
Loss of S1P Lyase Expression in Human Podocytes Causes a Reduction in Nephrin Expression That Involves PKCδ Activation


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
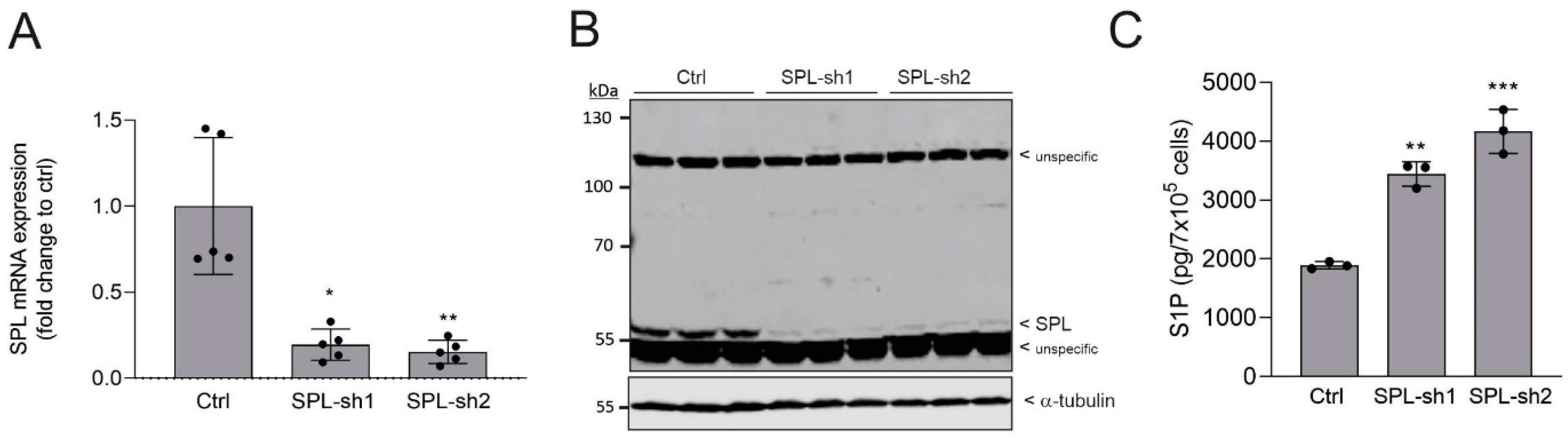
2.1. Generation and Characterization of SPL Knockdown in Human Podocytes
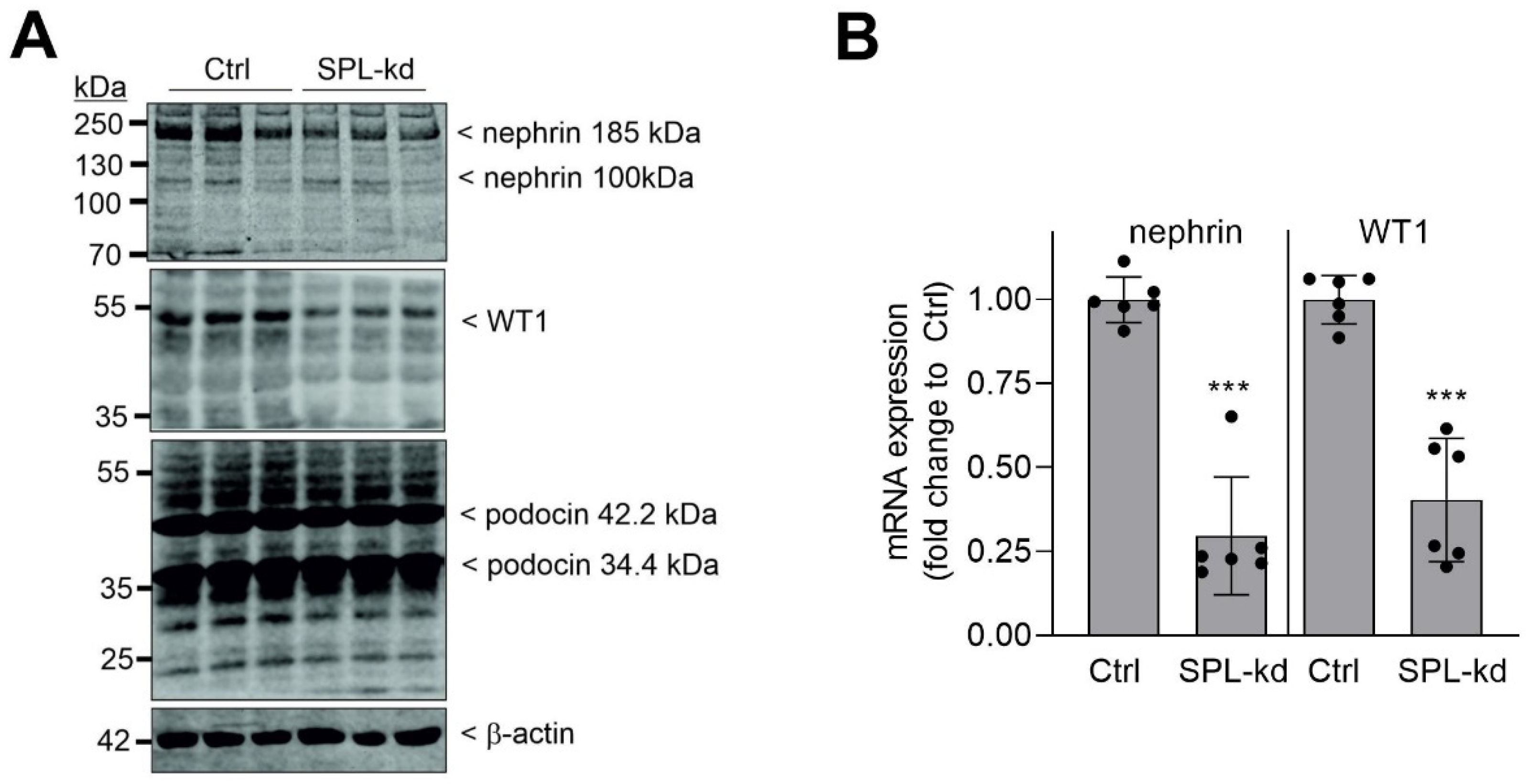
2.2. SPL Knockdown Downregulates WT1 and Nephrin Expression in Podocytes
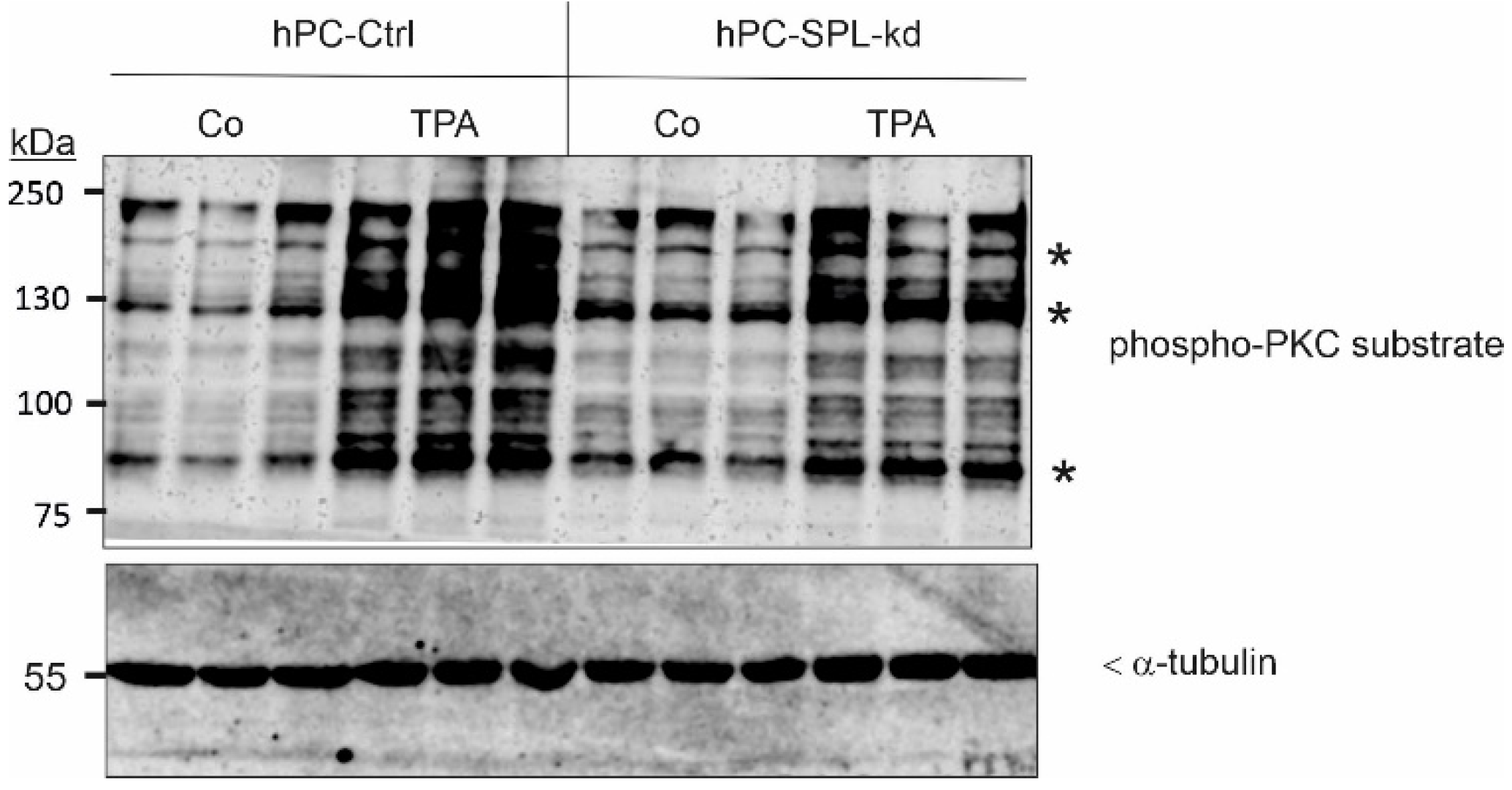
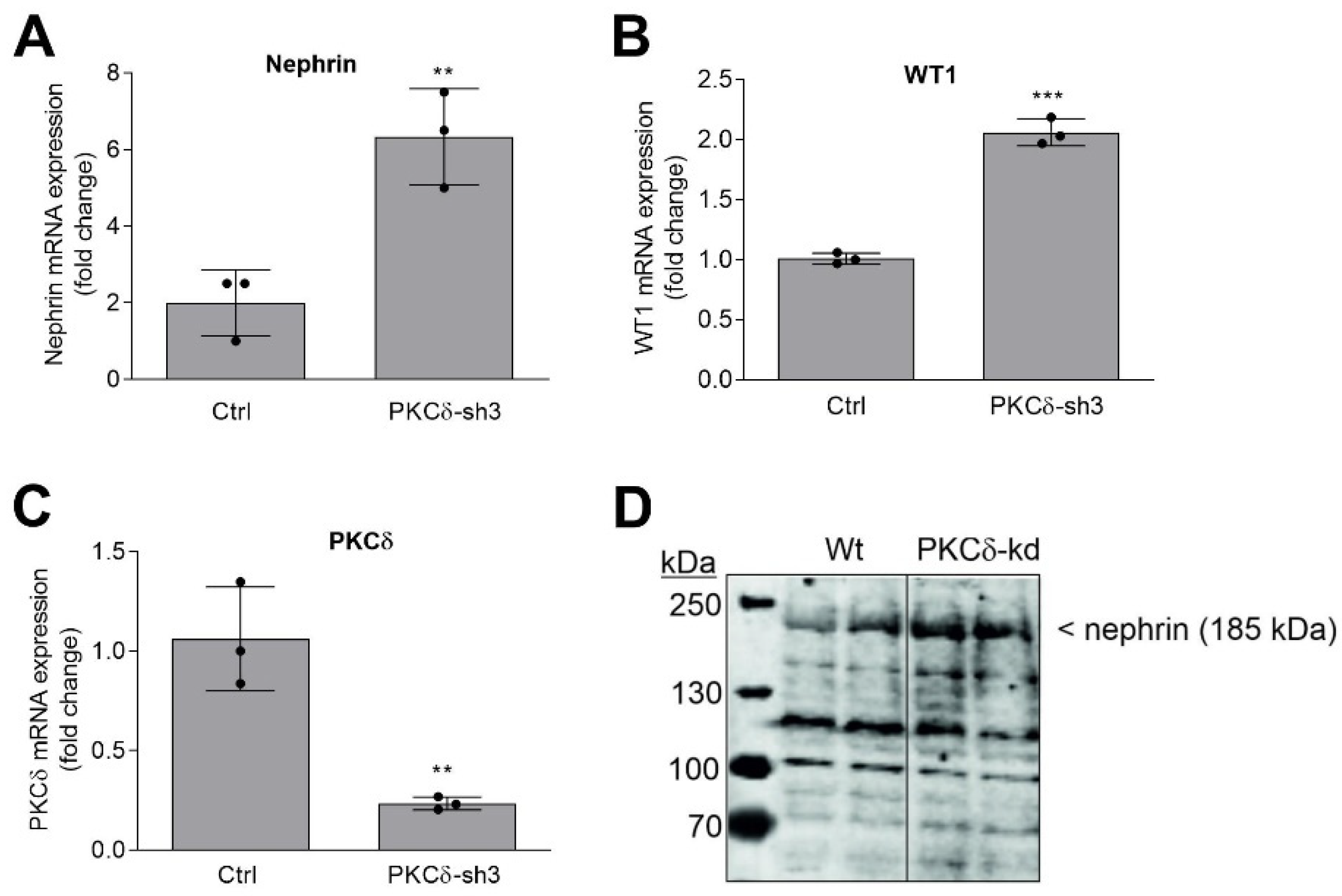
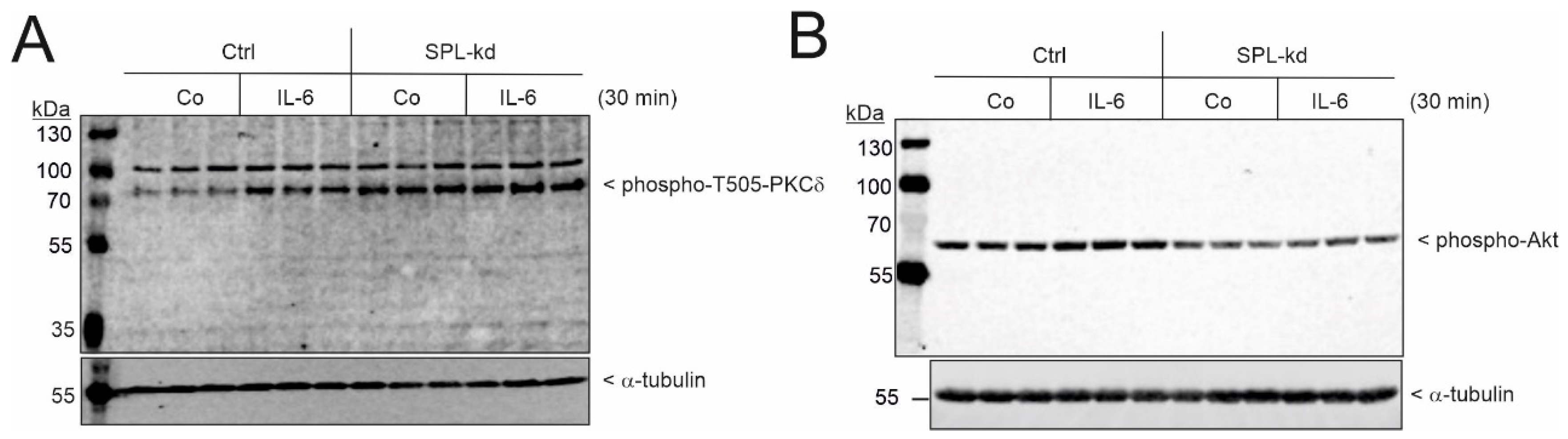
2.3. SPL Knockdown Stimulates Cellular PKC Activity in Podocytes
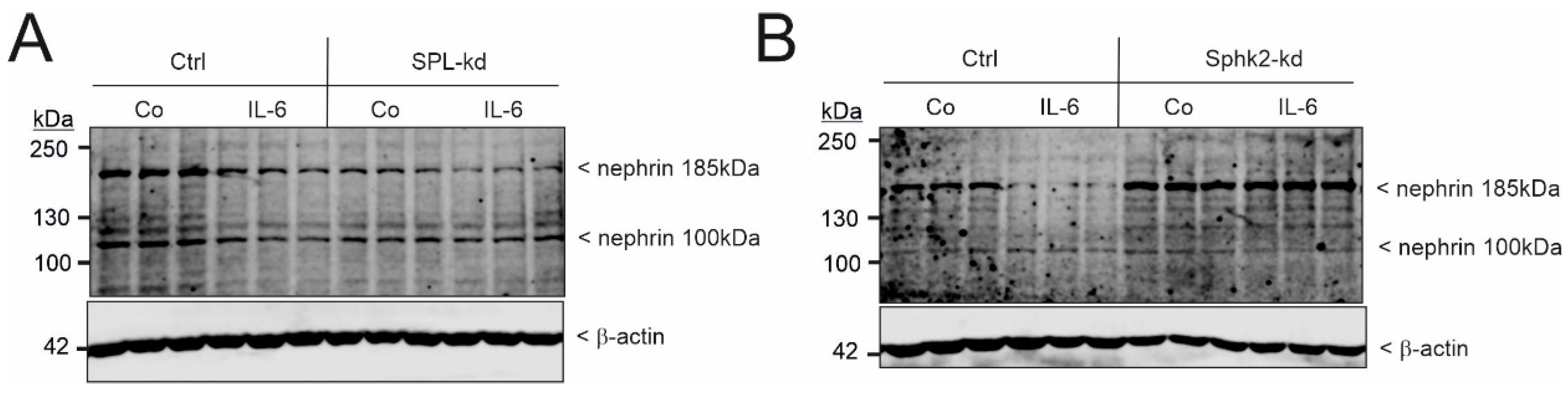
2.4. Interleukin 6 Accelerates Nephrin Downregulation in SPL Knockdown Podocytes
3. Discussion
4. Materials and Methods
4.1. Chemicals
4.2. Peptide Synthesis and Polyclonal Antibody Generation
4.3. Cell Culturing and Stable SPL and PKC Knockdown Generation
4.4. Western Blot Analysis
4.5. RNA Extraction and Quantitative PCR Analysis
4.6. Quantification of S1P by LC-MS/MS
4.7. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| AhR | Arylhydrocarbon receptor |
| DAG | Diacylglycerol |
| IL-6 | Interleukin 6 |
| Kd | knockdown |
| HIF | hypoxia-inducible factor |
| LC-MS | liquid chromatography-mass spectrometry |
| PDGF | platelet-derived growth factor |
| PKC | protein kinase C |
| S1P | sphingosine 1-phosphate |
| SDS-PAGE | sodium dodecyl sulfate-polyacrylamide gel electrophoresis |
| shRNA | small hairpin RNA |
| SphK | sphingosine kinase |
| SPL, Sgpl1 | S1P lyase |
| SPLIS | S1P lyase insufficiency syndrome |
| TPA | 12-O-tetradecanoyl-phorbol 13-acetate |
| WT1 | Wilms tumor suppressor gene 1 |
References
