.; Pozdeev, V.I.; Schmitz, M.; Rodriguez, F.; Ullmann, P.; Koncina, E.; Nurmik, M.; Frasquilho, S.; et al. Hypoxia-induced autophagy drives colorectal cancer initiation and progression by activating the PRKC/PKC-EZR (ezrin) pathway. Autophagy 2020, 16, 1436–1452. [Google Scholar] [CrossRef]
- Dallas, N.A.; Xia, L.; Fan, F.; Gray, M.J.; Gaur, P.; van Buren, G., 2nd; Samuel, S.; Kim, M.P.; Lim, S.J.; Ellis, L.M.; et al. Chemoresistant colorectal cancer cells, the cancer stem cell phenotype, and increased sensitivity to insulin-like growth factor-I receptor inhibition. Cancer Res. 2009, 69, 1951–1957. [Google Scholar] [CrossRef]
- Kasdagly, M.; Radhakrishnan, S.; Reddivari, L.; Veeramachaneni, D.N.; Vanamala, J. Colon carcinogenesis: Influence of Western diet-induced obesity and targeting stem cells using dietary bioactive compounds. Nutrition 2014, 30, 1242–1256. [Google Scholar] [CrossRef]
- Gao, T.; Liu, X.; He, B.; Pan, Y.; Wang, S. IGF2 loss of imprinting enhances colorectal cancer stem cells pluripotency by promoting tumor autophagy. Aging 2020, 12, 21236–21252. [Google Scholar] [CrossRef]
- Wen, Y.A.; Xing, X.; Harris, J.W.; Zaytseva, Y.Y.; Mitov, M.I.; Napier, D.L.; Weiss, H.L.; Mark Evers, B.; Gao, T. Adipocytes activate mitochondrial fatty acid oxidation and autophagy to promote tumor growth in colon cancer. Cell Death Dis. 2017, 8, e2593. [Google Scholar] [CrossRef] [PubMed]
- Del Cornò, M.; Baldassarre, A.; Calura, E.; Conti, L.; Martini, P.; Romualdi, C.; Varì, R.; Scazzocchio, B.; D’Archivio, M.; Masotti, A.; et al. Transcriptome Profiles of Human Visceral Adipocytes in Obesity and Colorectal Cancer Unravel the Effects of Body Mass Index and Polyunsaturated Fatty Acids on Genes and Biological Processes Related to Tumorigenesis. Front. Immunol. 2019, 10, 265. [Google Scholar] [CrossRef] [PubMed]
- Pan, J.; Kothan, S.; Liu, L.; Moe, A.T.M.; Dong, L.; Sun, Y.; Yang, Y. Autophagy participants in the dedifferentiation of mouse 3T3-L1 adipocytes triggered by hypofunction of insulin signaling. Cell. Signal. 2021, 80, 109911. [Google Scholar] [CrossRef]
- Fenton, J.I.; Birmingham, J.M. Adipokine regulation of colon cancer: Adiponectin attenuates interleukin-6-induced colon carcinoma cell proliferation via STAT-3. Mol. Carcinog. 2010, 49, 700–709. [Google Scholar] [CrossRef]
- Richards, C.H.; Roxburgh, C.S.; MacMillan, M.T.; Isswiasi, S.; Robertson, E.G.; Guthrie, G.K.; Horgan, P.G.; McMillan, D.C. The relationships between body composition and the systemic inflammatory response in patients with primary operable colorectal cancer. PLoS ONE 2012, 7, e41883. [Google Scholar] [CrossRef] [PubMed]
- Ye, L.; Zhang, T.; Kang, Z.; Guo, G.; Sun, Y.; Lin, K.; Huang, Q.; Shi, X.; Ni, Z.; Ding, N.; et al. Tumor-Infiltrating Immune Cells Act as a Marker for Prognosis in Colorectal Cancer. Front. Immunol. 2019, 10, 2368. [Google Scholar] [CrossRef]
