
Melatonin Activation by Cytochrome P450 Isozymes: How Does CYP1A2 Compare to CYP1A1?

Abstract
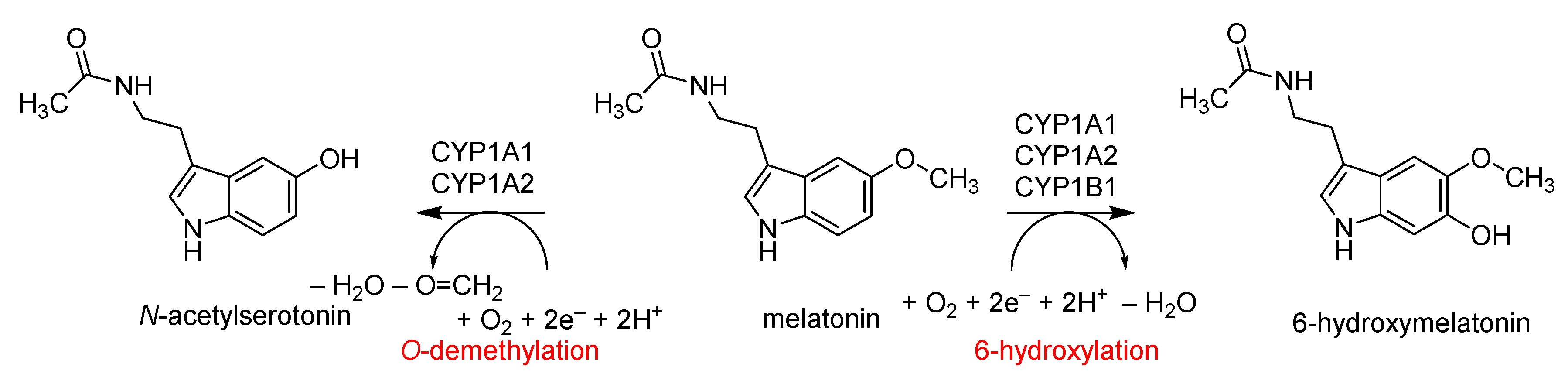
:1. Introduction
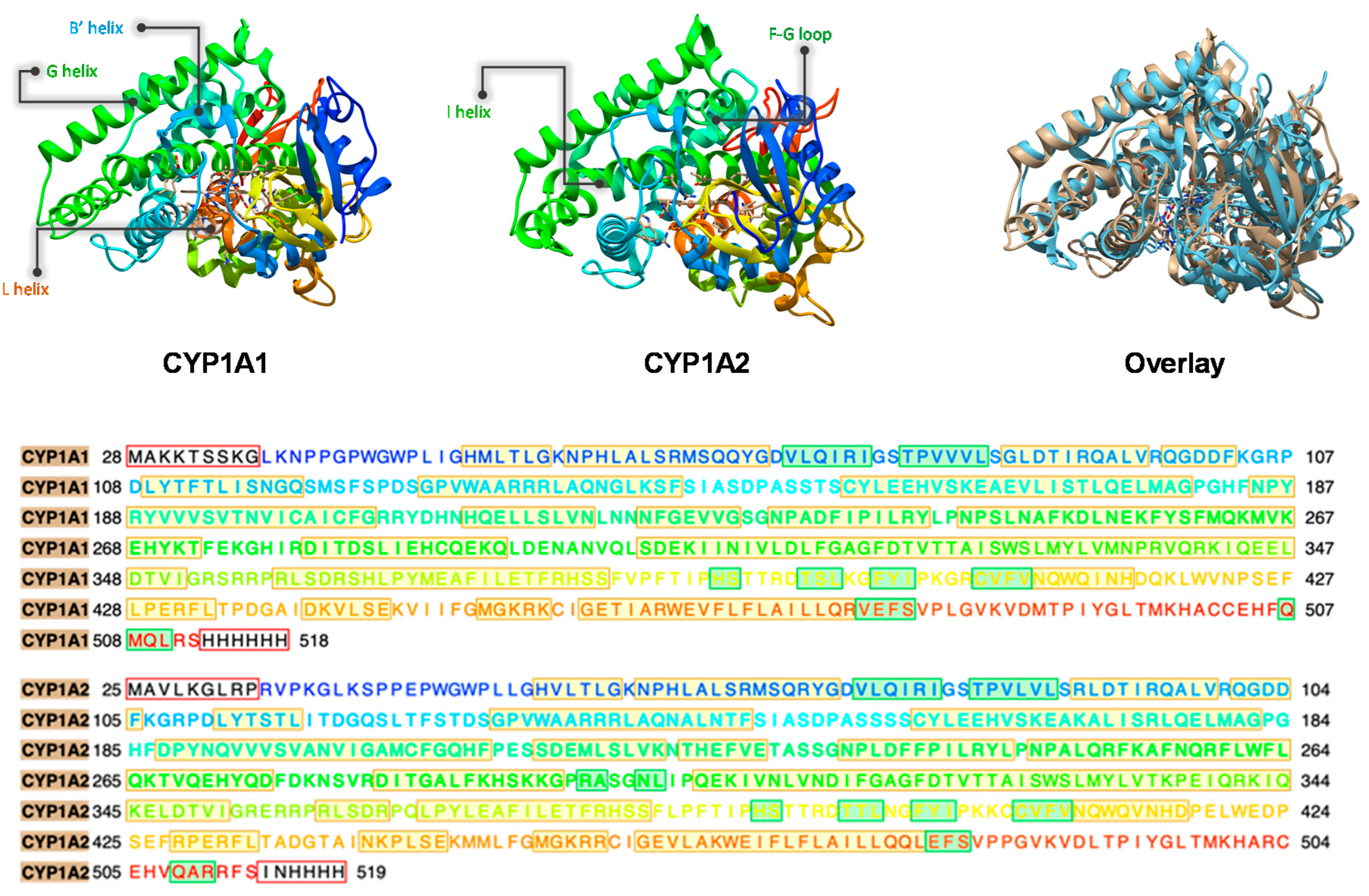
2. Results
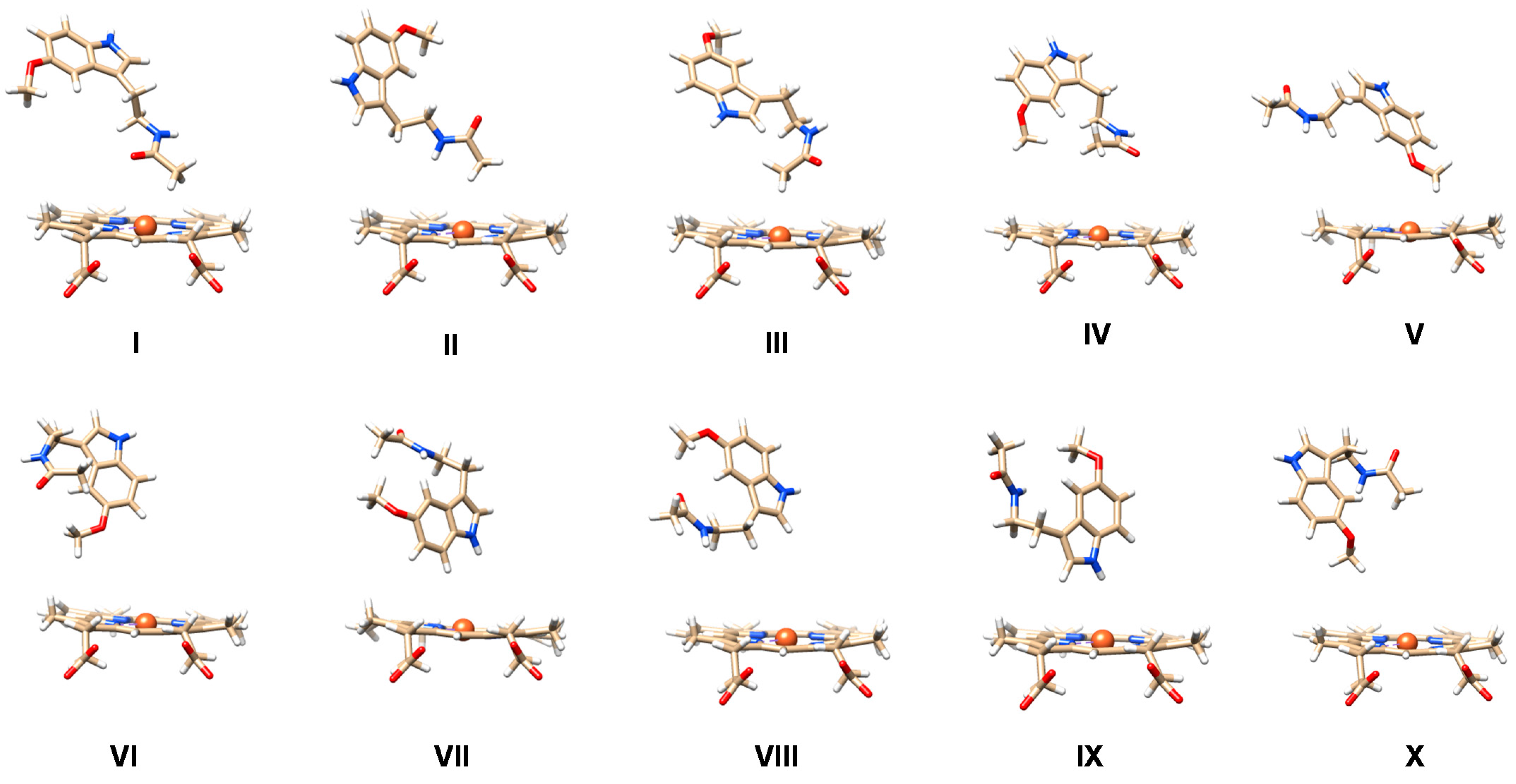
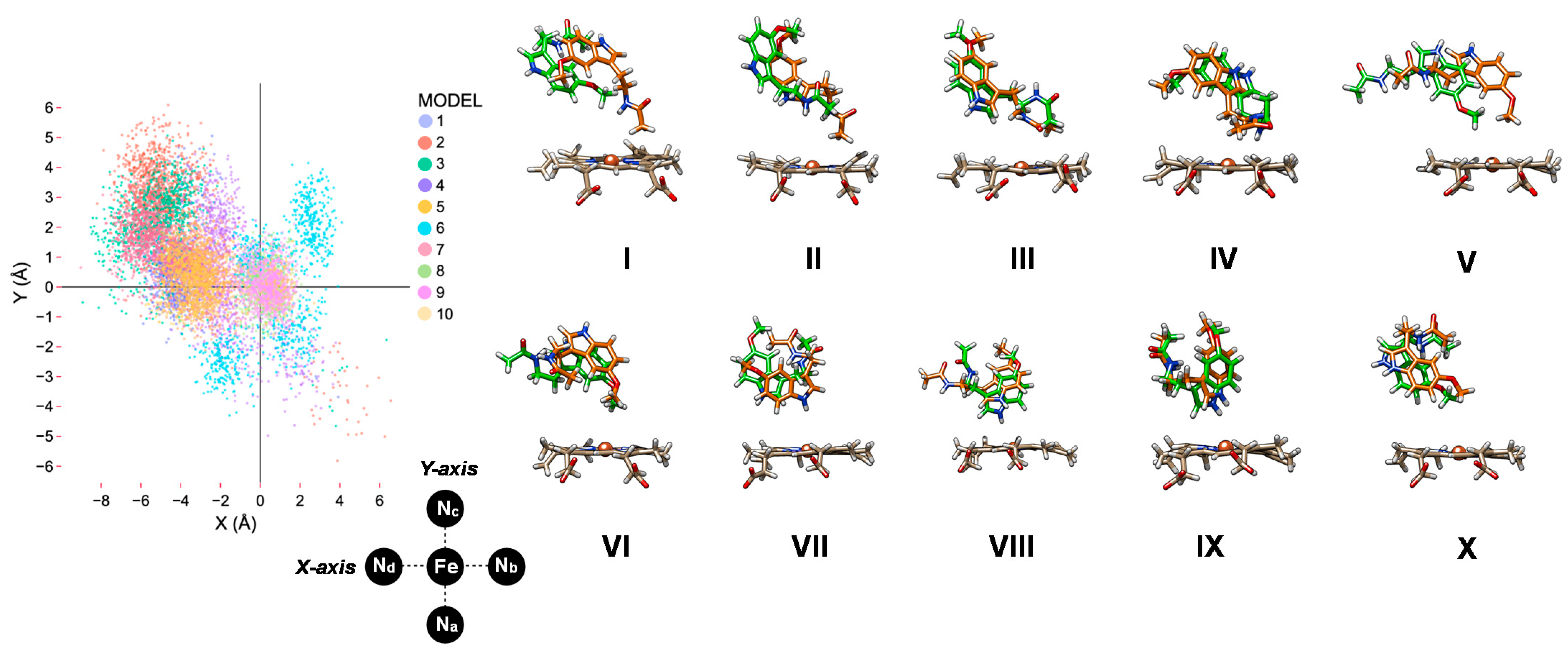
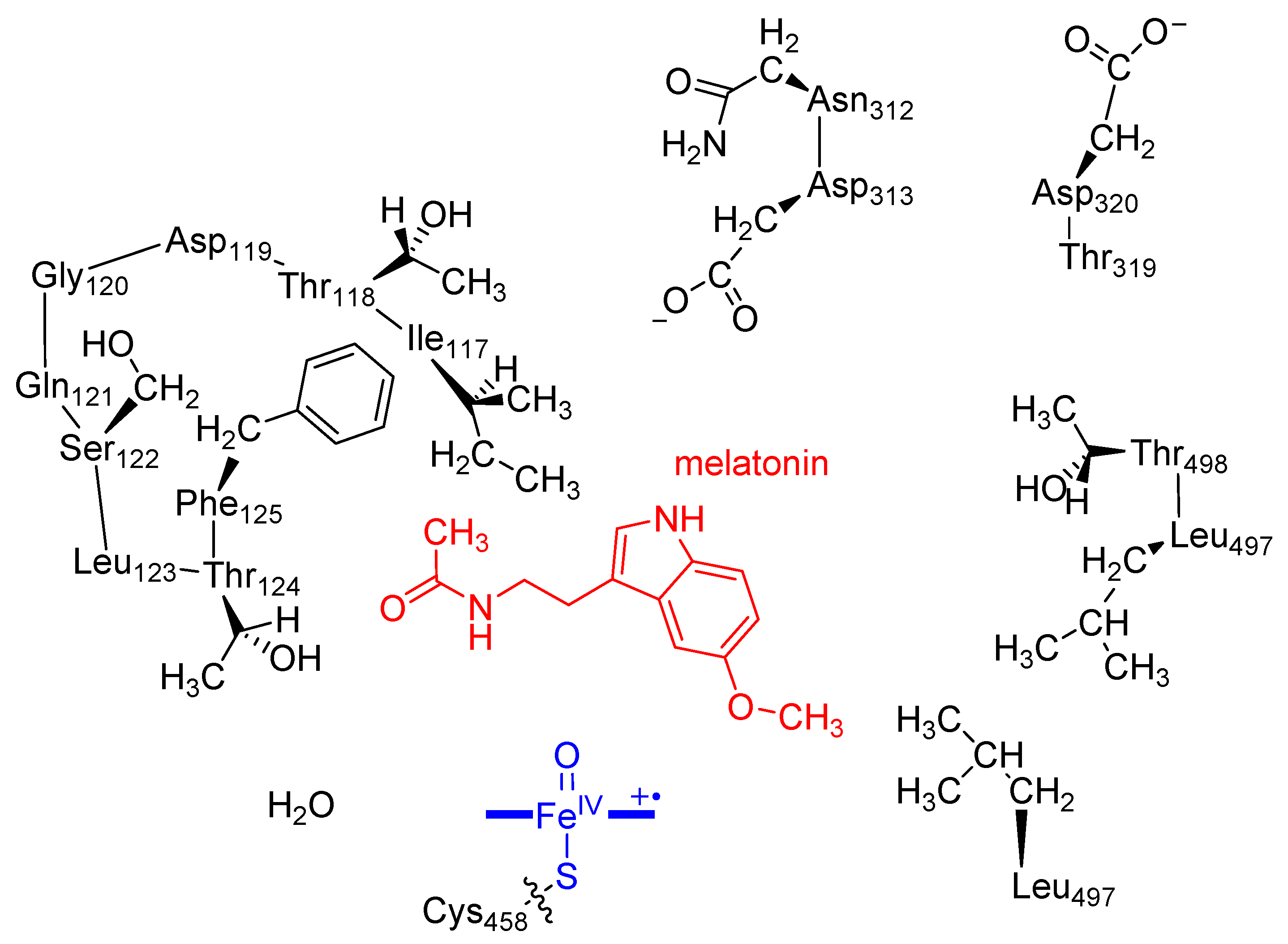
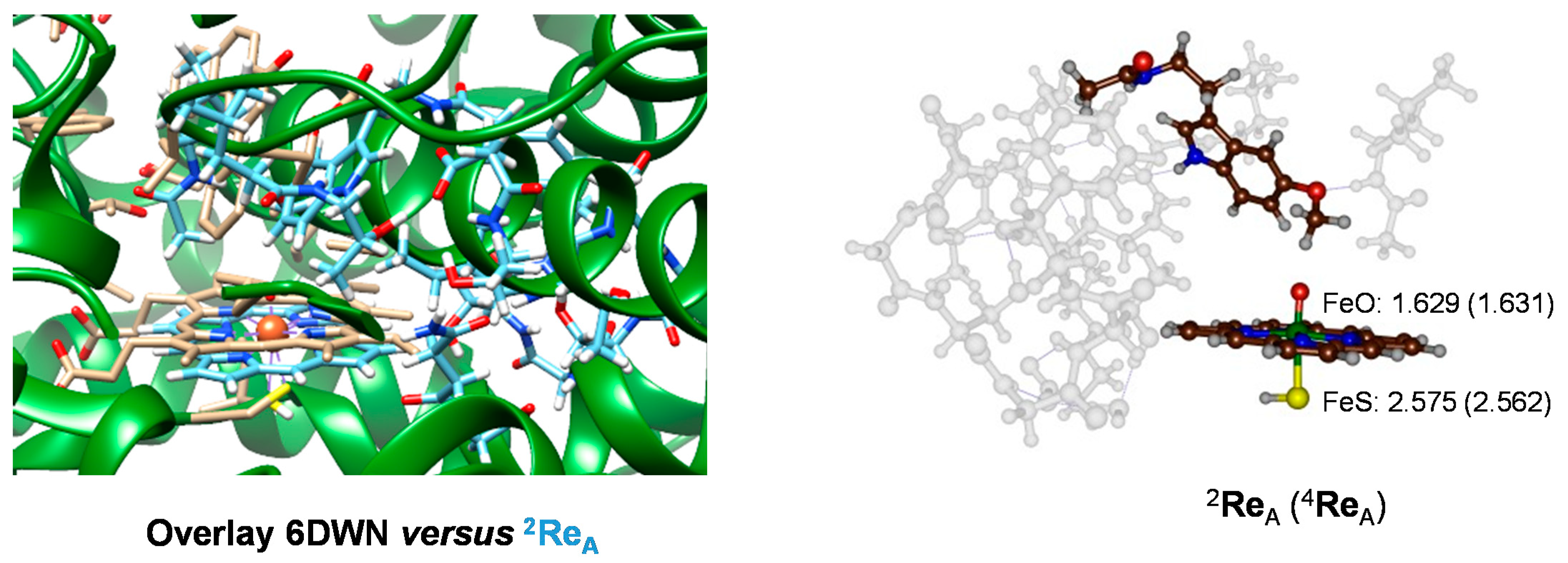
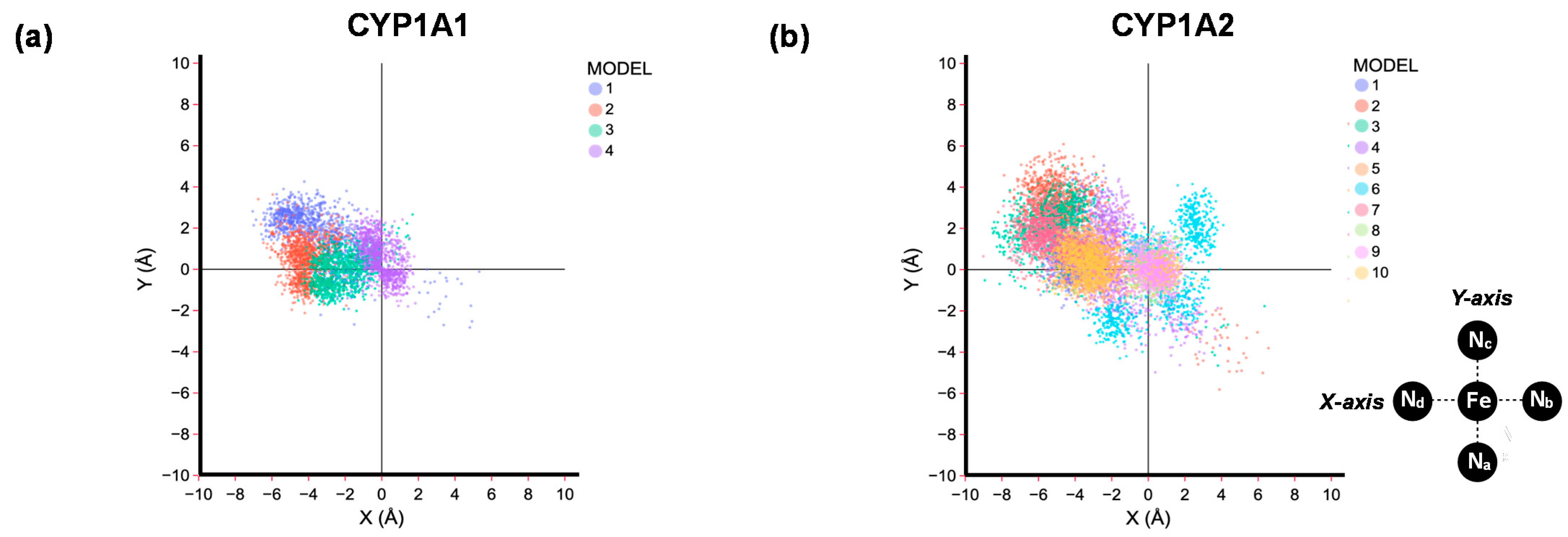
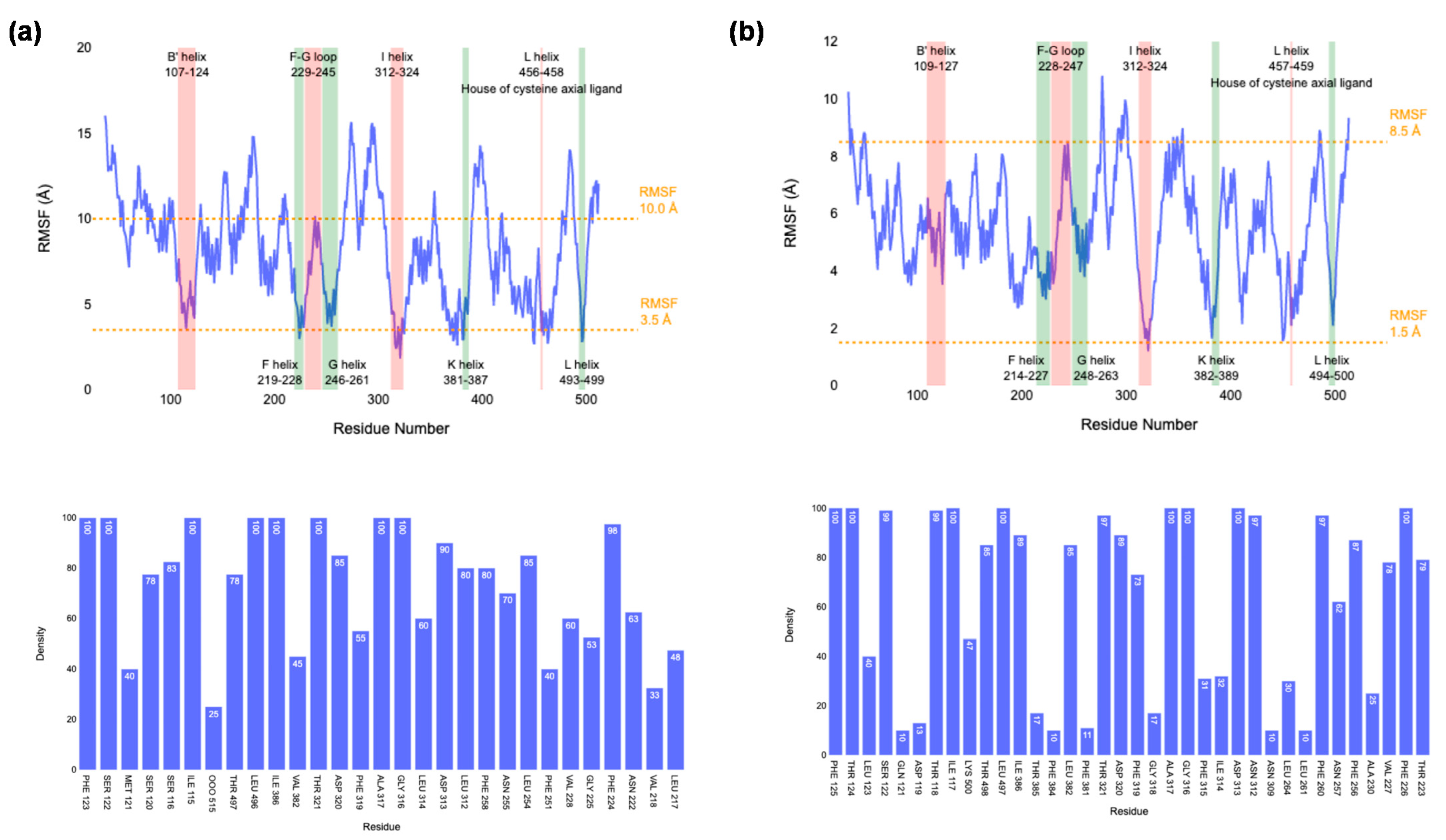
2.1. MD Simulations on Susbtrate Binding and Positioning in CYP1A2
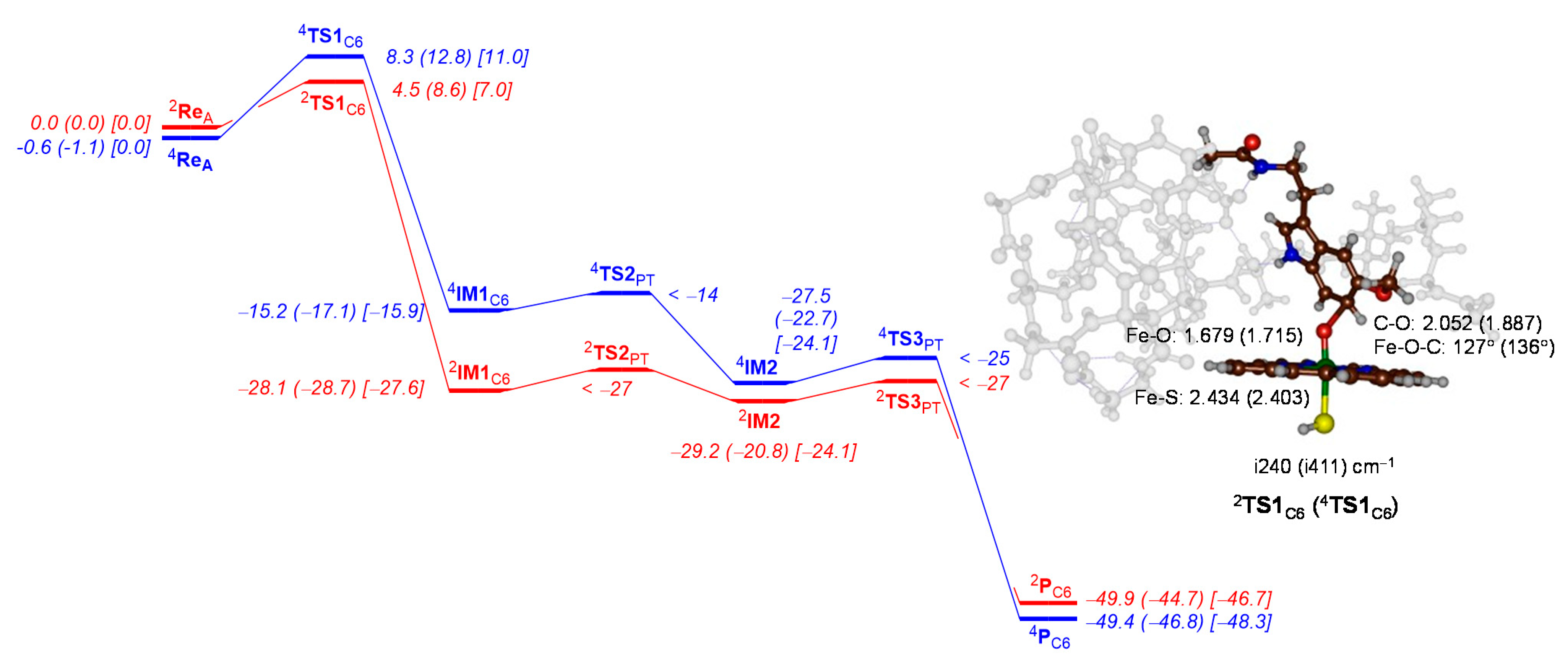
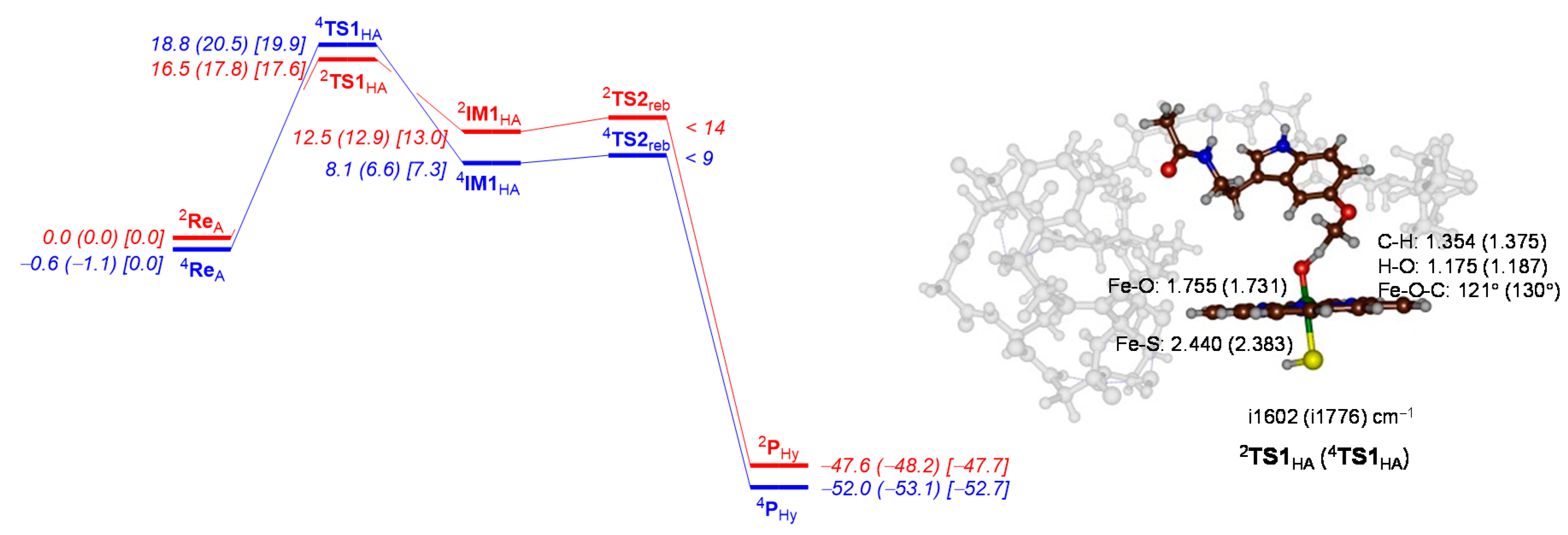
2.2. QM Cluster Calculations on CYP1A2 Mechanism of Melatonin Activation
3. Discussion
4. Materials and Methods
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Esteves, F.; Rueff, J.; Kranendonk, M. The central role of cytochrome P450 in xenobiotic metabolism-a brief review on a fascinating enzyme family. J. Xenobiot. 2021, 11, 94–114. [Google Scholar] [CrossRef] [PubMed]
- Sono, M.; Roach, M.P.; Coulter, E.D.; Dawson, J.H. Heme-containing oxygenases. Chem. Rev. 1996, 96, 2841–2888. [Google Scholar] [CrossRef] [PubMed]
- Meunier, B.; de Visser, S.P.; Shaik, S. Mechanism of oxidation reactions catalyzed by cytochrome P450 enzymes. Chem. Rev. 2004, 104, 3947–3980. [Google Scholar] [CrossRef] [PubMed]
- Denisov, I.G.; Makris, T.M.; Sligar, S.G.; Schlichting, I. Structure and chemistry of cytochrome P450. Chem. Rev. 2005, 105, 2253–2277. [Google Scholar] [CrossRef]
- Ortiz de Montellano, P.R. (Ed.) Cytochrome P450: Structure, Mechanism and Biochemistry, 3rd ed.; Kluwer Academic/Plenum Publishers: New York, NY, USA, 2005. [Google Scholar]
- Kadish, K.M.; Smith, K.M.; Guilard, R. (Eds.) Handbook of Porphyrin Science; World Scientific Publishing Co.: New Jersey, NJ, USA, 2010. [Google Scholar]
- Grogan, G. Cytochromes P450: Exploiting diversity and enabling application as biocatalysts. Curr. Opin. Chem. Biol. 2011, 15, 241–248. [Google Scholar] [CrossRef]
- Raunio, H.; Kuusisto, M.; Juvonen, R.O.; Pentikäinen, O.T. Modeling of interactions between xenobiotics and cytochrome P450 (CYP) enzymes. Front. Pharmacol. 2015, 6, 123. [Google Scholar] [CrossRef]
- Stavropoulou, E.; Pircalabioru, G.G.; Bezirtzoglou, E. The role of cytochromes P450 in infection. Front. Immunol. 2018, 9, 89. [Google Scholar] [CrossRef] [PubMed]
- Dunham, N.P.; Arnold, F.H. Nature’s machinery, repurposed: Expanding the repertoire of iron-dependent oxygenases. ACS Catal. 2020, 10, 12239–12255. [Google Scholar] [CrossRef]
- Poulos, T.L.; Follmer, A.H. Updating the paradigm: Redox partner binding and conformational dynamics in cytochromes P450. Acc. Chem. Res. 2022, 55, 373–380. [Google Scholar] [CrossRef]
- Nelson, D.R. The cytochrome P450 homepage. Hum. Genom. 2009, 4, 59–65. [Google Scholar] [CrossRef] [PubMed]
- Plant, N. The human cytochrome P450 sub-family: Transcriptional regulation, inter-individual variation and interaction networks. Biochim. Biophys. Acta 2007, 1770, 478–488. [Google Scholar] [CrossRef]
- Nebert, D.W.; Wikvall, K.; Miller, W.L. Human cytochromes P450 in health and disease. Philos. Trans. R. Soc. B Biol. Sci. 2013, 368, 20120431. [Google Scholar] [CrossRef]
- Mak, P.J.; Denisov, I.G. Spectroscopic studies of the cytochrome P450 reaction mechanism. Biochim. Biophys. Acta 2018, 1866, 178–204. [Google Scholar] [CrossRef]
- Whitehouse, C.J.C.; Bell, S.G.; Wong, L.-L. Desaturation of alkylbenzenes by cytochrome P450BM3 (CYP102A1). Chem. Eur. J. 2008, 14, 10905–10908. [Google Scholar] [CrossRef]
