
First Synthesis of DBU-Conjugated Cationic Carbohydrate Derivatives and Investigation of Their Antibacterial and Antifungal Activity
 , , and
, , and
Abstract
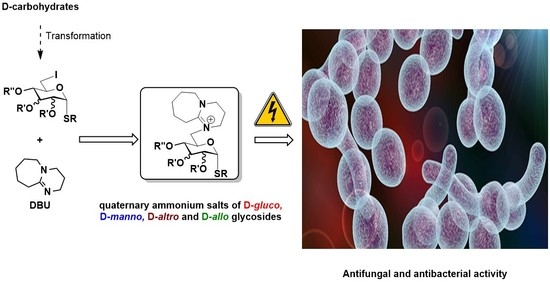
:
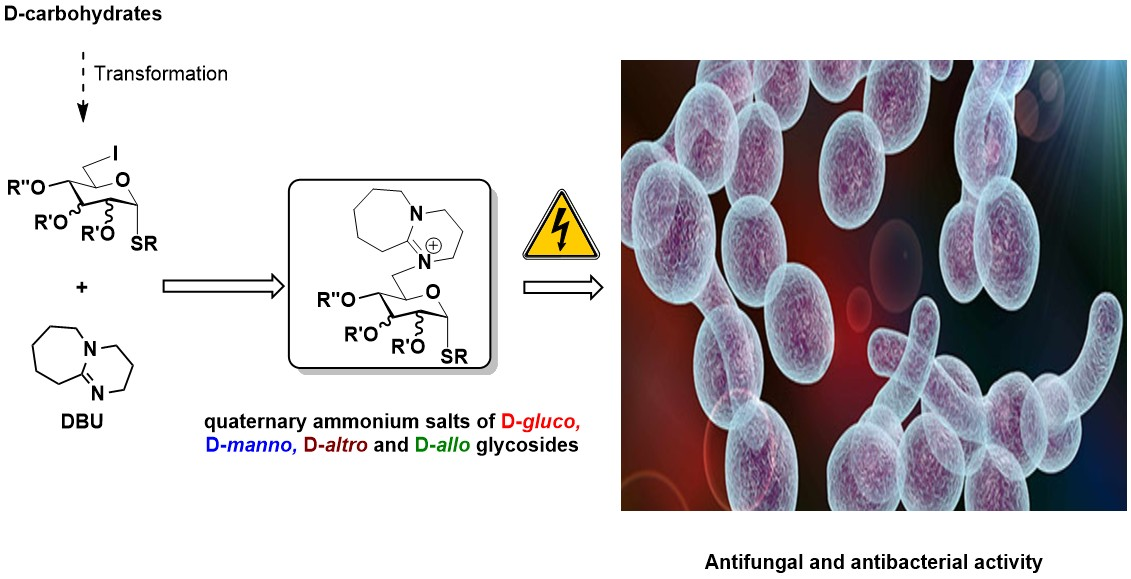
1. Introduction