- Zhu, S.; Wu, Q.; Zhang, B.; Wei, H.; Li, B.; Shi, W.; Fang, M.; Zhu, S.; Wang, L.; Lang, Z.Y.; et al. Autophagy-related gene expression classification defines three molecular subtypes with distinct clinical and microenvironment cell infiltration characteristics in colon cancer. Int. Immunopharmacol. 2020, 87, 106757. [Google Scholar] [CrossRef]
- Zhong, X.; Chen, B.; Yang, Z. The Role of Tumor-Associated Macrophages in Colorectal Carcinoma Progression. Cell. Physiol. Biochem. 2018, 45, 356–365. [Google Scholar] [CrossRef]
- Shao, L.N.; Zhu, B.S.; Xing, C.G.; Yang, X.D.; Young, W.; Cao, J.P. Effects of autophagy regulation of tumor-associated macrophages on radiosensitivity of colorectal cancer cells. Mol. Med. Rep. 2016, 13, 2661–2670. [Google Scholar] [CrossRef] [PubMed]
- Lin, X.T.; Wu, Q.N.; Qin, S.; Fan, D.J.; Lv, M.Y.; Chen, X.; Cai, J.W.; Weng, J.R.; Zou, Y.F.; Rong, Y.M.; et al. Identification of an Autophagy-Related Gene Signature for the Prediction of Prognosis in Early-Stage Colorectal Cancer. Front. Genet. 2021, 12, 755789. [Google Scholar] [CrossRef]
- Chen, S.; Wang, Y.; Wang, B.; Zhang, L.; Su, Y.; Xu, M.; Zhang, M. A signature based on 11 autophagy genes for prognosis prediction of colorectal cancer. PLoS ONE 2021, 16, e0258741. [Google Scholar] [CrossRef] [PubMed]
- Guffey, C.R.; Fan, D.; Singh, U.P.; Murphy, E.A. Linking obesity to colorectal cancer: Recent insights into plausible biological mechanisms. Curr. Opin. Clin. Nutr. Metab. Care 2013, 16, 595–600. [Google Scholar] [CrossRef]
- Bader, J.; Carson, M.; Enos, R.; Velazquez, K.; Sougiannis, A.; Singh, U.; Becker, W.; Nagarkatti, M.; Fan, D.; Murphy, A.; et al. High-fat diet-fed ovariectomized mice are susceptible to accelerated subcutaneous tumor growth potentially through adipose tissue inflammation, local insulin-like growth factor release, and tumor associated macrophages. Oncotarget 2020, 11, 4554–4569. [Google Scholar] [CrossRef]
- Wu, Y.; Yao, J.; Xie, J.; Liu, Z.; Zhou, Y.; Pan, H.; Han, W. The role of autophagy in colitis-associated colorectal cancer. Signal Transduct. Target. Ther. 2018, 3, 31. [Google Scholar] [CrossRef]
- Martinez-Outschoorn, U.E.; Lisanti, M.P.; Sotgia, F. Catabolic cancer-associated fibroblasts transfer energy and biomass to anabolic cancer cells, fueling tumor growth. Semin. Cancer Biol. 2014, 25, 47–60. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Sun, C.; Qin, Z. Metabolic reprogramming of cancer-associated fibroblasts and its effect on cancer cell reprogramming. Theranostics 2021, 11, 8322–8336. [Google Scholar] [CrossRef]
- Denton, A.E.; Roberts, E.W.; Fearon, D.T. Stromal Cells in the Tumor Microenvironment. In Advances in Experimental Medicine and Biology; Springer: Berlin/Heidelberg, Germany, 2018; Volume 1060. [Google Scholar] [CrossRef]