- Pickl, M.; Kurakin, S.; Cantú Reinhard, F.G.; Schmid, P.; Pöcheim, A.; Winkler, C.K.; Kroutil, W.; de Visser, S.P.; Faber, K. Mechanistic studies of fatty acid activation by CYP152 peroxygenases reveal unexpected desaturase activity. ACS Catal. 2019, 9, 565–577. [Google Scholar] [CrossRef]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The protein databank. Nucl. Acids Res. 2000, 28, 235–243. [Google Scholar] [CrossRef]
- Raag, R.; Swanson, B.A.; Poulos, T.L.; Ortiz de Montellano, P.R. Formation, crystal structure, and rearrangement of a cytochrome P-450cam iron-phenyl complex. Biochemistry 1990, 29, 8119–8126. [Google Scholar] [CrossRef]
- Scott, E.E.; He, Y.A.; Wester, M.R.; White, M.A.; Chin, C.C.; Halpert, J.R.; Johnson, E.F.; Stout, C.D. An open conformation of mammalian cytochrome P450 2B4 at 1.6-Å resolution. Proc. Natl. Acad. Sci. USA 2003, 100, 13196–13201. [Google Scholar] [CrossRef]
- Leys, D.; Mowat, C.G.; McLean, K.J.; Richmond, A.; Chapman, S.K.; Walkinshaw, M.D.; Munro, A.W. Atomic structure of Mycobacterium tuberculosis CYP121 to 1.06 A reveals novel features of cytochrome P450. J. Biol. Chem. 2003, 278, 5141–5147. [Google Scholar] [CrossRef]
- Rittle, J.; Green, M.T. Cytochrome P450 compound I: Capture, characterization, and C-H bond activation kinetics. Science 2010, 330, 933–937. [Google Scholar] [CrossRef]
- Huang, X.; Groves, J.T. Oxygen activation and radical transformations in heme proteins and metalloporphyrins. Chem. Rev. 2018, 118, 2491–2553. [Google Scholar] [CrossRef] [PubMed]
- Dubey, K.D.; Shaik, S. Cytochrome P450: The wonderful nanomachine revealed through dynamic simulations of the catalytic cycle. Acc. Chem. Res. 2019, 52, 389–399. [Google Scholar] [CrossRef] [PubMed]
- Shaik, S.; Kumar, D.; de Visser, S.P.; Altun, A.; Thiel, W. Theoretical perspective on the structure and mechanism of cytochrome P450 enzymes. Chem. Rev. 2005, 105, 2279–2328. [Google Scholar] [CrossRef] [PubMed]
- Wise, C.E.; Hsieh, C.H.; Poplin, N.L.; Makris, T.M. Dioxygen activation by the biofuel-generating cytochrome P450 OleT. ACS Catal. 2018, 8, 9342–9352. [Google Scholar] [CrossRef]
- Castro Martínez, F.M.; Páez López, D.; Sarmiento Pavía, P.D.; Sosa Torres, M.E.; Kroneck, P.M.H. Cytochrome P450. The dioxygen-activating heme thiolate. Met. Ions Life Sci. 2020, 20, 1. [Google Scholar]
- Mallinson, S.J.B.; Machovina, M.M.; Silveira, R.L.; Garcia-Borràs, M.; Gallup, N.; Johnson, C.W.; Allen, M.D.; Skaf, M.S.; Crowley, M.F.; Neidle, E.L.; et al. A promiscuous cytochrome P450 aromatic O-demethylase for lignin bioconversion. Nat. Commun. 2018, 9, 2487. [Google Scholar] [CrossRef]
- Relling, M.V.; Nemec, J.; Schuetz, E.G.; Schuetz, J.D.; Gonzalez, F.J.; Korzekwa, K.R. O-demethylation of epipodophyllotoxins is catalyzed by human cytochrome P450 3A4. Mol. Pharmacol. 1994, 45, 352–358. [Google Scholar]
- Kumar, D.; de Visser, S.P.; Shaik, S. Oxygen economy of cytochrome P450: What is the origin of the mixed functionality as a dehydrogenase–oxidase enzyme compared with its normal function? J. Am. Chem. Soc. 2004, 126, 5072–5073. [Google Scholar] [CrossRef]
- Kumar, D.; de Visser, S.P.; Sharma, P.K.; Hirao, H.; Shaik, S. Sulfoxidation mechanisms catalyzed by cytochrome P450 and horseradish peroxidase models: Spin selection induced by the ligand. Biochemistry 2005, 44, 8148–8158. [Google Scholar] [CrossRef]