2. Results and Discussion
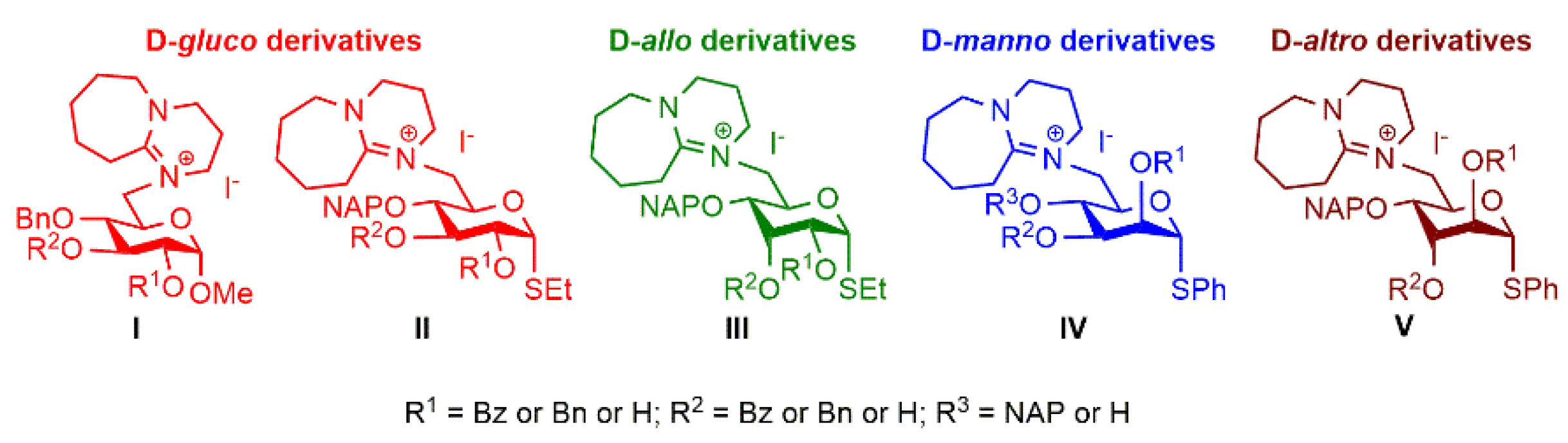
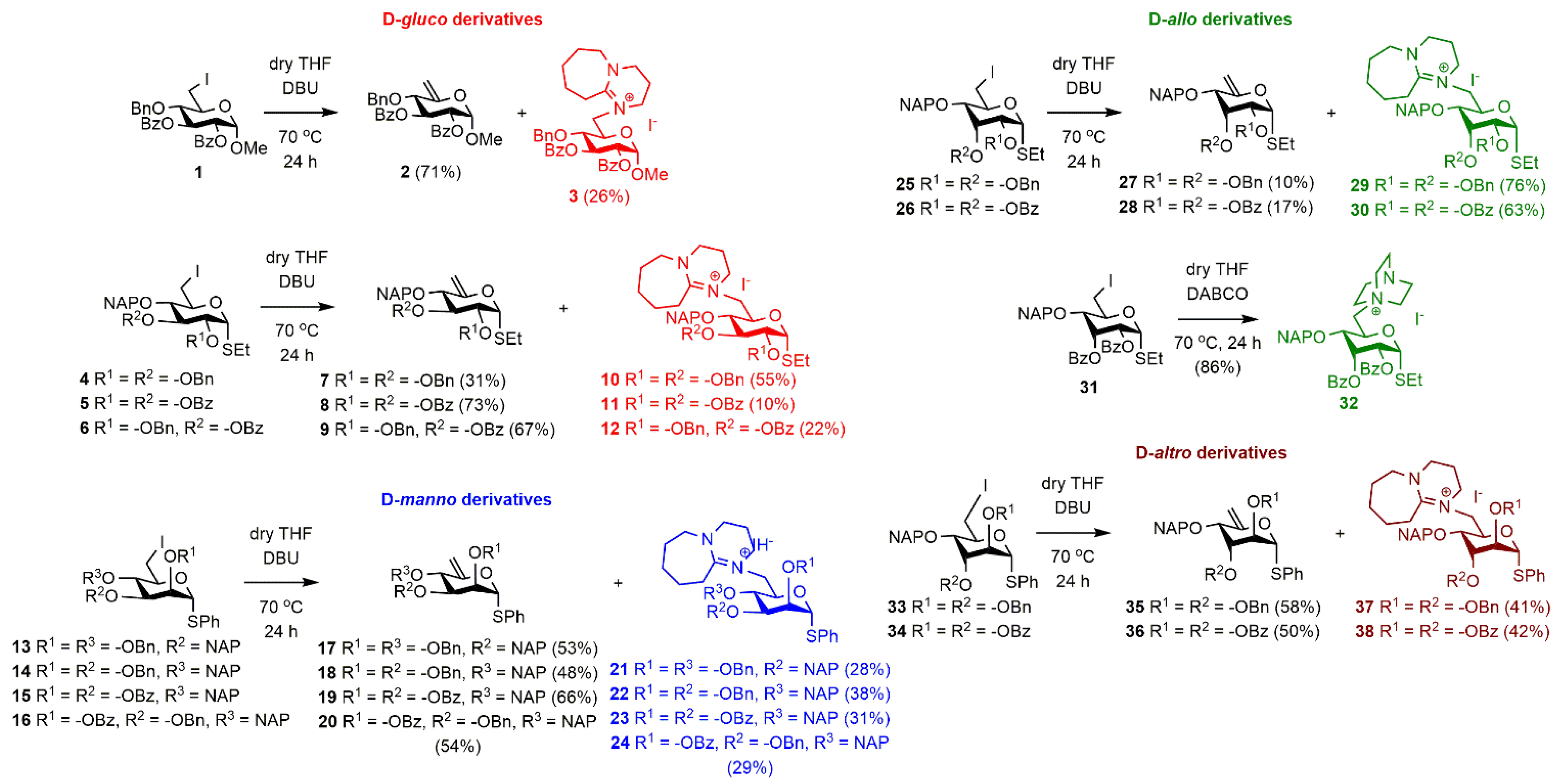
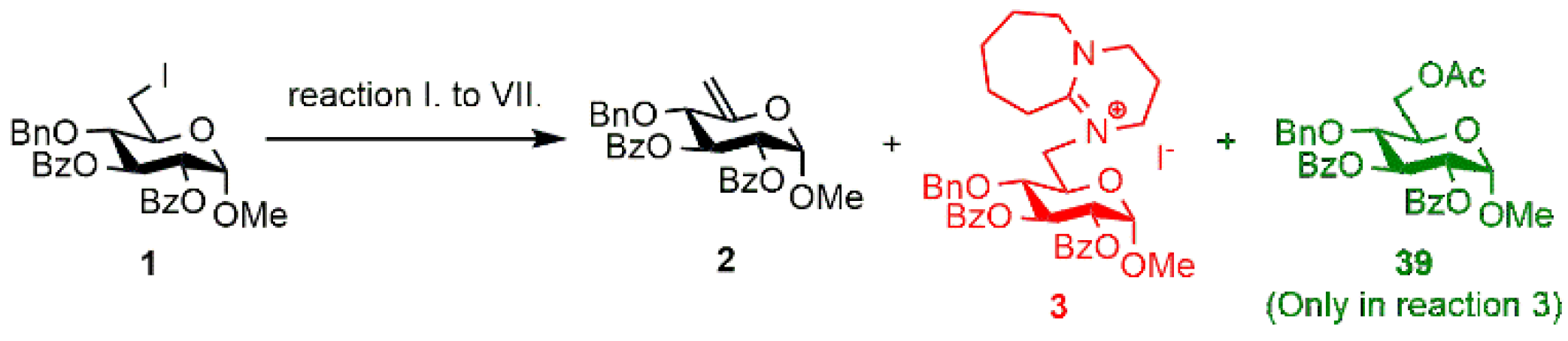
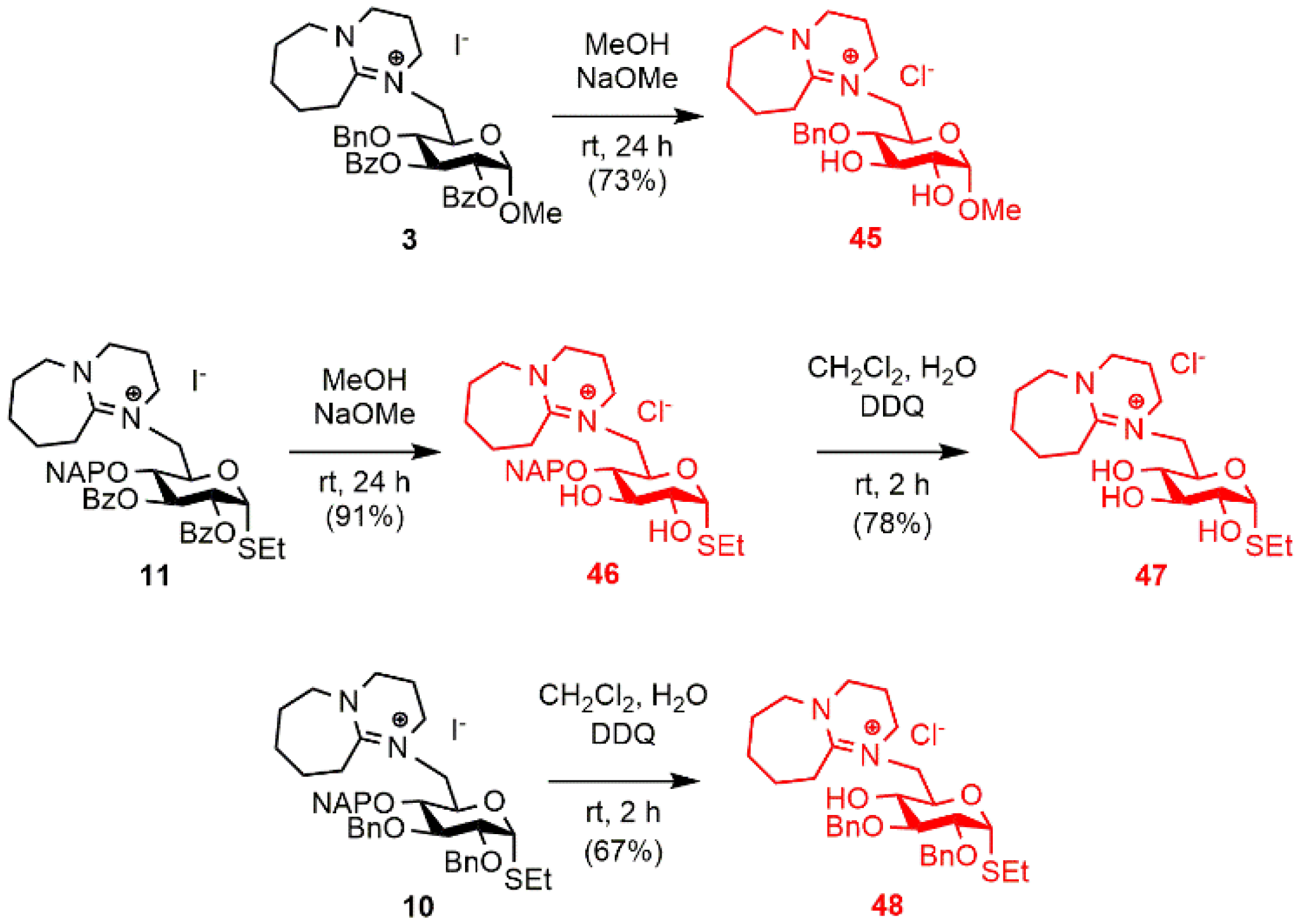
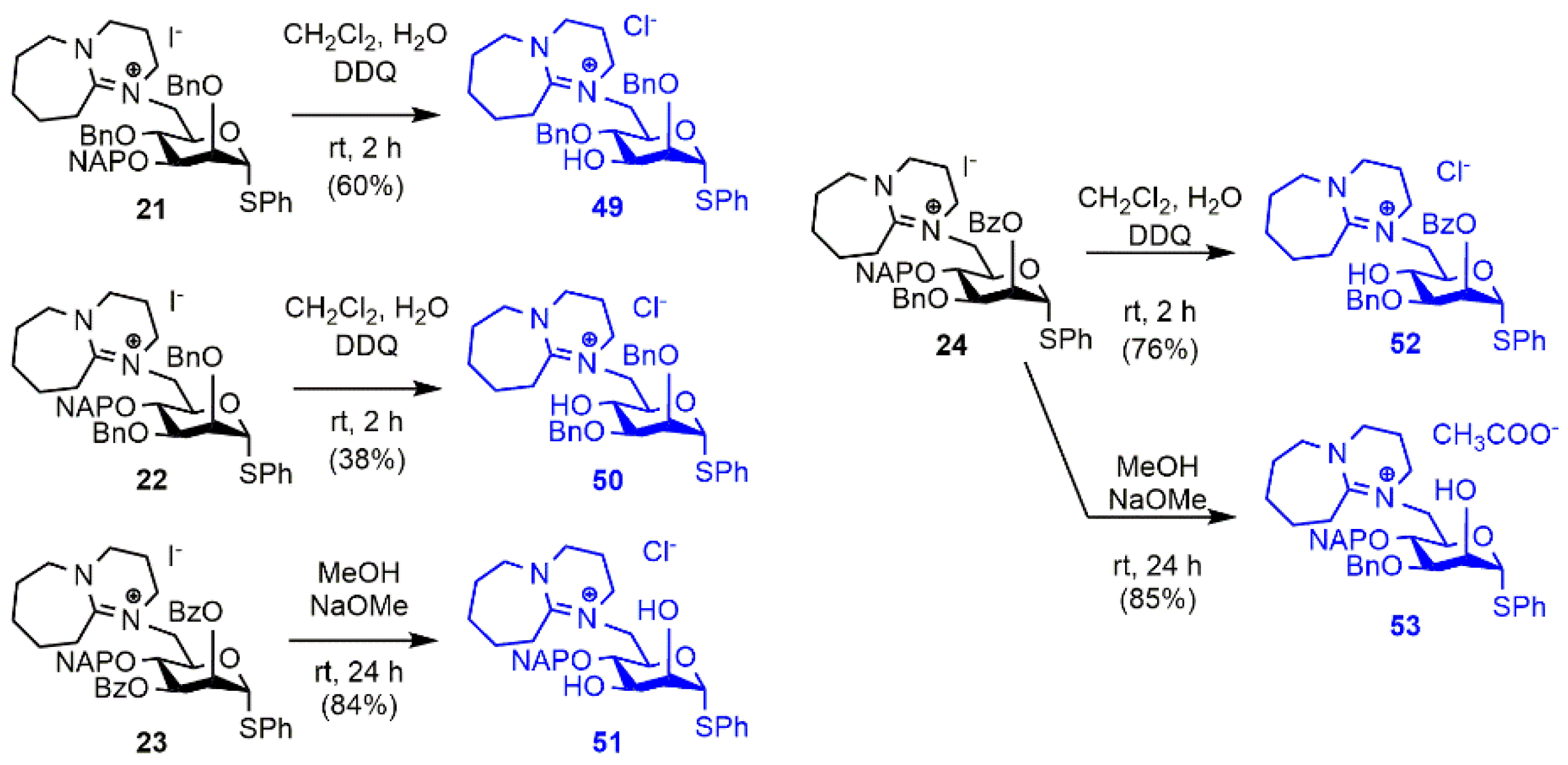