- LeBleu, V.S.; Kalluri, R. A peek into cancer-associated fibroblasts: Origins, functions and translational impact. Dis. Model Mech. 2018, 11, dmm029447. [Google Scholar] [CrossRef]
- Yan, Y.; Chen, X.; Wang, X.; Zhao, Z.; Hu, W.; Zeng, S.; Wei, J.; Yang, X.; Qian, L.; Zhou, S.; et al. The effects and the mechanisms of autophagy on the cancer-associated fibroblasts in cancer. J. Exp. Clin. Cancer Res. 2019, 38, 171. [Google Scholar] [CrossRef]
- Gong, J.; Lin, Y.; Zhang, H.; Liu, C.; Cheng, Z.; Yang, X.; Zhang, J.; Xiao, Y.; Sang, N.; Qian, X.; et al. Reprogramming of lipid metabolism in cancer-associated fibroblasts potentiates migration of colorectal cancer cells. Cell Death Dis. 2020, 11, 267. [Google Scholar] [CrossRef] [PubMed]
- Inoue, T.; Hayashi, Y.; Tsujii, Y.; Yoshii, S.; Sakatani, A.; Kimura, K.; Uema, R.; Kato, M.; Saiki, H.; Shinzaki, S.; et al. Suppression of autophagy promotes fibroblast activation in p53-deficient colorectal cancer cells. Sci. Rep. 2021, 11, 19524. [Google Scholar] [CrossRef] [PubMed]
- Zhou, W.; Xu, G.; Wang, Y.; Xu, Z.; Liu, X.; Xu, X.; Ren, G.; Tian, K. Oxidative stress induced autophagy in cancer associated fibroblast enhances proliferation and metabolism of colorectal cancer cells. Cell Cycle 2017, 16, 73–81. [Google Scholar] [CrossRef]
- Kobayashi, H.; Gieniec, K.A.; Lannagan, T.R.M.; Wang, T.; Asai, N.; Mizutani, Y.; Iida, T.; Ando, R.; Thomas, E.M.; Sakai, A.; et al. The Origin and Contribution of Cancer-Associated Fibroblasts in Colorectal Carcinogenesis. Gastroenterology 2022, 162, 890–906. [Google Scholar] [CrossRef] [PubMed]
- Tommelein, J.; de Vlieghere, E.; Verset, L.; Melsens, E.; Leenders, J.; Descamps, B.; Debucquoy, A.; Vanhove, C.; Pauwels, P.; Gespach, C.P.; et al. Radiotherapy-Activated Cancer-Associated Fibroblasts Promote Tumor Progression through Paracrine IGF1R Activation. Cancer Res. 2018, 78, 659–670. [Google Scholar] [CrossRef]
- Unger, C.; Kramer, N.; Unterleuthner, D.; Scherzer, M.; Burian, A.; Rudisch, A.; Stadler, M.; Schlederer, M.; Lenhardt, D.; Riedl, A.; et al. Stromal-derived IGF2 promotes colon cancer progression via paracrine and autocrine mechanisms. Oncogene 2017, 36, 5341–5355. [Google Scholar] [CrossRef]
- Amaravadi, R.K.; Kimmelman, A.C.; Debnath, J. Targeting Autophagy in Cancer: Recent Advances and Future Directions. Cancer Discov. 2019, 9, 1167–1181. [Google Scholar] [CrossRef]
- Li, Z.; Si, W.; Jin, W.; Yuan, Z.; Chen, Y.; Fu, L. Targeting autophagy in colorectal cancer: An update on pharmacological small-molecule compounds. Drug Discov. Today 2022, 27, 2373–2385. [Google Scholar] [CrossRef]
- Sousa-Squiavinato, A.C.M.; Arregui Ramos, D.A.; Wagner, M.S.; Tessmann, J.W.; de-Freitas-Junior, J.C.M.; Morgado-Díaz, J.A. Long-term resistance to 5-fluorouracil promotes epithelial-mesenchymal transition, apoptosis evasion, autophagy, and reduced proliferation rate in colon cancer cells. Eur. J. Pharmacol. 2022, 933, 175253. [Google Scholar] [CrossRef]