- Kumar, D.; Tahsini, L.; de Visser, S.P.; Kang, H.Y.; Kim, S.J.; Nam, W. The effect of porphyrin ligands on the regioselective dehydrogenation versus epoxidation of olefins by oxoiron(IV) mimics of cytochrome P450. J. Phys. Chem. A 2009, 113, 11713–11722. [Google Scholar] [CrossRef]
- Hagel, J.; Facchini, P. Biochemistry and occurrence of O-demethylation in plant metabolism. Front. Physiol. 2010, 1, 14. [Google Scholar] [CrossRef]
- Ali, H.S.; Henchman, R.H.; de Visser, S.P. Lignin biodegradation by a cytochrome P450 enzyme: A computational study into syringol activation by GcoA. Chem. Eur. J. 2020, 26, 13093–13102. [Google Scholar] [CrossRef]
- Zhao, Y.; Marschall, E.; Treisman, M.; McKay, A.; Padva, L.; Crüsemann, M.; Nelson, D.R.; Steer, D.L.; Schittenhelm, R.B.; Tailhades, J.; et al. Cytochrome P450Blt enables versatile peptide cyclisation to generate histidine- and tyrosine-containing crosslinked tripeptide building blocks. Angew. Chem. Int. Ed. 2022, 61, e202204957. [Google Scholar]
- Espinoza, R.V.; Maskeri, M.A.; Turlik, A.; Nangia, A.; Khatri, Y.; Montgomery, J.; Houk, K.N.; Sherman, D.H. Epoxidation and late-stage C−H functionalization by P450 TamI are mediated by variant heme-iron oxidizing species. ACS Catal. 2022, 12, 3731–3742. [Google Scholar] [CrossRef]
- Coleman, T.; Kirk, A.M.; Lee, J.H.Z.; Doherty, D.Z.; Bruning, J.B.; Krenske, E.H.; De Voss, J.J.; Bell, S.G. Different geometric requirements for cytochrome P450-catalyzed aliphatic versus aromatic hydroxylation results in chemoselective oxidation. ACS Catal. 2022, 12, 1258–1267. [Google Scholar] [CrossRef]
- Walsh, A.A.; Szklarz, G.D.; Scott, E.E. Human cytochrome P450 1A1 structure and utility in understanding drug and xenobiotic metabolism. J. Biol. Chem. 2013, 288, 12932–12943. [Google Scholar] [CrossRef]
- Kapelyukh, Y.; Henderson, C.J.; Scheer, N.; Rode, A.; Wolf, C.R. Defining the contribution of CYP1A1 and CYP1A2 to drug metabolism using humanized CYP1A1/1A2 and cyp1a1/cyp1a2 knockout mice. Drug Metab. Dispos. 2019, 47, 907–918. [Google Scholar] [CrossRef]
- Androutsopoulos, V.P.; Tsatsakis, A.M.; Spandidos, D.A. Cytochrome P450 CYP1A1: Wider roles in cancer progression and prevention. BMC Cancer 2009, 9, 187. [Google Scholar] [CrossRef]
- Riddick, D.S.; Ding, X.; Wolf, C.R.; Porter, T.D.; Pandey, A.V.; Zhang, Q.-Y.; Gu, J.; Finn, R.D.; Ronseaux, S.; McLaughlin, L.A.; et al. NADPH–cytochrome P450 oxidoreductase: Roles in physiology, pharmacology, and toxicology. Drug Metabol. Disp. 2013, 41, 12–23. [Google Scholar] [CrossRef]
- Alehaideb, Z.; Sheriffdeen, M.; Law, F.C.P. Inhibition of caffeine metabolism by Apiaceous and Rutaceae families of plant products in humans: In vivo and in vitro studies. Front. Pharmacol. 2021, 12, 641090. [Google Scholar] [CrossRef]
- White, C.M.; Sicignano, D.J.; Smith, K. Impact of interferons and biological drug inhibitors of IL-2 and IL-6 on small-molecule drug metabolism through the cytochrome P450 system. Ann. Pharmacother. 2022, 56, 170–180. [Google Scholar] [CrossRef]
- Grzegorzewski, J.; Bartsch, F.; Köller, A.; König, M. Pharmacokinetics of caffeine: A systematic analysis of reported data for application in metabolic phenotyping and liver function testing. Front. Pharmacol. 2022, 12, 752826. [Google Scholar] [CrossRef]
- Mak, P.J.; Duggal, R.; Denisov, I.G.; Gregory, M.C.; Sligar, S.G.; Kincaid, J.R. Human cytochrome CYP17A1: The structural basis for compromised lyase activity with 17-hydroxyprogesterone. J. Am. Chem. Soc. 2018, 140, 7324–7331. [Google Scholar] [CrossRef]