2.1. Synthesis of the DBU-Conjugated Derivatives
2.2. Biological Investigations of the DBU-Conjugated Derivatives
2.2.1. Antifungal and Antibacterial Studies
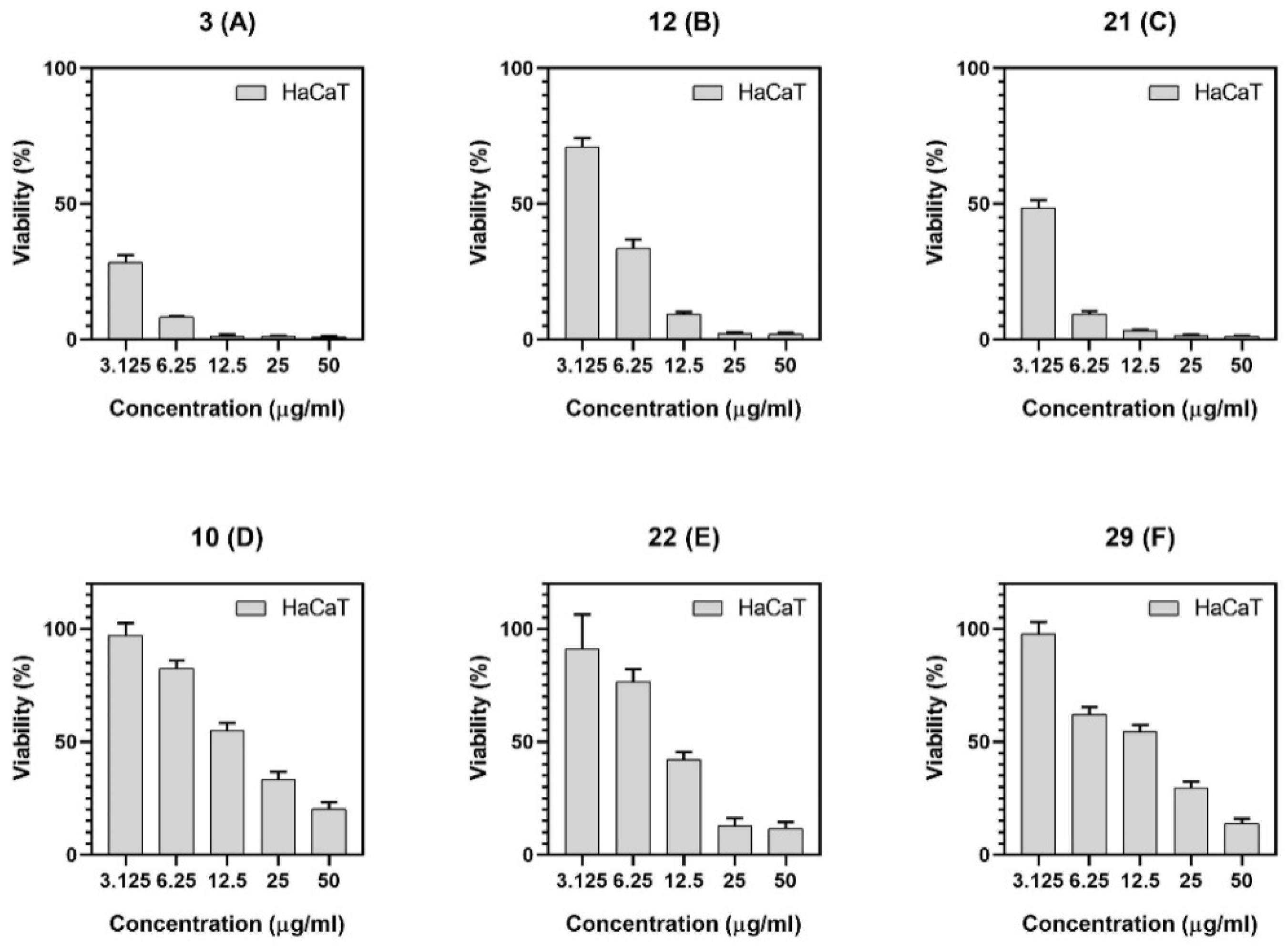
2.2.2. Cytotoxic Activity
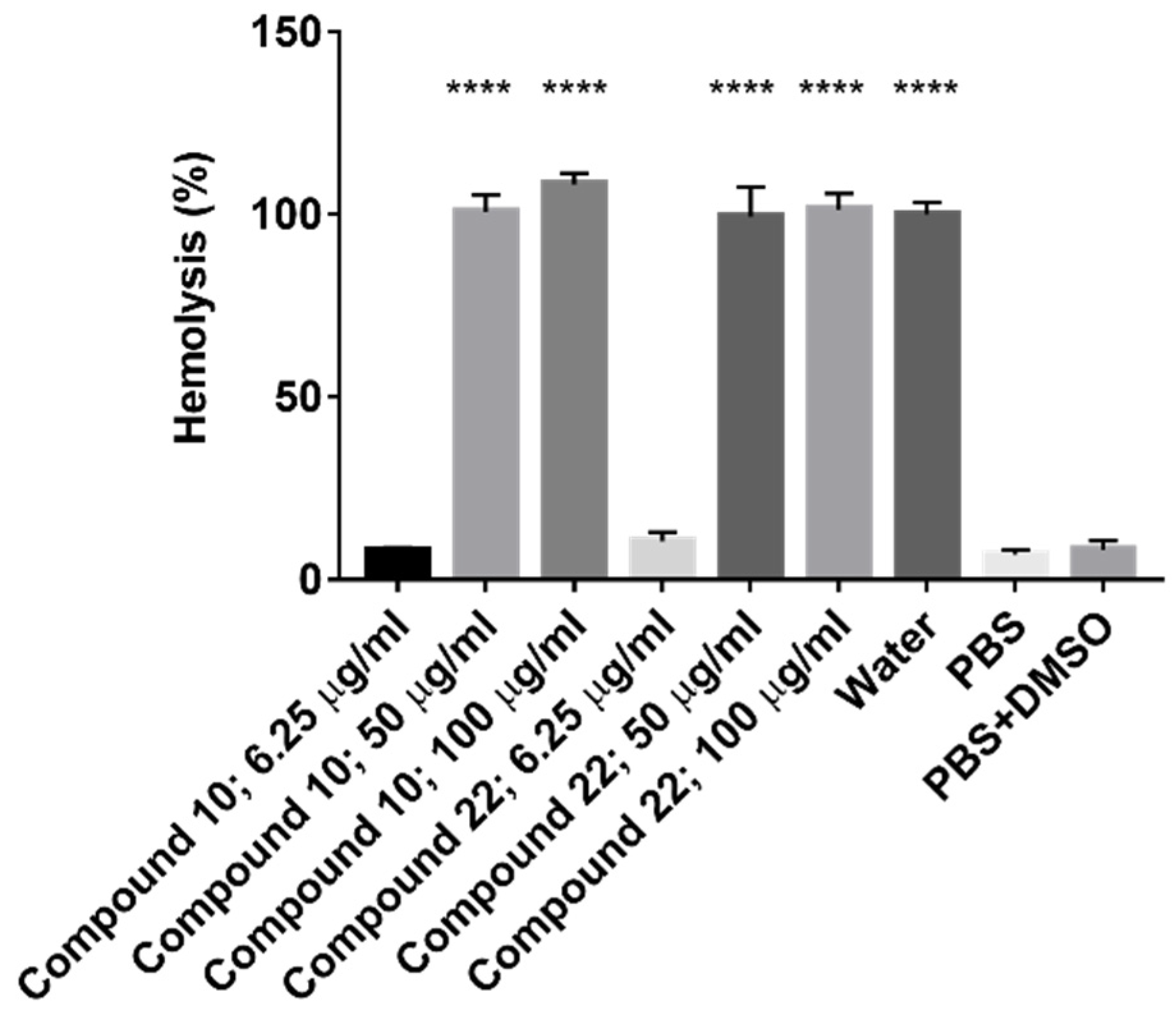
2.2.3. Hemolytic Activity