- Fu, Y.; Hong, L.; Xu, J.; Zhong, G.; Gu, Q.; Gu, Q.; Guan, Y.; Zheng, X.; Dai, Q.; Luo, X.; et al. Discovery of a small molecule targeting autophagy via ATG4B inhibition and cell death of colorectal cancer cells in vitro and in vivo. Autophagy 2019, 15, 295–311. [Google Scholar] [CrossRef]
- Wubetu, G.Y.; Utsunomiya, T.; Ishikawa, D.; Ikemoto, T.; Yamada, S.; Morine, Y.; Iwahashi, S.; Saito, Y.; Arakawa, Y.; Imura, S.; et al. Branched chain amino acid suppressed insulin-initiated proliferation of human cancer cells through induction of autophagy. Anticancer Res. 2014, 34, 4789–4796. [Google Scholar]
- Jing, Z.; Fei, W.; Zhou, J.; Zhang, L.; Chen, L.; Zhang, X.; Liang, X.; Xie, J.; Fang, Y.; Sui, X.; et al. Salvianolic acid B, a novel autophagy inducer, exerts antitumor activity as a single agent in colorectal cancer cells. Oncotarget 2016, 7, 61509–61519. [Google Scholar] [CrossRef] [PubMed]
- Fan, X.J.; Wang, Y.; Wang, L.; Zhu, M. Salidroside induces apoptosis and autophagy in human colorectal cancer cells through inhibition of PI3K/Akt/mTOR pathway. Oncol. Rep. 2016, 36, 3559–3567. [Google Scholar] [CrossRef] [PubMed]
- Ji, S.; Tang, S.; Li, K.; Li, Z.; Liang, W.; Qiao, X.; Wang, Q.; Yu, S.; Ye, M. Licoricidin inhibits the growth of SW480 human colorectal adenocarcinoma cells in vitro and in vivo by inducing cycle arrest, apoptosis and autophagy. Toxicol. Appl. Pharmacol. 2017, 326, 25–33. [Google Scholar] [CrossRef]
- Hu, S.; Yin, J.; Yan, S.; Hu, P.; Huang, J.; Zhang, G.; Wang, F.; Tong, Q.; Zhang, Y. Chaetocochin J, an epipolythiodioxopiperazine alkaloid, induces apoptosis and autophagy in colorectal cancer via AMPK and PI3K/AKT/mTOR pathways. Bioorganic Chem. 2021, 109, 104693. [Google Scholar] [CrossRef]
- Mogavero, A.; Maiorana, M.V.; Zanutto, S.; Varinelli, L.; Bozzi, F.; Belfiore, A.; Volpi, C.C.; Gloghini, A.; Pierotti, M.A.; Gariboldi, M.; et al. Metformin transiently inhibits colorectal cancer cell proliferation as a result of either AMPK activation or increased ROS production. Sci. Rep. 2017, 7, 15992. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Fan, Z. The epidermal growth factor receptor antibody cetuximab induces autophagy in cancer cells by downregulating HIF-1alpha and Bcl-2 and activating the beclin 1/hVps34 complex. Cancer Res. 2010, 70, 5942–5952. [Google Scholar] [CrossRef]
- Won, S.J.; Yen, C.H.; Liu, H.S.; Wu, S.Y.; Lan, S.H.; Jiang-Shieh, Y.F.; Lin, C.N.; Su, C.L. Justicidin A-induced autophagy flux enhances apoptosis of human colorectal cancer cells via class III PI3K and Atg5 pathway. J. Cell. Physiol. 2015, 230, 930–946. [Google Scholar] [CrossRef]
- Chen, Z.; Wang, C.; Dong, H.; Wang, X.; Gao, F.; Zhang, S.; Zhang, X. Aspirin has a better effect on PIK3CA mutant colorectal cancer cells by PI3K/Akt/Raptor pathway. Mol. Med. 2020, 26, 14. [Google Scholar] [CrossRef]