- Morlock, L.K.; Grobe, S.; Balke, K.; Mauersberger, S.; Böttcher, D.; Bornscheuer, U.T. Protein engineering of the progesterone hydroxylating P450-monooxygenase CYP17A1 alters its regioselectivity. ChemBioChem. 2018, 19, 1954–1958. [Google Scholar] [CrossRef]
- Labbè, L.; Abolfathi, Z.; Lessard, E.; Pakdel, H.; Beaune, P.; Turgeon, J. Role of specific cytochrome P450 enzymes in the N-oxidation of the antiarrhythmic agent mexiletine. Xenobiotica 2003, 33, 13–25. [Google Scholar] [CrossRef]
- Bart, A.G.; Scott, E.E. Structures of human cytochrome P450 1A1 with bergamottin and erlotinib reveal active-site modifications for binding of diverse ligands. J. Biol. Chem. 2018, 293, 19201–19210. [Google Scholar] [CrossRef]
- Sansen, S.; Yano, J.K.; Reynald, R.L.; Schoch, G.A.; Griffin, K.J.; Stout, C.D.; Johnson, E.F. Adaptations for the oxidation of polycyclic aromatic hydrocarbons exhibited by the structure of human P450 1A2. J. Biol. Chem. 2007, 282, 14348–14355. [Google Scholar] [CrossRef] [PubMed]
- Yoshigae, Y.; Kent, U.M.; Hollenberg, P.F. Role of the highly conserved threonine in cytochrome P450 2E1: Prevention of H2O2-induced inactivation during electron transfer. Biochemistry 2013, 52, 4636–4647. [Google Scholar] [CrossRef] [PubMed]
- Hamdane, D.; Zhang, H.; Hollenberg, P.F. Oxygen activation by cytochrome P450 monooxygenase. Photosynth. Res. 2008, 98, 657–666. [Google Scholar] [CrossRef]
- Winn, P.J.; Lüdemann, S.K.; Gauges, R.; Lounnas, V.; Wade, R.C. Comparison of the dynamics of substrate access channels in three cytochrome P450s reveals different opening mechanisms and a novel functional role for a buried arginine. Proc. Natl. Acad. Sci. USA 2002, 99, 5361–5366. [Google Scholar] [CrossRef]
- Mustafa, G.; Nandekar, P.P.; Camp, T.J.; Bruce, N.J.; Gregory, M.C.; Sligar, S.G.; Wade, R.C. Influence of Transmembrane Helix Mutations on Cytochrome P450-Membrane Interactions and Function. Biophys. J. 2019, 116, 419–432. [Google Scholar] [CrossRef] [PubMed]
- Sivaramakrishnan, S.; Ouellet, H.; Matsumura, H.; Guan, S.; Moënne-Loccoz, P.; Burlingame, A.L.; Ortiz de Montellano, P.R. Proximal ligand electron donation and reactivity of the cytochrome P450 ferric–peroxo anion. J. Am. Chem. Soc. 2012, 134, 6673–6684. [Google Scholar] [CrossRef] [PubMed]
- Ogliaro, F.; de Visser, S.P.; Shaik, S. The “push” effect of the thiolate ligand in cytochrome P450: A theoretical gauging. J. Inorg. Biochem. 2002, 91, 554–567. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, H.; Inabe, K.; Shirakawa, Y.; Umezawa, N.; Kato, N.; Higuchi, T. Role of thiolate ligand in spin state and redox switching in the cytochrome P450 catalytic cycle. Inorg. Chem. 2017, 56, 4245–4248. [Google Scholar] [CrossRef]
- Harris, D.; Loew, G.; Waskell, L. Structure and spectra of ferrous dioxygen and reduced ferrous dioxygen model cytochrome P450. J. Am. Chem. Soc. 1998, 120, 4308–4318. [Google Scholar] [CrossRef]
- Kondo, S.; Sakaki, T.; Ohkawa, H.; Inouye, K. Electrostatic interaction between cytochrome P450 and NADPH-P450 reductase: Comparison of mixed and fused systems consisting of rat cytochrome P450 1A1 and yeast NADPH-P450 reductase. Biochem. Biophys. Res. Commun. 1999, 257, 273–278. [Google Scholar] [CrossRef] [PubMed]
- Ogliaro, F.; de Visser, S.P.; Cohen, S.; Kaneti, J.; Shaik, S. The experimentally elusive oxidant of cytochrome P450: A theoretical “trapping” defining more closely the “real” species. ChemBioChem 2001, 2, 848–851. [Google Scholar] [CrossRef]
- Guengerich, F.P. Mechanisms of cytochrome P450-catalyzed oxidations. ACS Catal. 2018, 8, 10964–10976. [Google Scholar] [CrossRef]