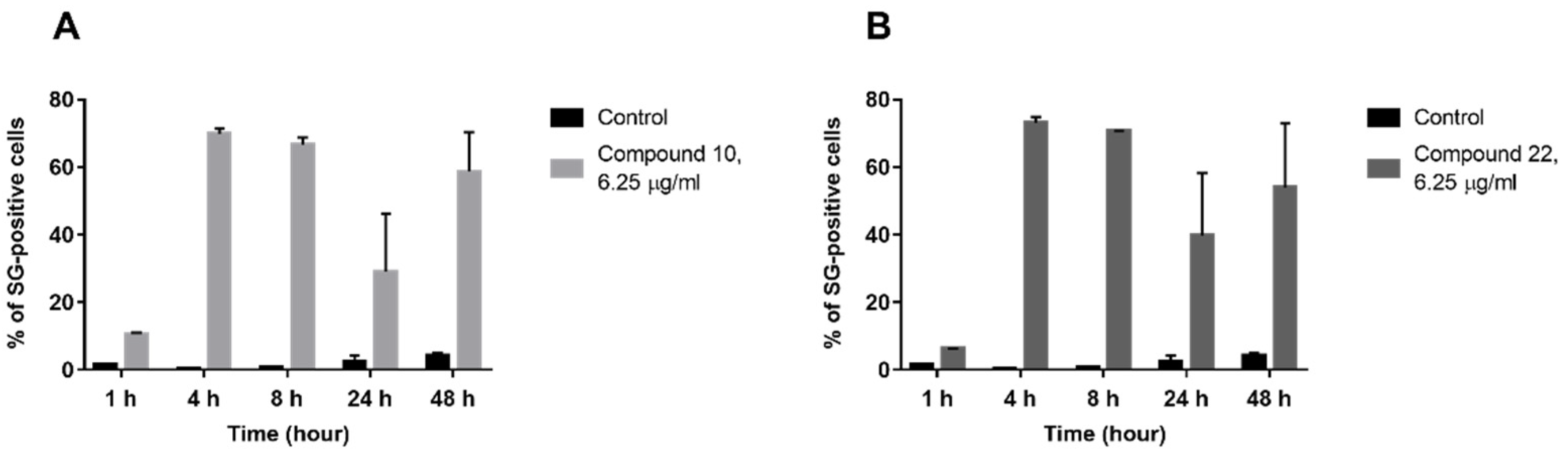
2.2.4. Membrane Permeabilization Study
3. Materials and Methods
3.1. General Information about the Syntheses
3.1.1. General Methods
- General Method A for Zemplén debenzoylation (45, 46, 51 and 53)
- 2.