- Zhang, C.D.; Wang, Y.L.; Zhou, D.M.; Zhu, M.Y.; Lv, Y.; Hao, X.Q.; Qu, C.F.; Chen, Y.; Gu, W.Z.; Wu, B.Q.; et al. A recombinant Chinese measles virus vaccine strain rMV-Hu191 inhibits human colorectal cancer growth through inducing autophagy and apoptosis regulating by PI3K/AKT pathway. Transl. Oncol. 2021, 14, 101091. [Google Scholar] [CrossRef]
- Wang, J.; Liang, D.; Zhang, X.P.; He, C.F.; Cao, L.; Zhang, S.Q.; Xiao, X.; Li, S.J.; Cao, Y.X. Novel PI3K/Akt/mTOR signaling inhibitor, W922, prevents colorectal cancer growth via the regulation of autophagy. Int. J. Oncol. 2021, 58, 70–82. [Google Scholar] [CrossRef]
- Sasaki, K.; Tsuno, N.H.; Sunami, E.; Tsurita, G.; Kawai, K.; Okaji, Y.; Nishikawa, T.; Shuno, Y.; Hongo, K.; Hiyoshi, M.; et al. Chloroquine potentiates the anti-cancer effect of 5-fluorouracil on colon cancer cells. BMC Cancer 2010, 10, 370. [Google Scholar] [CrossRef]
- Pasquier, B. Autophagy inhibitors. Cell. Mol. Life Sci. 2016, 73, 985–1001. [Google Scholar] [CrossRef] [PubMed]
- Zamame Ramirez, J.A.; Romagnoli, G.G.; Falasco, B.F.; Gorgulho, C.M.; Sanzochi Fogolin, C.; dos Santos, D.C.; Junior, J.P.A.; Lotze, M.T.; Ureshino, R.P.; Kaneno, R.; et al. Blocking drug-induced autophagy with chloroquine in HCT-116 colon cancer cells enhances DC maturation and T cell responses induced by tumor cell lysate. Int. Immunopharmacol. 2020, 84, 106495. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Wang, H.; Yu, M.; Ma, K.; Ning, L. Inhibition of autophagy by 3-methyladenine promotes migration and invasion of colon cancer cells through epithelial mesenchymal transformation. Transl. Cancer Res. 2022, 11, 2834–2842. [Google Scholar] [CrossRef]
- Wang, J.; Wang, J.; Li, L.; Feng, L.; Wang, Y.R.; Wang, Z.; Tan, N.H. RA-XII, a bicyclic hexapeptidic glucoside isolated from Rubia yunnanensis Diels, exerts antitumor activity by inhibiting protective autophagy and activating Akt-mTOR pathway in colorectal cancer cells. J. Ethnopharmacol. 2021, 266, 113438. [Google Scholar] [CrossRef]
- Chang, X.; Dong, R. Transcriptional regulation of autophagy-lysosomal pathway in cancer. Thorac. Cancer 2020, 11, 216–223. [Google Scholar] [CrossRef]
- Yang, X.; Niu, B.; Wang, L.; Chen, M.; Kang, X.; Wang, L.; Ji, Y.; Zhong, J. Autophagy inhibition enhances colorectal cancer apoptosis induced by dual phosphatidylinositol 3-kinase/mammalian target of rapamycin inhibitor NVP-BEZ235. Oncol. Lett. 2016, 12, 102–106. [Google Scholar] [CrossRef]
- Patergnani, S.; Missiroli, S.; Morciano, G.; Perrone, M.; Mantovani, C.M.; Anania, G.; Fiorica, F.; Pinton, P.; Giorgi, C. Understanding the Role of Autophagy in Cancer Formation and Progression Is a Real Opportunity to Treat and Cure Human Cancers. Cancers 2021, 13, 5622. [Google Scholar] [CrossRef]