- Pavenstädt, H.; Kriz, W.; Kretzler, M. Cell Biology of the Glomerular Podocyte. Physiol. Rev. 2003, 83, 253–307. [Google Scholar] [CrossRef]
- Garg, P. A Review of Podocyte Biology. Am. J. Nephrol. 2018, 47, 3–13. [Google Scholar] [CrossRef]
- Vivante, A.; Hildebrandt, F. Exploring the genetic basis of early-onset chronic kidney disease. Nat. Rev. Nephrol. 2016, 12, 133–146. [Google Scholar] [CrossRef]
- Kopp, J.B.; Anders, H.J.; Susztak, K.; Podesta, M.A.; Remuzzi, G.; Hildebrandt, F.; Romagnani, P. Podocytopathies. Nat. Rev. Dis. Prim. 2020, 6, 68. [Google Scholar] [CrossRef]
- Lovric, S.S.; Goncalves, S.; Gee, H.Y.; Oskouian, B.; Srinivas, H.; Choi, W.-I.; Shril, S.; Ashraf, S.; Tan, W.; Rao, J.; et al. Mutations in sphingosine-1-phosphate lyase cause nephrosis with ichthyosis and adrenal insufficiency. J. Clin. Investig. 2017, 127, 912–928. [Google Scholar] [CrossRef]
- Prasad, R.; Hadjidemetriou, I.; Maharaj, A.; Meimaridou, E.; Buonocore, F.; Saleem, M.; Hurcombe, J.; Bierzynska, A.; Barbagelata, E.; Bergadá, I.; et al. Sphingosine-1-phosphate lyase mutations cause primary adrenal insufficiency and steroid-resistant nephrotic syndrome. J. Clin. Investig. 2017, 127, 942–953. [Google Scholar] [CrossRef] [PubMed]
- Janecke, A.R.; Xu, R.; Steichen-Gersdorf, E.; Waldegger, S.; Entenmann, A.; Giner, T.; Krainer, I.; A Huber, L.; Hess, M.W.; Frishberg, Y.; et al. Deficiency of the sphingosine-1-phosphate lyase SGPL1 is associated with congenital nephrotic syndrome and congenital adrenal calcifications. Hum. Mutat. 2017, 38, 365–372. [Google Scholar] [CrossRef]
- Maceyka, M.; Harikumar, K.B.; Milstien, S.; Spiegel, S. Sphingosine-1-phosphate signaling and its role in disease. Trends Cell Biol. 2012, 22, 50–60. [Google Scholar] [CrossRef]
