
Excited State Intramolecular Proton Transfer Dynamics of Derivatives of the Green Fluorescent Protein Chromophore


Abstract
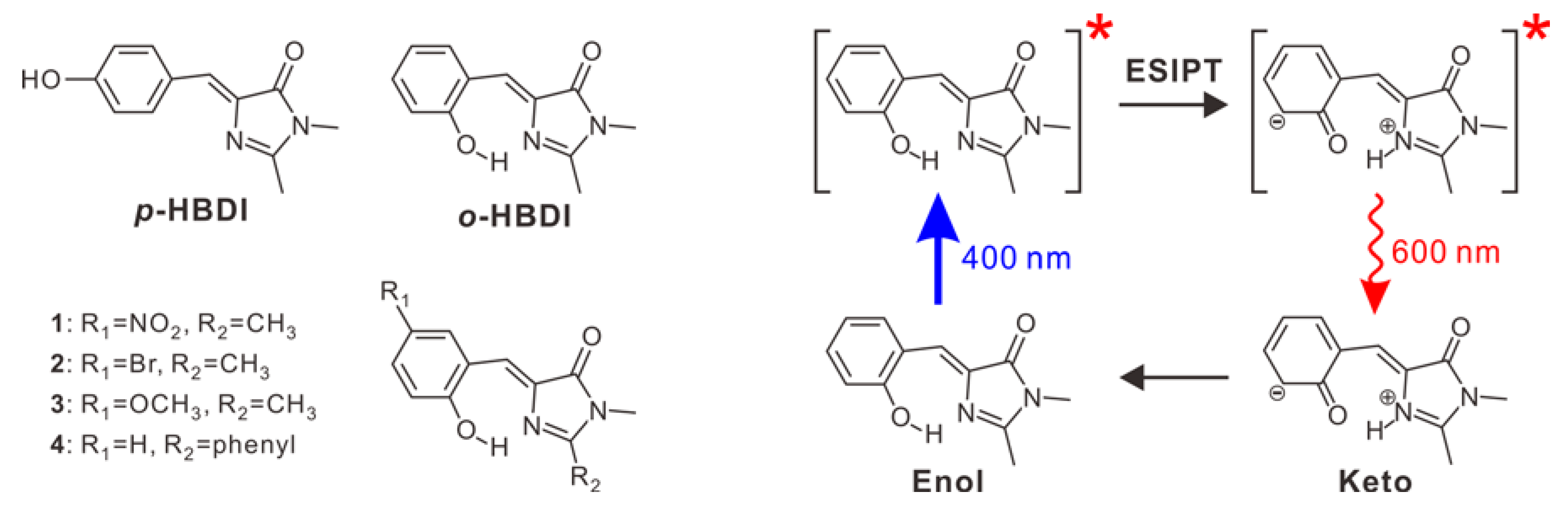
:1. Introduction
2. Results
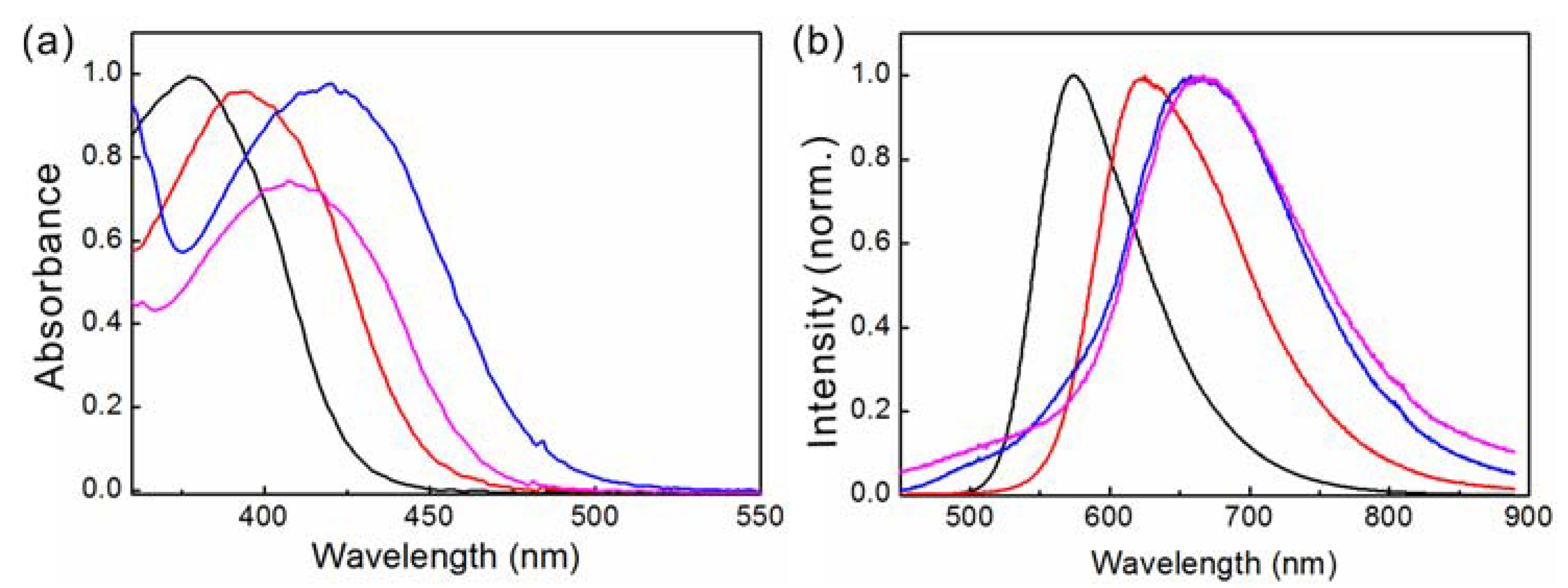
2.1. Steady-State Spectroscopy
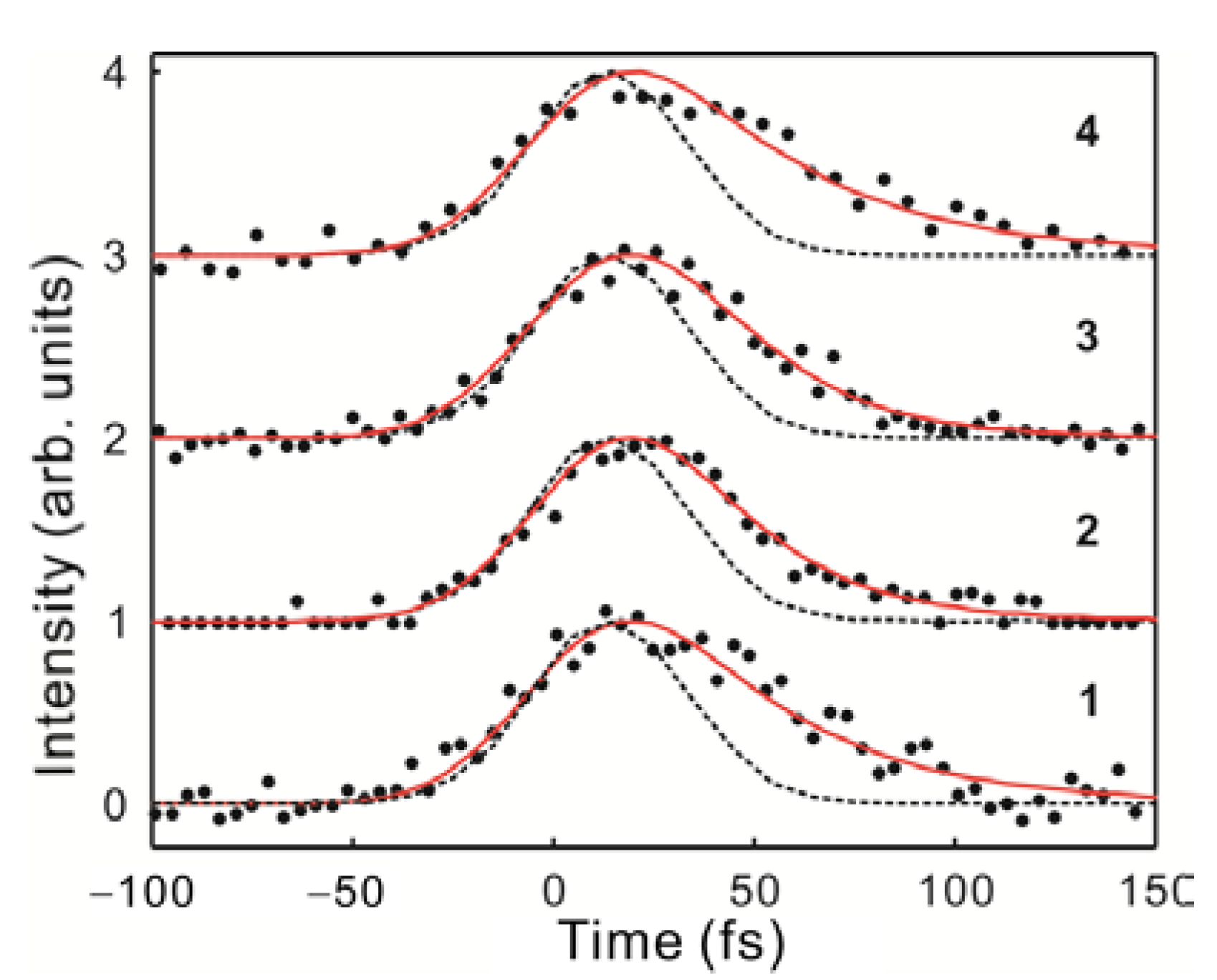
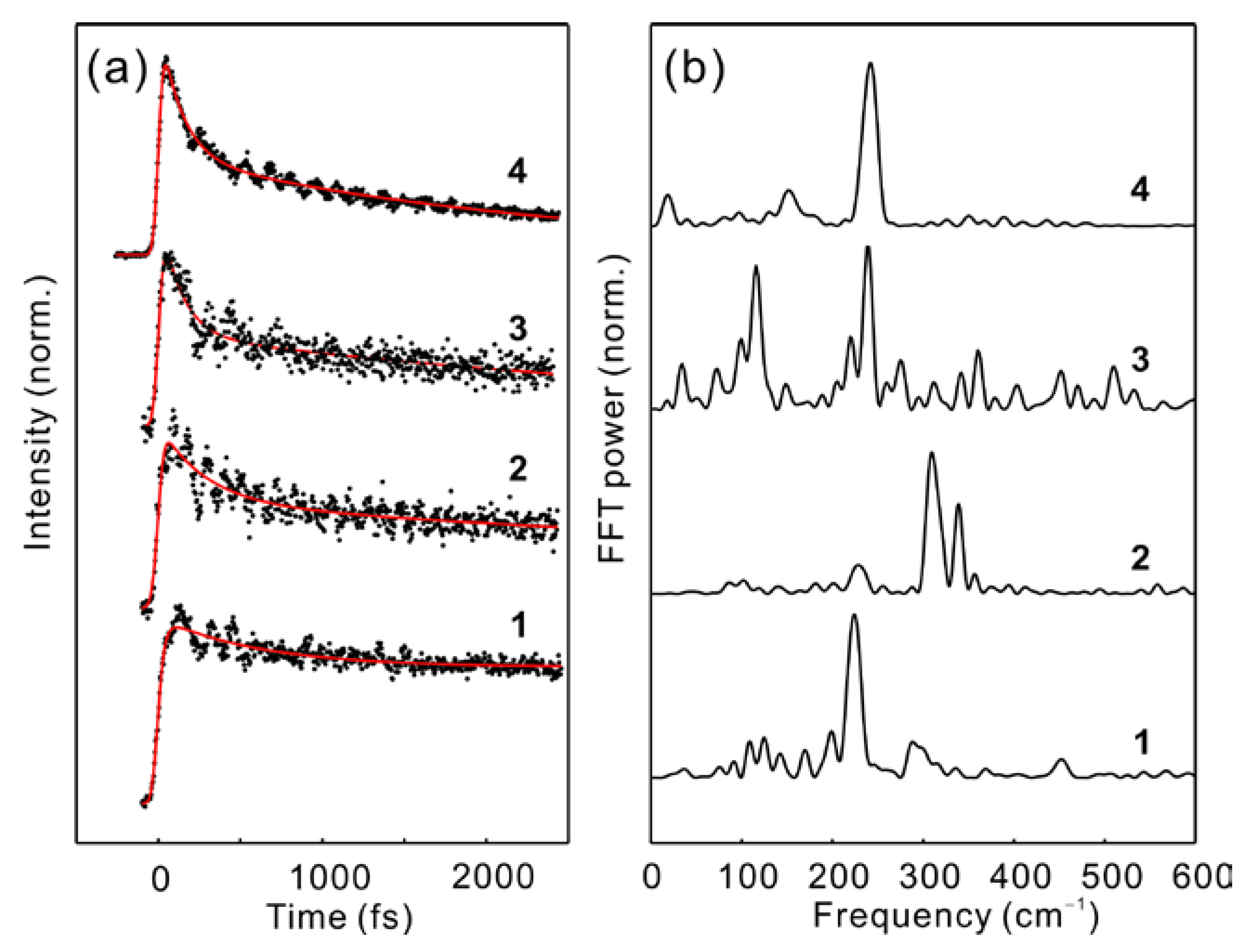
2.2. Time-Resolved Fluorescence
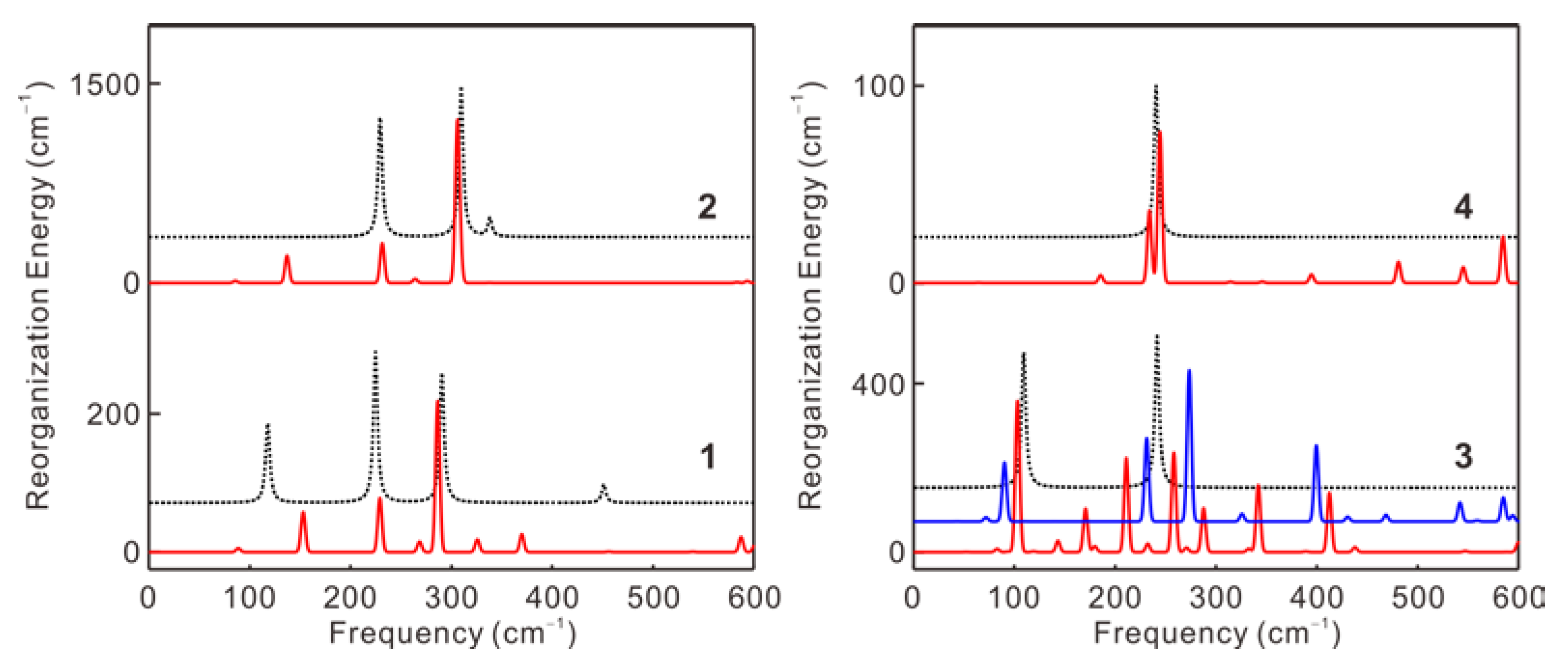
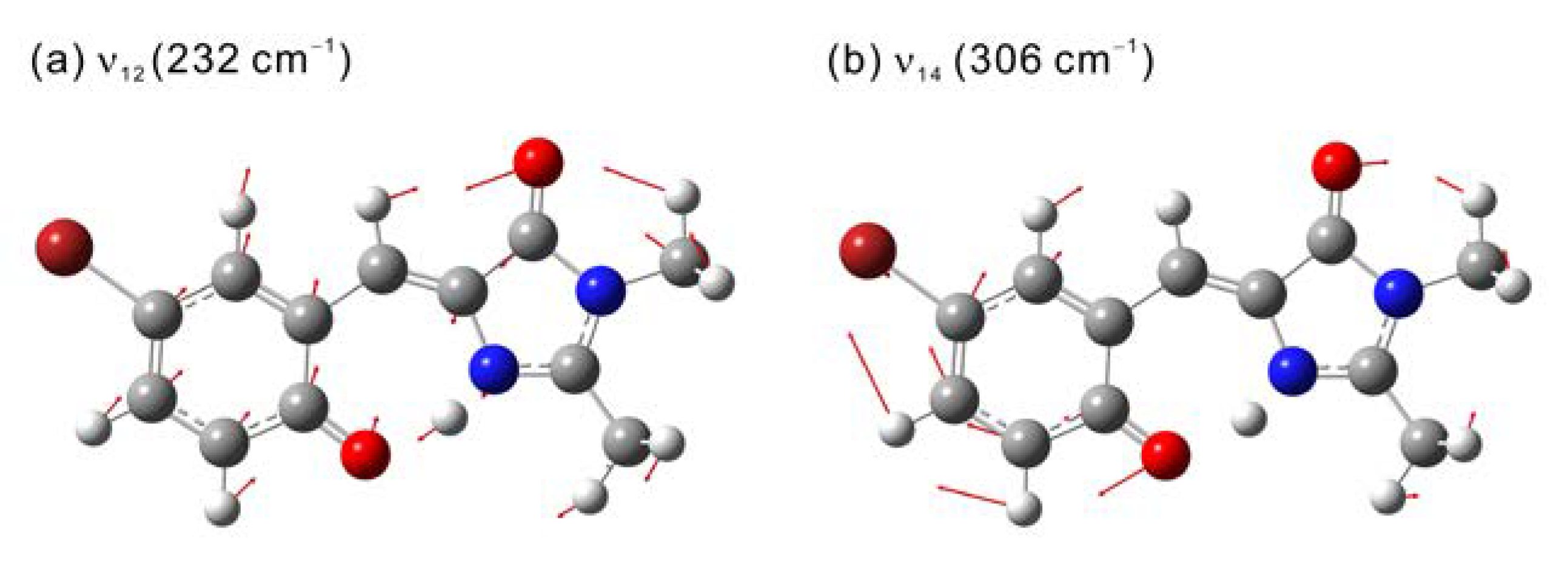
2.3. Quantum Chemical Calculations of CVSF
3. Discussion
4. Materials and Methods
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Chalfie, M.; Tu, Y.; Euskirchen, G.; Ward, W.; Prasher, D. Green Fluorescent Protein as a Marker for Gene Expression. Science 1994, 263, 802–805. [Google Scholar]
- Prasher, D.C.; Eckenrode, V.K.; Ward, W.W.; Prendergast, F.G.; Cormier, M.J. Primary Structure of the Aequorea Victoria Green-Fluorescent Protein. Gene 1992, 111, 229–233. [Google Scholar] [CrossRef]
- Ormö, M.; Cubitt, A.B.; Kallio, K.; Gross, L.A.; Tsien, R.Y.; Remington, S.J. Crystal Structure of the Aequorea Victoria Green Fluorescent Protein. Science 1996, 273, 1392–1395. [Google Scholar] [CrossRef]
- Reid, B.G.; Flynn, G.C. Chromophore Formation in Green Fluorescent Protein. Biochemistry 1997, 36, 6786–6791. [Google Scholar] [CrossRef]