- Wang, Y.; Lu, J.H.; Wu, Q.N.; Jin, Y.; Wang, D.S.; Chen, Y.X.; Liu, J.; Luo, X.J.; Meng, Q.; Pu, H.Y.; et al. LncRNA LINRIS stabilizes IGF2BP2 and promotes the aerobic glycolysis in colorectal cancer. Mol. Cancer. 2019, 18, 174. [Google Scholar] [CrossRef] [PubMed]
- Xia, C.; Li, Q.; Cheng, X.; Wu, T.; Gao, P.; Gu, Y. Insulin-like growth factor 2 mRNA-binding protein 2-stabilized long non-coding RNA Taurine up-regulated gene 1 (TUG1) promotes cisplatin-resistance of colorectal cancer via modulating autophagy. Bioengineered 2022, 13, 2450–2469. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Cell Type | Autophagy | |
|---|---|---|
| Normal Function | Effects of Functional Impairments | |
| Absorptive cell (colonocyte) |
|
|
| Goblet cell |
|
|
| Intestinal (colonic) stem cell |
|
|
| Paneth-like cell |
|
|
| Cell Type | Paracrine/Autocrine IGF-1 Signaling | |
|---|---|---|
| Normal Function | Effects of Functional Impairments | |
| Absorptive cell (colonocyte) |
|
|
| Goblet cell |
|
|
| Intestinal (colonic) stem cell |
|
|
| Paneth-like cell |
|
|
| Autophagic Activity | Agents | Mechanism of Action | References |
|---|---|---|---|
| Inducers | Cetuximab | Dual inhibitor of EGFR and class I PI3K/AKTt/mTOR | [248] |
| Adiponectin | Activator of AMPKα and PPARα and inhibitor of IGF-1/PI3K/AKT/mTOR | [200] | |
| JA | Increase expression of class III PI3K and Atg5 | [249] | |
| SAL B | Inhibitor of AKT/mTOR | [243] | |
| Salidroside | Inhibitor of PI3K/AKT/mTOR | [244] | |
| LCD | Activator of AMPK and inhibitor of AKT/mTOR | [245] | |
| Metformin | Activator of AMPK and inhibitor of mTOR and IGF-1R | [247] | |
| Calycosin | Activator of SIRT1/AMPK and inhibitor of AKT/mTOR | [88] | |
| Aspirin | Inhibitor of PI3K/AKT/Raptor | [250] | |
| CJ | Activator of AMPK and inhibitor of PI3K/AKT/mTOR | [246] | |
| rMV-Hu191 | Regulator of PI3K/AKT | [251] | |
| W922 | Inhibitor of PI3K/AKT/mTOR | [252] | |
| Inhibitors | 3-MA | Inhibitor of class III PI3K | [191,256] |
| Wortmannin | Inhibitor of PI3K | [191,254] | |
| IGF-1 | Activator of AKT/mTOR in 5-FU-resistant cells | [202] | |
| RA-XII | Regulator of mTOR and NF-κB | [257] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kasprzak, A. Autophagy and the Insulin-like Growth Factor (IGF) System in Colonic Cells: Implications for Colorectal Neoplasia. Int. J. Mol. Sci. 2023, 24, 3665. https://doi.org/10.3390/ijms24043665
Kasprzak A. Autophagy and the Insulin-like Growth Factor (IGF) System in Colonic Cells: Implications for Colorectal Neoplasia. International Journal of Molecular Sciences. 2023; 24(4):3665. https://doi.org/10.3390/ijms24043665
Chicago/Turabian StyleKasprzak, Aldona. 2023. "Autophagy and the Insulin-like Growth Factor (IGF) System in Colonic Cells: Implications for Colorectal Neoplasia" International Journal of Molecular Sciences 24, no. 4: 3665. https://doi.org/10.3390/ijms24043665