- Huwiler, A.; Pfeilschifter, J. Sphingolipid signaling in renal fibrosis. Matrix Biol. 2018, 68–69, 230–247. [Google Scholar] [CrossRef]
- Stepanovska, B.; Huwiler, A. Targeting the S1P receptor signaling pathways as a promising approach for treatment of au-toimmune and inflammatory diseases. Pharmacol. Res. 2020, 154, 104170. [Google Scholar] [CrossRef]
- Blaho, V.A.; Hla, T. An update on the biology of sphingosine 1-phosphate receptors. J. Lipid Res. 2014, 55, 1596–1608. [Google Scholar] [CrossRef]
- Kihara, Y.; Maceyka, M.; Spiegel, S.; Chun, J. Lysophospholipid receptor nomenclature review: IUPHAR Review 8. Br. J. Pharmacol. 2014, 171, 3575–3594. [Google Scholar] [CrossRef]
- Huwiler, A.; Pfeilschifter, J. New players on the center stage: Sphingosine 1-phosphate and its receptors as drug targets. Biochem. Pharmacol. 2008, 75, 1893–1900. [Google Scholar] [CrossRef] [PubMed]
- Alemany, R.; Van Koppen, C.J.; Danneberg, K.; Ter Braak, M.; Zu Heringdorf, D.M. Regulation and functional roles of sphingosine kinases. Naunyn-Schmiedeberg’s Arch. Pharmacol. 2007, 374, 413–428. [Google Scholar] [CrossRef]
- Haddadi, N.; Lin, Y.G.; Simpson, A.M.; Nassif, N.T.; McGowan, E.M. “Dicing and Splicing” Sphingosine Kinase and Rel-evance to Cancer. Int. J. Mol. Sci. 2017, 18, 1891. [Google Scholar] [CrossRef]
- Bourquin, F.; Capitani, G.; Grutter, M.G. PLP-dependent enzymes as entry and exit gates of sphingolipid metabolism. Protein. Sci. 2011, 20, 1492–1508. [Google Scholar] [CrossRef]
- Saba, J.D. Fifty years of lyase and a moment of truth: Sphingosine phosphate lyase from discovery to disease[S]. J. Lipid Res. 2019, 60, 456–463. [Google Scholar] [CrossRef]