- General Method B for cleavage of the 2-naphthylmethyl-group (NAP) (47–50 and 52)
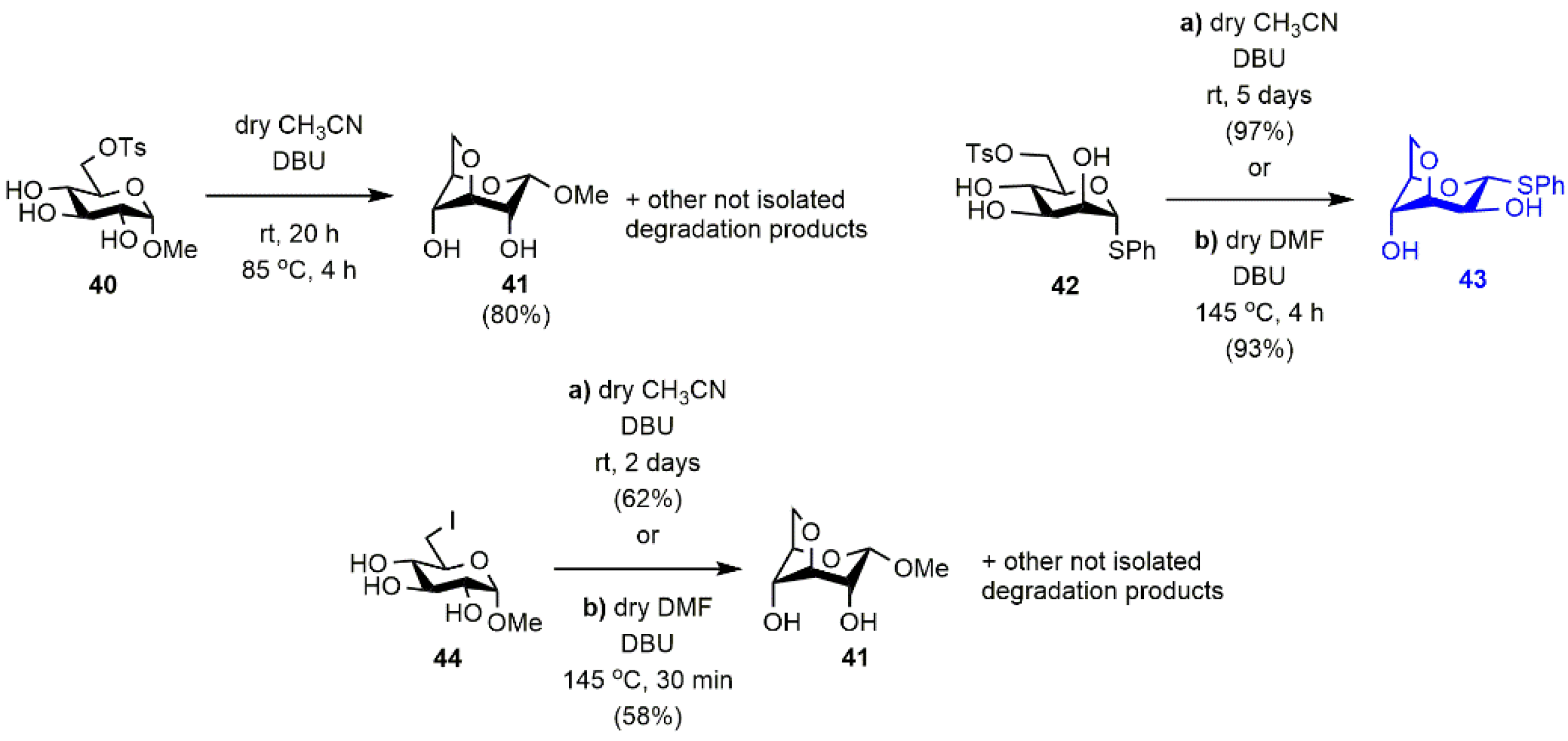
3.1.2. Optimization of the Synthesis of the DBU-Conjugated Methyl α-d-glucopyranoside derivative 3
- Methyl 3,6-anhydro-α-d-glucopyranoside (41) [38].