- Chen, K.-Y.; Hsieh, C.-C.; Cheng, Y.-M.; Lai, C.-H.; Chou, P.-T. Extensive Spectral Tuning of the Proton Transfer Emission from 550 to 675 nm via a Rational Derivatization of 10-Hydroxybenzo[H]Quinoline. Chem. Commun. 2006, 42, 4395–4397. [Google Scholar] [CrossRef]
- Meech, S.R. Excited State Reactions in Fluorescent Proteins. Chem. Soc. Rev. 2009, 38, 2922–2934. [Google Scholar]
- Chatterjee, S.; Ahire, K.; Karuso, P. Room-Temperature Dual Fluorescence of a Locked Green Fluorescent Protein Chromophore Analogue. J. Am. Chem. Soc. 2020, 142, 738–749. [Google Scholar]
- Vengris, M.; van Stokkum, I.H.M.; He, X.; Bell, A.F.; Tonge, P.J.; van Grondelle, R.; Larsen, D.S. Ultrafast Excited and Ground-State Dynamics of the Green Fluorescent Protein Chromophore in Solution. J. Phys. Chem. A 2004, 108, 4587–4598. [Google Scholar]
- Stavrov, S.S.; Solntsev, K.M.; Tolbert, L.M.; Huppert, D. Probing the Decay Coordinate of the Green Fluorescent Protein: Arrest of Cis−Trans Isomerization by the Protein Significantly Narrows the Fluorescence Spectra. J. Am. Chem. Soc. 2006, 128, 1540–1546. [Google Scholar]
- Litvinenko, K.L.; Webber, N.M.; Meech, S.R. Internal Conversion in the Chromophore of the Green Fluorescent Protein: Temperature Dependence and Isoviscosity Analysis. J. Phys. Chem. A 2003, 107, 2616–2623. [Google Scholar]
- Mandal, D.; Tahara, T.; Meech, S.R. Excited-State Dynamics in the Green Fluorescent Protein Chromophore. J. Phys. Chem. B 2003, 108, 1102–1108. [Google Scholar] [CrossRef]