- Schmahl, J.; Raymond, C.; Soriano, P. PDGF signaling specificity is mediated through multiple immediate early genes. Nat. Genet. 2006, 39, 52–60. [Google Scholar] [CrossRef]
- Schumann, J.; Grevot, A.; Ledieu, D.; Wolf, A.; Schubart, A.; Piaia, A.; Sutter, E.; Cote, S.; Beerli, C.; Pognan, F.; et al. Reduced Activity of Sphingosine-1-Phosphate Lyase Induces Podocyte-related Glomerular Pro-teinuria, Skin Irritation, and Platelet Activation. Toxicol. Pathol. 2015, 43, 694–703. [Google Scholar] [CrossRef] [PubMed]
- Martin, C.E.; Jones, N. Nephrin Signaling in the Podocyte: An Updated View of Signal Regulation at the Slit Diaphragm and Beyond. Front. Endocrinol. 2018, 9, 302. [Google Scholar] [CrossRef] [PubMed]
- Patrakka, J.; Tryggvason, K. Nephrin—A unique structural and signaling protein of the kidney filter. Trends Mol. Med. 2007, 13, 396–403. [Google Scholar] [CrossRef] [PubMed]
- Ahola, H.; Wang, S.-X.; Luimula, P.; Solin, M.-L.; Holzman, L.B.; Holthöfer, H. Cloning and Expression of the Rat Nephrin Homolog. Am. J. Pathol. 1999, 155, 907–913. [Google Scholar] [CrossRef] [PubMed]
- Rinschen, M.M.; Hoppe, A.-K.; Grahammer, F.; Kann, M.; Völker, L.A.; Schurek, E.-M.; Binz, J.; Höhne, M.; Demir, F.; Malisic, M.; et al. N-Degradomic Analysis Reveals a Proteolytic Network Processing the Podocyte Cytoskeleton. J. Am. Soc. Nephrol. 2017, 28, 2867–2878. [Google Scholar] [CrossRef]
- Wagner, N.; Wagner, K.D.; Xing, Y.; Scholz, H.; Schedl, A. The major podocyte protein nephrin is transcriptionally activated by the Wilms’ tumor suppressor WT1. J. Am. Soc. Nephrol. 2004, 15, 3044–3051. [Google Scholar] [CrossRef] [PubMed]