- Phenyl 3,6-anhydro-1-thio-α-d-mannopyranoside (43)
3.1.3. Synthesis and Transformation of the DBU-Conjugated Derivatives
3.2. Antimicrobial Investigations of the DBU-Conjugated Derivatives
3.2.1. Materials
3.2.2. Methods
3.2.3. Evaluation
3.3. Viability Tests
3.4. Hemolytic Activity
3.5. Membrane Permeabilization Study of Lactobacillus plantarum by SYTOX Green Staining
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Uçkay, I.; Pittet, D.; Vaudaux, P.; Sax, H.; Lew, D.; Waldvogel, F. Foreign body infections due to Staphylococcus epidermidis. Ann. Med. 2009, 41, 109–119. [Google Scholar] [CrossRef]
- Magill, S.S.; Edwards, J.R.; Bamberg, W.; Beldavs, Z.G.; Dumyati, G.; Kainer, M.A.; Lynfield, R.; Maloney, M.; McAllister-Hollod, L.; Nadle, J.; et al. Multistate point-prevalence survey of health care-associated infections. N. Engl. J. Med. 2014, 370, 1198–1208. [Google Scholar] [CrossRef]
- Zhang, Q.; Lambert, G.; Liao, D.; Kim, H.; Robin, K.; Tung, C.-K.; Pourmand, N.; Austin, R.H. Acceleration of Emergence of Bacterial Antibiotic Resistance in Connected Microenvironments. Science 2011, 333, 1764–1767. [Google Scholar] [CrossRef]
- Martínez, J.L. Antibiotics and Antibiotic Resistance Genes in Natural Environments. Science 2008, 321, 365–367. [Google Scholar] [CrossRef]
- Boucher, H.W.; Talbot, G.H.; Bradley, J.S.; Edwards, J.E.; Gilbert, D.; Rice, L.B.; Scheld, M.; Spellberg, B.; Bartlett, J. Bad Bugs, No Drugs: No Eskape! An Update from the Infectious Diseases Society of America. Clin. Infect. Dis. 2009, 48, 1–12. [Google Scholar] [CrossRef]
- Loeffler, J.; Steven, D.A. Antifungal Drug Resistance. Clin. Infect. Dis. 2003, 36, S31–S41. [Google Scholar] [CrossRef]
- Stokes, J.M.; Lopatkin, A.J.; Lobritz, M.A.; Collins, J.J. Bacterial Metabolism and Antibiotic Efficacy. Cell Metab. 2019, 30, 251–259. [Google Scholar] [CrossRef]
- Baracho, M.S.; Baracho, I.R. An analysis of the spontaneous mutation rate measurement in filamentous fungi. Genet. Mol. Biol. 2003, 26, 83–87. [Google Scholar] [CrossRef]
- Ernst, E.J.; Rogers, P.D. Antifungal Agents: Methods and Protocols (Methods in Molecular Medicine), 1st ed.; Human Press: Totowa, NJ, USA, 2005; pp. 1–210. [Google Scholar]
- Malayeri, F.A.; Rezaei, A.A.; Raiesi, O. Antifungal agents: Polyene, azole, antimetabolite, other and future agents. J. Bas. Res. Med. Sci. 2018, 5, 48–55. [Google Scholar] [CrossRef]
- Sawyer, P.R.; Brogden, R.N.; Pinder, K.M.; Speight, T.M.; Avery, G.S. Clotrimazole: A Review of its Antifungal Activity and Therapeutic Efficacy. Drugs 1975, 9, 424–447. [Google Scholar] [CrossRef]
- Pelletier, R.; Peter, J.; Antin, C.; Gonzalez, C.; Wood, L.; Walsh, T.J. Emergence of Resistance of Candida albicans to Clotrimazole in Human Immunodeficiency Virus-Infected Children: In Vitro and Clinical Correlations. J. Clin. Microbiol. 2000, 38, 1563–1568. [Google Scholar] [CrossRef]
- Grant, S.M.; Clissold, S.P. Fluconazole. A review of its pharmacodynamic and pharmacokinetic properties, and therapeutic potential in superficial and systemic mycoses. Drugs 1990, 39, 877–916. [Google Scholar] [CrossRef]
- Rex, J.H.; Rinaldi, M.G.; Pfaller, M.A. Resistance of Candida Species to Fluconazole. Antimicrob. Agents Chemother. 1995, 39, 1–8. [Google Scholar] [CrossRef]
- Polak, A.; Scholer, H.J. Mode of action of 5-fluorocytosine and mechanisms of resistance. Chemotherapy 1975, 21, 113–130. [Google Scholar] [CrossRef]
- Vermes, A.; Guchelaar, H.-J.; Dankert, J. Flucytosine: A review of its pharmacology, clinical indications, pharmacokinetics, toxicity and drug interactions. J. Antimicrob. Chemother. 2000, 46, 171–179. [Google Scholar] [CrossRef]
- Balfour, J.A.; Faulds, D. Terbinafine. A review of its pharmacodynamic and pharmacokinetic properties, and therapeutic potential in superficial mycoses. Drugs 1992, 43, 259–284. [Google Scholar] [CrossRef]
- Abdel-Rahman, S.M.; Nahata, M.C. Oral terbinafine: A new antifungal agent. Ann. Pharmacother. 1997, 31, 445–456. [Google Scholar] [CrossRef]
- Brajtburg, J.; Powderly, W.G.; Kobayashi, G.S.; Medoff, G. Amphotericin B: Current understanding of mechanisms of action. Antimicrob. Agents Chemother. 1990, 34, 183–188. [Google Scholar] [CrossRef]
- Ostrosky-Zeichner, L.; Marr, K.A.; Rex, J.H.; Cohen, S.H. Amphotericin B: Time for a new “gold standard”. Clin. Infect. Dis. 2003, 37, 415–425. [Google Scholar]
- Zhou, C.; Wang, F.; Chen, H.; Li, M.; Qiao, F.; Liu, Z.; Hou, Y.; Wu, C.; Fan, Y.; Liu, L.; et al. Selective Antimicrobial Activities and Action Mechanism of Micelles Self-assembled by Cationic Oligomeric Surfactants. ACS Appl. Mater. Interfaces 2016, 8, 4242–4249. [Google Scholar] [CrossRef]
- Abel, T.; Cohen, J.L.I.; Engel, R.; Filshtinskaya, M.; Melkonian, A.; Melkonian, K. Preparation and investigation of antibacterial carbohydrate-based surfaces. Carbohydr. Res. 2002, 337, 2495–2499. [Google Scholar] [CrossRef] [PubMed]
- Engel, R.; Ghani, I.; Montenegro, D.; Thomas, M.; Klaritch-Vrana, B.; Castaño, A.; Friedman, L.; Leb, J.; Rothman, L.; Lee, H.; et al. Polycationic Glycosides. Molecules 2011, 16, 1508–1518. [Google Scholar] [CrossRef] [PubMed]
- Nikfarjam, N.; Ghomi, M.; Agarwal, T.; Hassanpour, M.; Sharifi, E.; Khorsandi, D.; Khan, M.A.; Rossi, F.; Rossetti, A.; Zare, E.N.; et al. Antimicrobial Ionic Liquid-based Materials for Biomedical Applications. Adv. Funct. Mater. 2021, 31, 2104148. [Google Scholar] [CrossRef]
- Kwasniewska, D.; Chen, Y.-L.; Wieczorek, D. Biological Activity of Quaternary Ammonium Salts and Their Derivatives. Pathogens 2020, 9, 459. [Google Scholar] [CrossRef] [PubMed]
- Jennings, M.C.; Minbiole, K.P.C.; Wuest, W.M. Quaternary Ammonium Compounds: An Antimicrobial Mainstay and Platform for Innovation to Address Bacterial Resistance. ACS Infect. Dis. 2015, 1, 288–303. [Google Scholar] [CrossRef]