- Chatterjee, T.; Roy, D.; Das, A.; Ghosh, A.; Bag, P.P.; Mandal, P.K. Chemical Tweaking of a Non-Fluorescent GFP Chromophore to a Highly Fluorescent Coumarinic Fluorophore: Application Towards Photo-Uncaging and Stem Cell Imaging. RSC Adv. 2013, 3, 24021–24024. [Google Scholar]
- List, N.H.; Jones, C.M.; Martínez, T.J. Internal Conversion of the Anionic GFP Chromophore: In and out of the I-Twisted S1/S0 Conical Intersection Seam. Chem. Sci. 2022, 13, 373–385. [Google Scholar]
- Chen, K.-Y.; Cheng, Y.-M.; Lai, C.-H.; Hsu, C.-C.; Ho, M.-L.; Lee, G.-H.; Chou, P.-T. Ortho Green Fluorescence Protein Synthetic Chromophore; Excited-State Intramolecular Proton Transfer via a Seven-Membered-Ring Hydrogen-Bonding System. J. Am. Chem. Soc. 2007, 129, 4534–4535. [Google Scholar] [CrossRef]
- Chuang, W.T.; Hsieh, C.C.; Lai, C.H.; Shih, C.W.; Chen, K.Y.; Hung, W.Y.; Hsu, Y.H.; Chou, P.T. Excited-State Intramolecular Proton Transfer Molecules Bearing o-Hydroxy Analogues of Green Fluorescent Protein Chromophore. J. Org. Chem. 2011, 76, 8189–8202. [Google Scholar]
- Liu, Z.-Y.; Hu, J.-W.; Chen, C.-L.; Chen, Y.-A.; Chen, K.-Y.; Chou, P.-T. Correlation among Hydrogen Bond, Excited-State Intramolecular Proton-Transfer Kinetics and Thermodynamics for −OH Type Proton-Donor Molecules. J. Phys. Chem. C 2018, 122, 21833–21840. [Google Scholar] [CrossRef]