- Guo, G.; Morrison, D.J.; Licht, J.D.; Quaggin, S.E. WT1 Activates a Glomerular-Specific Enhancer Identified from the Human Nephrin Gene. J. Am. Soc. Nephrol. 2004, 15, 2851–2856. [Google Scholar] [CrossRef] [PubMed]
- Stepanovska, B.; Lange, A.I.; Schwalm, S.; Pfeilschifter, J.; Coldewey, S.M.; Huwiler, A. Downregulation of S1P Lyase Im-proves Barrier Function in Human Cerebral Microvascular Endothelial Cells Following an Inflammatory Challenge. Int. J. Mol. Sci. 2020, 21, 1240. [Google Scholar] [CrossRef] [PubMed]
- Antal, C.E.; Newton, A.C. Tuning the signalling output of protein kinase C. Biochem. Soc. Trans. 2014, 42, 1477–1483. [Google Scholar] [CrossRef]
- Parker, P.J.; Murray-Rust, J. PKC at a glance. J. Cell. Sci. 2004, 117, 131–132. [Google Scholar] [CrossRef]
- Menne, J.; Meier, M.; Park, J.-K.; Boehne, M.; Kirsch, T.; Lindschau, C.; Ociepka, R.; Leitges, M.; Rinta-Valkama, J.; Holthofer, H.; et al. Nephrin loss in experimental diabetic nephropathy is prevented by deletion of protein kinase C alpha signaling in-vivo. Kidney Int. 2006, 70, 1456–1462. [Google Scholar] [CrossRef]
- Mima, A.; Kitada, M.; Geraldes, P.; Li, Q.; Matsumoto, M.; Mizutani, K.; Qi, W.; Li, C.; Leitges, M.; Rask-Madsen, C.; et al. Glomerular VEGF resistance induced by PKCdelta/SHP-1 activation and contribution to diabetic nephropathy. FASEB J. 2012, 26, 2963–2974. [Google Scholar] [CrossRef] [Green Version]
- Imeri, F.; Stepanovska Tanturovska, B.; Schwalm, S.; Saha, S.; Zeng-Brouwers, J.; Pavenstadt, H.; Pfeilschifter, J.; Schaefer, L.; Huwiler, A. Loss of sphingosine kinase 2 enhances Wilm’s tumor suppressor gene 1 and nephrin expression in podocytes and protects from streptozotocin-induced podocytopathy and albuminuria in mice. Matrix Biol. 2021, 98, 32–48. [Google Scholar] [CrossRef]