- Escribano, B.; Colín-González, A.; Santamaria, A.; Túnez, I. The role of melatonin in multiple sclerosis, Huntington’s disease and cerebral ischemia. CNS Neurol. Disord. Drug Targets 2014, 13, 1096–1119. [Google Scholar] [CrossRef]
- Slominski, A.T.; Semak, I.; Fischer, T.W.; Kim, T.-K.; Kleszczyński, K.; Hardeland, R.; Reiter, R.J. Metabolism of melatonin in the skin: Why is it important? Exp. Dermatol. 2017, 26, 563–568. [Google Scholar] [CrossRef]
- Hardeland, R. Taxon- and site-specific melatonin catabolism. Molecules 2017, 22, 2015. [Google Scholar] [CrossRef]
- Salehi, B.; Sharopov, F.; Fokou, P.V.T.; Kobylinska, A.; de Jonge, L.; Tadio, K.; Sharifi-Rad, J.; Posmyk, M.M.; Martorell, M.; Martins, N.; et al. Melatonin in medicinal and food plants: Occurrence, bioavailability, and health potential for humans. Cells 2019, 8, 681. [Google Scholar] [CrossRef]
- Mokkawes, T.; Lim, Z.Q.; de Visser, S.P. Mechanism of melatonin metabolism by CYP1A1. What determines the bifurcation pathways of hydroxylation versus deformylation? J. Phys. Chem. B 2022, 126, 9591–9606. [Google Scholar] [CrossRef]
- Schyman, P.; Usharani, D.; Wang, Y.; Shaik, S. Brain chemistry: How does P450 catalyze the O-demethylation reaction of 5-methoxytryptamine to yield serotonin? J. Phys. Chem. B 2010, 114, 7078–7089. [Google Scholar] [CrossRef]
- Oláh, J.; Mulholland, A.J.; Harvey, J.N. Understanding the determinants of selectivity in drug metabolism through modeling of dextromethorphan oxidation by cytochrome P450. Proc. Natl. Acad. Sci. USA 2011, 108, 6050–6055. [Google Scholar] [CrossRef]
- Rydberg, P.; Ryde, U.; Olsen, L. Sulfoxide, sulfur, and nitrogen oxidation and dealkylation by cytochrome P450. J. Chem. Theory Comput. 2008, 4, 1369–1377. [Google Scholar] [CrossRef]
- Li, D.; Wang, Y.; Yang, C.; Han, K. Theoretical study of N-dealkylation of N-cyclopropyl-N-methylaniline catalyzed by cytochrome P450: Insight into the origin of the regioselectivity. Dalton Trans. 2009, 38, 291–297. [Google Scholar] [CrossRef]
- de Visser, S.P.; Shaik, S. A proton-shuttle mechanism mediated by the porphyrin in benzene hydroxylation by cytochrome P450 enzymes. J. Am. Chem. Soc. 2003, 125, 7413–7424. [Google Scholar] [CrossRef]
- Bathelt, C.M.; Mulholland, A.J.; Harvey, J.N. QM/MM modelling of benzene hydroxylation in human cytochrome P450 2C9. J. Phys. Chem. A 2008, 112, 13149–13156. [Google Scholar] [CrossRef]
- Shaik, S.; Milko, P.; Schyman, P.; Usharani, D.; Chen, H. Trends in aromatic oxidation reactions catalyzed by cytochrome P450 enzymes: A valence bond modeling. J. Chem. Theory Comput. 2011, 7, 327–339. [Google Scholar] [CrossRef]
- Colomban, C.; Tobing, A.H.; Mukherjee, G.; Sastri, C.V.; Sorokin, A.B.; de Visser, S.P. Mechanism of oxidative activation of fluorinated aromatic compounds by N-bridged diiron-phthalocyanine. What determines the reactivity? Chem. Eur. J. 2019, 25, 14320–14331. [Google Scholar] [CrossRef]
- de Visser, S.P. Substitution of hydrogen by deuterium changes the regioselectivity of ethylbenzene hydroxylation by an oxo-iron-porphyrin catalyst. Chem. Eur. J. 2006, 12, 8168–8177. [Google Scholar] [CrossRef]
- Kumar, D.; Sastry, G.N.; de Visser, S.P. Axial ligand effect on the rate constant of aromatic hydroxylation by iron(IV)-oxo complexes mimicking cytochrome P450 enzymes. J. Phys. Chem. B 2012, 116, 718–730. [Google Scholar] [CrossRef]