- Ma, D.; Liang, F.; Wang, L.; Lee, S.T.; Hung, L.S. Blue Organic Light-Emitting Devices with an Oxadiazole-Containing Emitting Layer Exhibiting Excited State Intramolecular Proton Transfer. Chem. Phys. Lett. 2002, 358, 24–28. [Google Scholar] [CrossRef]
- Kim, S.; Seo, J.; Jung, H.K.; Kim, J.J.; Park, S.Y. White Luminescence from Polymer Thin Films Containing Excited-State Intramolecular Proton-Transfer Dyes. Adv. Mater. 2005, 17, 2077–2082. [Google Scholar]
- Park, S.; Kwon, J.E.; Kim, S.H.; Seo, J.; Chung, K.; Park, S.-Y.; Jang, D.-J.; Medina, B.a.M.n.; Gierschner, J.; Park, S.Y. A White-Light-Emitting Molecule: Frustrated Energy Transfer between Constituent Emitting Centers. J. Am. Chem. Soc. 2009, 131, 14043–14049. [Google Scholar]
- Douhal, A.; Lahmani, F.; Zewail, A.H. Proton-Transfer Reaction Dynamics. Chem. Phys. 1996, 207, 477–498. [Google Scholar] [CrossRef]
- Furukawa, K.; Yamamoto, N.; Hino, K.; Sekiya, H. Excited-State Intramolecular Proton Transfer and Conformational Relaxation in 4′-N,N-Dimethylamino-3-Hydroxyflavone Doped in Acetonitrile Crystals. Phys. Chem. Chem. Phys. 2016, 18, 28564–28575. [Google Scholar] [CrossRef]
- Joshi, H.C.; Antonov, L. Excited-State Intramolecular Proton Transfer: A Short Introductory Review. Molecules 2021, 26, 1475. [Google Scholar] [CrossRef]