- Allende, M.L.; Bektas, M.; Lee, B.G.; Bonifacino, E.; Kang, J.; Tuymetova, G.; Chen, W.; Saba, J.D.; Proia, R.L. Sphingo-sine-1-phosphate lyase deficiency produces a pro-inflammatory response while impairing neutrophil trafficking. J. Biol. Chem. 2011, 286, 7348–7358. [Google Scholar] [CrossRef] [PubMed]
- Su, H.; Lei, C.-T.; Zhang, C. Interleukin-6 Signaling Pathway and Its Role in Kidney Disease: An Update. Front. Immunol. 2017, 8, 405. [Google Scholar] [CrossRef] [PubMed]
- Parekh, D.B.; Katso, R.M.; Leslie, N.R.; Downes, C.P.; Procyk, K.J.; Waterfield, M.D.; Parker, P.J. Beta1-integrin and PTEN control the phosphorylation of protein kinase C. Biochem. J. 2000, 352, 425–433. [Google Scholar] [CrossRef]
- Palmer, R.E.; Kotsianti, A.; Cadman, B.; Boyd, T.; Gerald, W.; Haber, D.A. WT1 regulates the expression of the major glo-merular podocyte membrane protein Podocalyxin. Curr. Biol. 2001, 11, 1805–1809. [Google Scholar] [CrossRef]
- Fraizer, G.C.; Wu, Y.J.; Hewitt, S.M.; Maity, T.; Ton, C.C.; Huff, V.; Saunders, G.F. Transcriptional regulation of the human Wilms’ tumor gene (WT1). Cell type-specific enhancer and promiscuous promoter. J. Biol. Chem. 1994, 269, 8892–8900. [Google Scholar] [CrossRef]
- Wagner, K.D.; Wagner, N.; Wellmann, S.; Schley, G.; Bondke, A.; Theres, H.; Scholz, H. Oxygen-regulated expression of the Wilms’ tumor suppressor Wt1 involves hypoxia-inducible factor-1 (HIF-1). FASEB J. 2003, 17, 1364–1366. [Google Scholar] [CrossRef] [PubMed]