- Cantú Reinhard, F.G.; Sainna, M.A.; Upadhyay, P.; Balan, G.A.; Kumar, D.; Fornarini, S.; Crestoni, M.E.; de Visser, S.P. A systematic account on aromatic hydroxylation by a cytochrome P450 model Compound I: A low-pressure mass spectrometry and computational study. Chem. Eur. J. 2016, 22, 18608–18619. [Google Scholar] [CrossRef]
- Ma, X.; Idle, J.; Krausz, K.; Gonzalez, F. Metabolism of melatonin by human cytochromes P450. Drug Metab. Dispos. 2005, 33, 489–494. [Google Scholar] [CrossRef]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef]
- de Visser, S.P. Second-coordination sphere effects on selectivity and specificity of heme and nonheme iron enzymes. Chem. Eur. J. 2020, 26, 5308–5327. [Google Scholar] [CrossRef]
- Mukherjee, G.; Satpathy, J.K.; Bagha, U.K.; Mubarak, M.Q.E.; Sastri, C.V.; de Visser, S.P. Inspiration from Nature: Influence of engineered ligand scaffolds and auxiliary factors on the reactivity of biomimetic oxidants. ACS Catal. 2021, 11, 9761–9797. [Google Scholar] [CrossRef]
- Siegbahn, P.E.M.; Himo, F. The quantum chemical cluster approach for modeling enzyme reactions. WIRES 2011, 1, 323–336. [Google Scholar] [CrossRef]
- Himo, F.; de Visser, S.P. Status report on the quantum chemical cluster approach for modeling enzyme reactions. Commun. Chem. 2022, 5, 29. [Google Scholar] [CrossRef]
- Green, M.T. Evidence for Sulfur-Based Radicals in Thiolate Compound I Intermediates. J. Am. Chem. Soc. 1999, 121, 7939–7940. [Google Scholar] [CrossRef]
- Schöneboom, J.C.; Lin, H.; Reuter, N.; Thiel, W.; Cohen, S.; Ogliaro, F.; Shaik, S. The elusive oxidant species of cytochrome P450 enzymes: Characterization by combined quantum mechanical/molecular mechanical (QM/MM) calculations. J. Am. Chem. Soc. 2002, 124, 8142–8151. [Google Scholar] [CrossRef]
- De Visser, S.P.; Shaik, S.; Sharma, P.K.; Kumar, D.; Thiel, W. Active species of horseradish peroxidase (HRP) and cytochrome P450: Two electronic chameleons. J. Am. Chem. Soc. 2003, 125, 15779–15788. [Google Scholar] [CrossRef]
- Bathelt, C.M.; Zurek, J.; Mulholland, A.J.; Harvey, J.N. Electronic structure of compound I in human isoforms of cytochrome P450 from QM/MM modeling. J. Am. Chem. Soc. 2005, 127, 12900–12908. [Google Scholar] [CrossRef]
- Porro, C.S.; Sutcliffe, M.J.; de Visser, S.P. Quantum mechanics/molecular mechanics studies on the sulfoxidation of dimethyl sulfide by Compound I and Compound 0 of cytochrome P450: Which is the better oxidant? J. Phys. Chem. A 2009, 113, 11635–11642. [Google Scholar] [CrossRef]
- Radoń, M.; Broclawik, E.; Pierloot, K. DFT and ab initio study of iron-oxo porphyrins: May they have a low-lying iron(V)-oxo electromer? J. Chem. Theory Comput. 2011, 7, 898–908. [Google Scholar] [CrossRef]
- Lonsdale, R.; Oláh, J.; Mulholland, A.J.; Harvey, J.N. Does compound I vary significantly between isoforms of cytochrome P450? J. Am. Chem. Soc. 2011, 133, 15464–15474. [Google Scholar] [CrossRef]
- Quesne, M.G.; Senthilnathan, D.; Singh, D.; Kumar, D.; Maldivi, P.; Sorokin, A.B.; de Visser, S.P. Origin of the enhanced reactivity of μ-nitrido-bridged diiron(IV)-oxo porphyrinoid complexes over cytochrome P450 compound I. ACS Catal. 2016, 6, 2230–2243. [Google Scholar] [CrossRef]
- Kepp, K.P. Heme isomers substantially affect heme’s electronic structure and function. Phys. Chem. Chem. Phys. 2017, 19, 22355–22362. [Google Scholar] [CrossRef]
- Li, X.-X.; Postils, V.; Sun, W.; Faponle, A.S.; Solà, M.; Wang, Y.; Nam, W.; de Visser, S.P. Reactivity patterns of (protonated) compound II and compound I of cytochrome P450: Which is the better oxidant? Chem. Eur. J. 2017, 23, 6406–6418. [Google Scholar] [CrossRef]