- Hsieh, C.-C.; Chou, P.-T.; Shih, C.-W.; Chuang, W.-T.; Chung, M.-W.; Lee, J.; Joo, T. Comprehensive Studies on an Overall Proton Transfer Cycle of the Ortho-Green Fluorescent Protein Chromophore. J. Am. Chem. Soc. 2011, 133, 2932–2943. [Google Scholar] [CrossRef]
- Cui, G.; Lan, Z.; Thiel, W. Intramolecular Hydrogen Bonding Plays a Crucial Role in the Photophysics and Photochemistry of the GFP Chromophore. J. Am. Chem. Soc. 2011, 134, 1662–1672. [Google Scholar] [CrossRef]
- Barkhuijsen, H.; de Beer, R.; van Ormondt, D. Improved Algorithm for Noniterative Time-Domain Model Fitting to Exponentially Damped Magnetic Resonance Signals. J. Magn. Reson. 1987, 73, 553–557. [Google Scholar] [CrossRef]
- Wise, F.W.; Rosker, M.J.; Millhauser, G.L.; Tang, C.L. Application of Linear Prediction Least-Squares Fitting to Time-Resolved Optical Spectroscopy. IEEE J. Quantum Electron. 1987, 23, 1116–1121. [Google Scholar] [CrossRef]
- Millhauser, G.L.; Freed, J.H. Linear Prediction and Resolution Enhancement of Complex Line Shapes in Two-Dimensional Electron-Spin-Echo Spectroscopy. J. Chem. Phys. 1986, 85, 63–67. [Google Scholar] [CrossRef]
- Kim, C.H.; Joo, T. Coherent Excited State Intramolecular Proton Transfer Probed by Time-Resolved Fluorescence. Phys. Chem. Chem. Phys. 2009, 11, 10266–10269. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16, Revision C.01; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Merrick, J.P.; Moran, D.; Radom, L. An Evaluation of Harmonic Vibrational Frequency Scale Factors. J. Phys. Chem. A 2007, 111, 11683–11700. [Google Scholar] [CrossRef]