- Demeter, F.; Bereczki, I.; Borbás, A.; Herczeg, M. Synthesis of Four Orthogonally Protected Rare l-Hexose Thioglycosides from d-Mannose by C-5 and C-4 Epimerization. Molecules 2022, 27, 3422. [Google Scholar] [CrossRef]
- Demeter, F.; Bényei, A.; Borbás, A.; Herczeg, M. Synthesis of the three most expensive l-hexose thioglycosides from d-glucose. Synthesis 2023. [Google Scholar] [CrossRef]
- Baidya, M.; Mayr, H. Nucleophilicities and carbon basicities of DBU and DBN. Chem. Commun. 2008, 15, 1792–1794. [Google Scholar] [CrossRef]
- Maji, B.; Breugst, M.; Mayr, H. N-Heterocyclic Carbenes: Organocatalysts with Moderate Nucleophilicity but Extraordinarily High Lewis Basicity. Angew. Chem. Int. Ed. Engl. 2011, 50, 6915–6919. [Google Scholar] [CrossRef]
- Li, Z.; Ji, P.; Cheng, J.-P. Brönsted Basicities and Nucleophilicities of N-Heterocyclic Olefins in Solution: N-Heterocyclic Carbene versus N-Heterocyclic Olefin. Which Is More Basic, and Which Is More Nucleophilic? J. Org. Chem. 2021, 86, 2974–2985. [Google Scholar] [CrossRef]
- Lammers, H.; Cohen-Fernandes, P.; Habraken, C.L. Nucleophilic behaviour of DBU and DBN in reactions with 4-halo-3,5-di-methyl-1-nitro-1H-pyrazoles. Tetrahedron 1994, 50, 865–870. [Google Scholar] [CrossRef]
- Johnson, M.G.; Foglesong, R.J. Nucleophilic behavior of DBU in a conjugate addition reaction. Tetrahedron Lett. 1997, 38, 7003–7006. [Google Scholar] [CrossRef]
- Németh, A.G.; Szabó, R.; Domján, A.; Keserű, G.M.; Ábrányi-Balogh, P. Chromatography-Free Multicomponent Synthesis of Thioureas Enabled by Aqueous Solution of Elemental Sulfur. ChemistryOpen 2021, 10, 16–27. [Google Scholar] [CrossRef] [PubMed]
- Thomas, M.; Montenegro, D.; Castaño, A.; Friedman, L.; Leb, J.; Huang, M.L.; Rothman, L.; Lee, H.; Capodiferro, C.; Ambinder, D.; et al. Polycations. 17. Synthesis and properties of polycationic derivatives of carbohydrates. Carbohydr. Res. 2009, 344, 1620–1627. [Google Scholar] [CrossRef]
- Suwinski, J.; Walczak, K. Salts of 5-Substituted Uracils with 1,8-Diazabicyclo[5.4.0]-undec-7-ene: Convenient Reagents for Nucleophilic Addition and Substitution Reactions. Synthesis 2001, 2, 225–228. [Google Scholar] [CrossRef]
- Cramer, F.; Otterbach, H.; Springmann, H. Eine Synthese der 6-Desoxy-6-amino-glucose. Chem. Ber. 1959, 92, 384–391. [Google Scholar] [CrossRef]
- Eskew, N.A.; Evans, S.A., Jr. Oxaphosphoranylation of Methyl α-d-Glucopyranoside with Diethoxytriphenylphosphorane. A Highly Stereoselective Route to Anhydropyranosides. J. Chem. Soc. Chem. Commun. 1990, 706–708. [Google Scholar] [CrossRef]
- Kerékgyártó, J.; Szurmai, Z.; Lipták, A. Synthesis of p-trifluoroacetamidophenyl 6-deoxy-2-O-(3-O-[2-O-methyl-3-O-(2-O-methyl-α-d-rhamnopyranosyl)-α-l-fucopyranosyl]-α-l-rhamnopyranosyl}-α-l-talopyranoside: A spacer armed tetrasaccharide glycopeptidolipid antigen of Mycobacterium avium serovar 20. Carbohydr. Res. 1993, 245, 65–80. [Google Scholar] [CrossRef]
- Gómez, A.M.; Casillas, M.; Rodríguez, B.; Valverde, S.; López, J.C. Synthesis of furanoid and pyranoid C-1 aryl glycals by reaction of glycosyl chlorides with organolithium reagents. Arkivoc 2010, 3, 288–302. [Google Scholar] [CrossRef]
- Skaanderup, P.R.; Poulsen, C.S.; Hyldtoft, L.; Jørgensen, M.R.; Madsen, R. Regioselective Conversion of Primary Alcohols into Iodides in Unprotected Methyl Furanosides and Pyranosides. Synthesis 2002, 12, 1721–1727. [Google Scholar] [CrossRef]
- Benedict, K.M.; Gow, S.P.; Checkley, S.; Booker, C.W.; McAllister, T.A.; Morley, P.S. Methodological comparisons for antimicrobial resistance surveillance in feedlot cattle. BMC Vet. Res. 2013, 9, 216. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clinical and Laboratory Standards Institute (CLSI). Performance Standards for Antimicrobial Susceptibility Testing: Twenty-Second Informational Supplement; M100-S22; Clinical and Laboratory Standards Institute (CLSI): Wayne, PA, USA, 2012. [Google Scholar]
- European Committee on Antimicrobial Susceptibility Testing (Eucast) Documents: Document Version 2.0 2012-01-01. 2012. Available online: http://eucast.org/clinical_breakpoints (accessed on 2 January 2023).
- Marchese, A.; Esposito, S.; Barbieri, R.; Bassetti, M.; Debbia, E. Does the adoption of EUCAST susceptibility breakpoints affect the selection of antimicrobials to treat acute community-acquired respiratory tract infections? BMC Inf. Dis. 2012, 12, 181. [Google Scholar] [CrossRef] [PubMed]
- Berkow, E.L.; Lockhart, S.R.; Ostrosky-Zeichner, L. Antifungal Susceptibility Testing: Current Approaches. Clin. Microbiol. Rev. 2020, 33, e00069-19. [Google Scholar] [CrossRef] [PubMed]
- Koehling, H.L.; Willinger, B.; Buer, J.; Rath, P.M.; Steinmann, J. Comparative Evaluation of a New Commercial Colorimetric Microdilution Assay (SensiQuattro Candida EU) with MIC Test Strip and EUCAST Broth Microdilution Methods for Susceptibility Testing of Invasive Candida Isolates. J. Clin. Microbiol. 2015, 53, 255–261. [Google Scholar] [CrossRef]
- Wikler, M.A. Methods for Dilution Antimicrobial Susceptibility Tests for Bacteria That Grow Aerobically: Approved Standard, 10th ed.; Clinical and Laboratory Standards Institute: Wayne, PA, USA, 2006. [Google Scholar]
- Kőszeginé, S.H.; Török, I.; Ráfliné, R.Z.; Nagy, A.; Fejér, I.; Kertész, P.; Posgayné, K.E.; Vankó, É. Microbiological testing of non-sterile products, Chapter 2.6.12–2.6.13. In Hungarian Pharmacopoeia, 8th ed.; Vincze, J., Ed.; Medicina: Budapest, Hungary, 2003; Volume I, pp. 159–169. [Google Scholar]
- Available online: https://www.graphpad.com/guides/prism/latest/curve-fitting/reg_fitting_lines_to_semilog.htm (accessed on 2 January 2023).