- Su, H.; Ma, G.; Liu, Y. Theoretical insights into the mechanism and stereoselectivity of olefin cyclopropanation catalyzed by two engineered cytochrome P450 enzymes. Inorg. Chem. 2018, 57, 11738–11745. [Google Scholar] [CrossRef] [PubMed]
- Spinello, A.; Pavlin, M.; Casalino, L.; Magistrato, A. A Dehydrogenase dual hydrogen abstraction mechanism promotes estrogen biosynthesis: Can we expand the functional annotation of the aromatase enzyme? Chem. Eur. J. 2018, 24, 10840–10849. [Google Scholar] [CrossRef] [PubMed]
- Phung, Q.M.; Pierloot, K. Low-lying electromeric states in chloro-ligated iron(IV)-oxo porphyrin as a model for compound I, studied with second-order perturbation theory based on density matrix renormalization group. J. Chem. Theory Comput. 2019, 15, 3033–3043. [Google Scholar] [CrossRef]
- Cheng, Q.; DeYonker, N.J. QM-cluster model study of the guaiacol hydrogen atom transfer and oxygen rebound with cytochrome P450 enzyme GcoA. J. Phys. Chem. B 2021, 125, 3296–3306. [Google Scholar] [CrossRef] [PubMed]
- de Visser, S.P.; Kumar, D.; Cohen, S.; Shacham, R.; Shaik, S. A predictive pattern of computed barriers for C–H hydroxylation by Compound I of cytochrome P450. J. Am. Chem. Soc. 2004, 126, 8362–8363. [Google Scholar] [CrossRef]
- Shaik, S.; Kumar, D.; de Visser, S.P. A valence bond modeling of trends in hydrogen abstraction barriers and transition states of hydroxylation reactions catalyzed by cytochrome P450 enzymes. J. Am. Chem. Soc. 2008, 130, 10128–10140. [Google Scholar] [CrossRef]
- Faponle, A.S.; Quesne, M.G.; Sastri, C.V.; Banse, F.; de Visser, S.P. Differences and comparisons of the properties and reactivities of iron(III)-hydroperoxo complexes with saturated coordination sphere. Chem. Eur. J. 2015, 21, 1221–1236. [Google Scholar] [CrossRef]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef]
- Case, D.A.; Cheatham III, T.E.; Darden, T.; Gohlke, H.; Luo, R.; Merz, K.M., Jr.; Onufriev, A.; Simmerling, C.; Wang, B.; Woods, R.J. The Amber biomolecular simulation programs. J. Comput. Chem. 2005, 26, 1668–1688. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09, Revision D.01; Gaussian, Inc.: Wallingford, CT, USA, 2013. [Google Scholar]
- Becke, A.D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar] [CrossRef] [PubMed]
- Li, P.; Merz, K.M. MCPB.py: A python based metal center parameter builder. J. Chem. Inf. Model. 2016, 56, 599–604. [Google Scholar] [CrossRef] [PubMed]
- Price, D.J.; Brooks, C.L. A modified TIP3P water potential for simulation with Ewald summation. J. Chem. Phys. 2004, 121, 10096–10103. [Google Scholar] [CrossRef] [PubMed]
- Maier, J.A.; Martinez, C.; Kasavajhala, K.; Wickstrom, L.; Hauser, K.E.; Simmerling, C. ff14SB: Improving the accuracy of protein side chain and backbone parameters from ff99SB. J. Chem. Theory Comput. 2015, 11, 3696–3713. [Google Scholar] [CrossRef]
- Ghafoor, S.; Mansha, A.; de Visser, S.P. Selective hydrogen atom abstraction from dihydroflavonol by a nonheme iron center is the key step in the enzymatic flavonol synthesis and avoids byproducts. J. Am. Chem. Soc. 2019, 141, 20278–20292. [Google Scholar] [CrossRef]
- Louka, S.; Barry, S.M.; Heyes, D.J.; Mubarak, M.Q.E.; Ali, H.S.; Alkhalaf, L.M.; Munro, A.W.; Scrutton, N.S.; Challis, G.L.; de Visser, S.P. The catalytic mechanism of aromatic nitration by cytochrome P450 TxtE: Involvement of a ferric-peroxynitrite intermediate. J. Am. Chem. Soc. 2020, 142, 15764–15779. [Google Scholar] [CrossRef]