- Lee, S.N.; Ahn, J.; Joo, T. Coherent Vibrational Spectrum Via Time-Resolved Fluorescence for Molecular Dynamics and Identification of Emitting Species–Application to Excited-State Intramolecular Proton Transfer. J. Phys. Chem. A 2022, 126, 4962–4968. [Google Scholar] [CrossRef]
- Reimers, J.R. A Practical Method for the Use of Curvilinear Coordinates in Calculations of Normal-Mode-Projected Displacements and Duschinsky Rotation Matrices for Large Molecules. J. Chem. Phys. 2001, 115, 9103–9109. [Google Scholar] [CrossRef]
- Kim, J.; Kang, D.-g.; Kim, S.K.; Joo, T. Role of Coherent Nuclear Motion in the Ultrafast Intersystem Crossing of Ruthenium Complexes. Phys. Chem. Chem. Phys. 2020, 22, 25811–25818. [Google Scholar] [CrossRef]
- Takeuchi, S.; Tahara, T. Coherent Nuclear Wavepacket Motions in Ultrafast Excited-State Intramolecular Proton Transfer: Sub-30-Fs Resolved Pump-Probe Absorption Spectroscopy of 10-Hydroxybenzo[H]Quinoline in Solution. J. Phys. Chem. A 2005, 109, 10199–10207. [Google Scholar] [CrossRef]
- Schriever, C.; Barbatti, M.; Stock, K.; Aquino, A.J.A.; Tunega, D.; Lochbrunner, S.; Riedle, E.; de Vivie-Riedle, R.; Lischka, H. The Interplay of Skeletal Deformations and Ultrafast Excited-State Intramolecular Proton Transfer: Experimental and Theoretical Investigation of 10-Hydroxybenzo[H]Quinoline. Chem. Phys. 2008, 347, 446–461. [Google Scholar] [CrossRef]