- Dame, C.; Kirschner, K.M.; Bartz, K.V.; Wallach, T.; Hussels, C.S.; Scholz, H. Wilms tumor suppressor, Wt1, is a transcrip-tional activator of the erythropoietin gene. Blood 2006, 107, 4282–4290. [Google Scholar] [CrossRef]
- Kim, H.; Na, Y.-R.; Kim, S.Y.; Yang, E.G. Protein Kinase C Isoforms Differentially Regulate Hypoxia-Inducible Factor-1α Accumulation in Cancer Cells. J. Cell. Biochem. 2015, 117, 647–658. [Google Scholar] [CrossRef] [PubMed]
- Falahatpisheh, M.H.; Nanez, A.; Ramos, K.S. AHR Regulates WT1 Genetic Programming during Murine Nephrogenesis. Mol. Med. 2011, 17, 1275–1284. [Google Scholar] [CrossRef]
- Ichii, O.; Otsuka-Kanazawa, S.; Nakamura, T.; Ueno, M.; Kon, Y.; Chen, W.; Rosenberg, A.Z.; Kopp, J.B. Podocyte Injury Caused by Indoxyl Sulfate, a Uremic Toxin and Aryl-Hydrocarbon Receptor Ligand. PLoS ONE 2014, 9, e108448. [Google Scholar] [CrossRef]
- Han, E.H.; Kim, H.G.; Lee, E.J.; Jeong, H.G. Endosulfan Induces CYP1A1 Expression Mediated through Aryl Hydrocarbon Receptor Signal Transduction by Protein Kinase C. Toxicol. Res. 2015, 31, 339–345. [Google Scholar] [CrossRef]
- Ye, Y.; Raychaudhuri, B.; Gurney, A.; Campbell, C.E.; Williams, B.R. Regulation of WT1 by phosphorylation: Inhibition of DNA binding, alteration of transcriptional activity and cellular translocation. EMBO J. 1996, 15, 5606–5615. [Google Scholar] [CrossRef] [PubMed]
- Arellano-Rodríguez, M.; Zapata-Benavides, P.; Arellano-Rodríguez, N.C.; Izaguirre-Álvarez, J.M.; Franco-Molina, M.A.; Muro, F.D.J.T.D.; Mendoza-Gamboa, E.; Soto-Domínguez, A.; Saavedra-Alonso, S.; Rodríguez-Padilla, C. The Inflammatory Process Modulates the Expression and Localization of WT1 in Podocytes Leading to Kidney Damage. Vivo 2021, 35, 3137–3146. [Google Scholar] [CrossRef]