- Boukamp, P.; Petrussevska, R.T.; Breitkreutz, D.; Hornung, J.; Markham, A.; Fusenig, N.E. Normal keratinization in a spontaneously immortalized aneuploid human keratinocyte cell line. J. Cell Biol. 1988, 106, 761–771. [Google Scholar] [CrossRef]
- Boukamp, P.; Popp, S.; Altmeyer, S.; Hülsen, A.; Fasching, C.; Cremer, T.; Fusenig, N.E. Sustained nontumorigenic phenotype correlates with a largely stable chromosome content during long-term culture of the human keratinocyte line HaCaT. Genes Chromosomes Cancer 1997, 19, 201–214. [Google Scholar] [CrossRef]
- Malanga, M.; Szemán, J.; Fenyvesi, É.; Puskás, I.; Csabai, K.; Gyémánt, G.; Fenyvesi, F.; Szente, J. “Back to the Future”: A New Look at Hydroxypropyl Beta-Cyclodextrins. J. Pharm. Sci. 2016, 105, 2921–2931. [Google Scholar] [CrossRef]
- Kovács, R.; Erdélyi, L.; Fenyvesi, F.; Balla, N.; Kovács, F.; Vámosi, G.; Klusóczki, Á.; Gyöngyösi, A.; Bácskay, I.; Vecsernyés, M.; et al. Concentration-Dependent Antibacterial Activity of Chitosan on Lactobacillus plantarum. Pharmaceutics 2023, 15, 18. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry | Solvent | Reagent | Temp. (°C) | Time | Yield |
|---|---|---|---|---|---|
| 1. 1 | THF | DBU (2.5 equiv.) | 70 | 24 | 2: 71% |
| 3: 26% | |||||
| 2. | CH3CN | DBU (1.0 equiv.) | rt | 16 days | 1: 9% |
| 2: 63% | |||||
| 3: 11% | |||||
| 3. | CH3CN | DBU (1.5 equiv.) | rt | 4 days | 2: 72% |
| 3: 14% | |||||
| 4. | CH3CN | DBU (3.0 equiv.) + Glacial acetic acid (3.0 equiv.) | rt | 3 days | 2: 10% |
| 3: 0% | |||||
| 39: 87% | |||||
| 5. | CH3CN | DBU (6.0 equiv.) + p-TSA (6.0 equiv.) | rt to 85 °C | 21 h at rt, 5 h 85 °C | 1: 84% |
| 6. | CH3CN | DBU (6.0 equiv.) | rt | 6 days | 2: 70% |
| 3: 18% | |||||
| 7. | - | DBU (31.25 equiv.) | rt | 24 h | 2: 32% |
| 3: 11% 2 | |||||
| 8. | DMF | DBU (3.0 equiv.) | 145 | 45 min | 1: 2% |
| 2: 58% | |||||
| 3: 33% |
| Compound | MIC in µg/mL | clogP | ||
|---|---|---|---|---|
| Escherichia coli (ATCC 8739) | Staphylococcus aureus (ATCC 6538) | Candida albicans (ATCC 10231) | ||
| 3 | >100 | ≥6.25 | ≥50 | 7.413 |
| 10 | ≥100 | 6.25 | ≤6.25 | 9.672 |
| 11 | >100 | >100 | >100 | 9.542 |
| 12 | >100 | 6.25 | ≥50 | 9.547 |
| 21 | >100 | ≥6.25 | ≥50 | 10.683 |
| 22 | ≥100 | 6.25 | ≤6.25 | 10.683 |
| 23 | ≥100 | ≥100 | ≥100 | 10.618 |
| 24 | ≥100 | ≥6.25 | ≥50 | 10.635 |
| 29 | >100 | <12.5 | <6.25 | 9.673 |
| 30 | ≥100 | ≥25 | ≥25 | 9.542 |
| 32 | ≥100 | ≥6.25 | ≥100 | 6.373 |
| 37 | >100 | ≥50 | >100 | 10.683 |
| 38 | >100 | >100 | >100 | 10.618 |
| 45 | >100 | >100 | >100 | 2.661 |
| 46 | >100 | >100 | >100 | 4.790 |
| 47 | >100 | >100 | >100 | 1.826 |
| 48 | >100 | ≥25 | ≥50 | 6.052 |
| 49 | >100 | >100 | >100 | 7.070 |
| 50 | >100 | ≥50 | ≥25 | 7.715 |
| 51 | >100 | ≥12.5 | ≥50 | 5.800 |
| 52 | ≥100 | ≥100 | >100 | 7.667 |
| 53 | >100 | ≥6.25 | ≥6.25 | 8.240 |
| Ceftazidime | 8 [42] | 8 [43,44,45] | - | - |
| Gentamicin | 8 [42] | 8 [43,44,45] | - | - |
| Amphotericin B | - | - | <0.5 [46,47] | - |
| Compound | MIC in µg/mL Candida albicans (ATCC 10231) |
|---|---|
| 10 | 5.0 |
| 22 | 2.5 |
| 29 | 2.5 |
| Compound | HaCaT IC50 (µg/mL) |
|---|---|
| 3 | 0.13 ± 0.06 |
| 10 | 15.3 ± 0.78 |
| 12 | 4.26 ± 0.16 |
| 21 | 1.02 ± 0.06 |
| 22 | 10.3 ± 0.41 |
| 29 | 11.9 ± 0.25 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Demeter, F.; Török, P.; Kiss, A.; Kovásznai-Oláh, R.; Szigeti, Z.M.; Baksa, V.; Kovács, F.; Balla, N.; Fenyvesi, F.; Váradi, J.; et al. First Synthesis of DBU-Conjugated Cationic Carbohydrate Derivatives and Investigation of Their Antibacterial and Antifungal Activity. Int. J. Mol. Sci. 2023, 24, 3550. https://doi.org/10.3390/ijms24043550
Demeter F, Török P, Kiss A, Kovásznai-Oláh R, Szigeti ZM, Baksa V, Kovács F, Balla N, Fenyvesi F, Váradi J, et al. First Synthesis of DBU-Conjugated Cationic Carbohydrate Derivatives and Investigation of Their Antibacterial and Antifungal Activity. International Journal of Molecular Sciences. 2023; 24(4):3550. https://doi.org/10.3390/ijms24043550
Chicago/Turabian StyleDemeter, Fruzsina, Patrik Török, Alexandra Kiss, Richárd Kovásznai-Oláh, Zsuzsa Máthéné Szigeti, Viktória Baksa, Fruzsina Kovács, Noémi Balla, Ferenc Fenyvesi, Judit Váradi, and et al. 2023. "First Synthesis of DBU-Conjugated Cationic Carbohydrate Derivatives and Investigation of Their Antibacterial and Antifungal Activity" International Journal of Molecular Sciences 24, no. 4: 3550. https://doi.org/10.3390/ijms24043550