- Ali, H.S.; de Visser, S.P. Electrostatic perturbations in the substrate-binding pocket of taurine/α-ketoglutarate dioxygenase determine its selectivity. Chem. Eur. J. 2022, 28, e202104167. [Google Scholar] [CrossRef]
- Hay, P.J.; Wadt, W.R. Ab initio effective core potentials for molecular calculations. Potentials for the transition metal atoms Sc to Hg. J. Chem. Phys. 1985, 82, 270–272. [Google Scholar] [CrossRef]
- Kendall, R.A.; Dunning, T.H., Jr.; Harrison, R.J. Electron affinities of the first-row atoms revisited. Systematic basis sets and wave functions. J. Chem. Phys. 1992, 96, 6796–6806. [Google Scholar] [CrossRef]
- Luchini, G.; Alegre-Requena, J.V.; Funes-Ardoiz, I.; Paton, R.S. GoodVibes version 3.2. F1000Research 2020, 9, 291. [Google Scholar] [CrossRef]
- İşci, Ü.; Faponle, A.S.; Afanasiev, P.; Albrieux, F.; Briois, V.; Ahsen, V.; Dumoulin, F.; Sorokin, A.B.; de Visser, S.P. Site-selective formation of an iron(IV)-oxo species at the more electron-rich iron atom of heteroleptic μ-nitrido diiron phthalocyanines. Chem. Sci. 2015, 6, 5063–5075. [Google Scholar] [CrossRef] [PubMed]
- Cheaib, K.; Mubarak, M.Q.E.; Sénéchal-David, K.; Herrero, C.; Guillot, R.; Clémancey, M.; Latour, J.-M.; de Visser, S.P.; Mahy, J.-P.; Banse, F.; et al. Selective formation of an FeIVO or an FeIIIOOH intermediate from FeII-H2O2: Controlled heterolytic vs homolytic O–O bond cleavage by the second coordination sphere. Angew. Chem. Int. Ed. 2019, 58, 854–858. [Google Scholar] [CrossRef] [PubMed]
- Dias, A.H.S.; Yadav, R.; Mokkawes, T.; Kumar, A.; Skaf, M.S.; Sastri, C.V.; Kumar, D.; de Visser, S.P. Biotransformation of bisphenol by human cytochrome P450 2C9 enzymes: A density functional theory study. Inorg. Chem. 2023, 62, 2244–2256. [Google Scholar] [CrossRef] [PubMed]
- Sliwiak, J.; Sikorski, M.; Jaskolski, M. PR-10 proteins as potential mediators of melatonin-cytokinin cross-talk in plants: Crystallographic studies of LlPR-10.2B isoform from yellow lupine. FEBS J. 2018, 185, 1907–1922. [Google Scholar] [CrossRef]
- Wang, J.; Wang, W.; Kollman, P.; Case, D. Antechamber: An accessory software package for molecular mechanical calculations. J. Chem. Inf. Comput. Sci. 2000, 222. [Google Scholar]
- Roe, D.R.; Cheatham, T.E., III. PTRAJ and CPPTRAJ: Software for Processing and Analysis of Molecular Dynamics Trajectory Data. J. Chem. Theory Comput. 2013, 9, 3084–3095. [Google Scholar] [CrossRef]
- ChemDraw Professional, PerkinElmer Informatics. Available online: http://www.perkinelmer.co.uk/category/chemdraw (accessed on 10 January 2023).











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Structure | CYP1A1 2 | CYP1A1 2 | CYP1A2 3 |
|---|---|---|---|
| Model II | Model I | ||
| TS1HA | 3.3 (6.7) | 17.8 (22.3) | 16.5 (18.8) |
| TS1C6 | 6.5 (12.0) | 10.0 (13.7) | 4.5 (8.3) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mokkawes, T.; de Visser, S.P. Melatonin Activation by Cytochrome P450 Isozymes: How Does CYP1A2 Compare to CYP1A1? Int. J. Mol. Sci. 2023, 24, 3651. https://doi.org/10.3390/ijms24043651
Mokkawes T, de Visser SP. Melatonin Activation by Cytochrome P450 Isozymes: How Does CYP1A2 Compare to CYP1A1? International Journal of Molecular Sciences. 2023; 24(4):3651. https://doi.org/10.3390/ijms24043651
Chicago/Turabian StyleMokkawes, Thirakorn, and Sam P. de Visser. 2023. "Melatonin Activation by Cytochrome P450 Isozymes: How Does CYP1A2 Compare to CYP1A1?" International Journal of Molecular Sciences 24, no. 4: 3651. https://doi.org/10.3390/ijms24043651