- Lochbrunner, S.; Wurzer, A.J.; Riedle, E. Microscopic Mechanism of Ultrafast Excited-State Intramolecular Proton Transfer: A 30-fs Study of 2-(2′-Hydroxyphenyl)Benzothiazole. J. Phys. Chem. A 2003, 107, 10580–10590. [Google Scholar] [CrossRef]
- Rhee, H.; Joo, T. Noncollinear Phase Matching in Fluorescence Upconversion. Opt. Lett. 2005, 30, 96–98. [Google Scholar] [CrossRef]
- Manoj, P.; Min, C.-K.; Aravindakumar, C.T.; Joo, T. Ultrafast Charge Transfer Dynamics in 2-Aminopurine Modified Double Helical DNA. Chem. Phys. 2008, 352, 333–338. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compounds | Stokes Shift (cm−1) | τavg. (ps) | λdet (nm) | A1 | τ1 (fs) | A2 | τ2 (fs) | A3 | τ3 (ps) |
|---|---|---|---|---|---|---|---|---|---|
| 1 | 9100 | 480 a | 430 | 1.0 | 36 | ||||
| 550 | −1.0 | 37 | 0.24 | 235 | 0.76 | 15 | |||
| 2 | 9380 | 10 a | 440 | 1.0 | 24 | ||||
| 580 | −1.0 | 37 | 0.47 | 236 | 0.53 | 7.9 | |||
| 3 | 8560 | 2 b | 450 | 1.0 | 24 | ||||
| 630 | −1.0 | 23 | 0.61 | 105 | 0.39 | 5.1 | |||
| 4 | 9370 | 1 b | 450 | 1.0 | 38 | ||||
| 630 | −1.0 | 16 | 0.62 | 131 | 0.38 | 2.4 |
| Compounds | Amplitude | Freq. (cm−1) | Dephasing Time (fs) | Vib. Mode | Cal Freq. (cm−1) |
|---|---|---|---|---|---|
| 1 | 0.54 | 118 | 520 | ν10 | 153(?) |
| 1.0 | 225 | 690 | ν13 | 229 | |
| 0.88 | 291 | 360 | ν15 | 287 | |
| 0.12 | 451 | 2500 | ν21 | 456 | |
| 2 | 0.78 | 229 | 510 | ν12 | 232 |
| 1.0 | 310 | 1100 | ν14 | 306 | |
| 0.12 | 338 | 3200 | ν17 | 338 | |
| 3 | 0.9 | 109 | 530 | ν6 | 103 |
| 1.0 | 242 | 370 | ν13 | 232 | |
| 4 | 1.0 | 241 | 1500 | ν12 | 245 |
| Compound | λ (nm) | d (O-H) | d (N-H) | d (O-N) |
|---|---|---|---|---|
| 1 | 574 (0.171) | 1.6606 | 1.0415 | 2.6106 |
| 2 | 595 (0.155) | 1.5138 | 1.0776 | 2.5381 |
| 3 | 581 (0.090) | 1.0728 | 1.4438 | 2.5042 |
| 3 a | 669 (0.151) | 1.6281 | 1.0000 | 2.5622 |
| 4 | 672 (0.185) | 1.6206 | 1.0570 | 2.5999 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lee, J.; Shin, P.; Chou, P.-T.; Joo, T. Excited State Intramolecular Proton Transfer Dynamics of Derivatives of the Green Fluorescent Protein Chromophore. Int. J. Mol. Sci. 2023, 24, 3448. https://doi.org/10.3390/ijms24043448
Lee J, Shin P, Chou P-T, Joo T. Excited State Intramolecular Proton Transfer Dynamics of Derivatives of the Green Fluorescent Protein Chromophore. International Journal of Molecular Sciences. 2023; 24(4):3448. https://doi.org/10.3390/ijms24043448
Chicago/Turabian StyleLee, Junghwa, Pyoungsik Shin, Pi-Tai Chou, and Taiha Joo. 2023. "Excited State Intramolecular Proton Transfer Dynamics of Derivatives of the Green Fluorescent Protein Chromophore" International Journal of Molecular Sciences 24, no. 4: 3448. https://doi.org/10.3390/ijms24043448