- Tossidou, I.; Teng, B.; Menne, J.; Shushakova, N.; Park, J.-K.; Becker, J.U.; Modde, F.; Leitges, M.; Haller, H.; Schiffer, M. Podocytic PKC-Alpha Is Regulated in Murine and Human Diabetes and Mediates Nephrin Endocytosis. PLoS ONE 2010, 5, e10185. [Google Scholar] [CrossRef]
- Zhu, J.; Zhang, G.; Song, Z.; Xiang, X.; Shu, S.; Liu, Z.; Yang, D.; Wei, Q.; Dong, Z. Protein Kinase C-delta Mediates Kidney Tubular Injury in Cold Storage-Associated Kidney Transplantation. J. Am. Soc. Nephrol. 2020, 31, 1050–1065. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Pabla, N.; Wei, Q.; Dong, G.; Messing, R.O.; Wang, C.Y.; Dong, Z. PKC-delta promotes renal tubular cell apoptosis associated with proteinuria. J. Am. Soc. Nephrol. 2010, 21, 1115–1124. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Zhang, J.; Chen, H.; Wang, Z.; Hou, J.; Wang, L. Dimethylamine enhances platelet hyperactivity in chronic kidney disease model. J. Bioenerg. Biomembr. 2021, 53, 585–595. [Google Scholar] [CrossRef]
- Neehus, A.L.; Moriya, K.; Nieto-Patlan, A.; Le Voyer, T.; Levy, R.; Ozen, A.; Karakoc-Aydiner, E.; Baris, S.; Yildiran, A.; Altundag, E.; et al. Impaired respiratory burst contributes to infections in PKCdelta-deficient patients. J. Exp. Med. 2021, 218, e20210501. [Google Scholar] [CrossRef] [PubMed]
- Bektas, M.; Allende, M.L.; Lee, B.G.; Chen, W.; Amar, M.J.; Remaley, A.T.; Saba, J.D.; Proia, R.L. Sphingosine 1-Phosphate Lyase Deficiency Disrupts Lipid Homeostasis in Liver*. J. Biol. Chem. 2010, 285, 10880–10889. [Google Scholar] [CrossRef] [Green Version]
- Gerl, M.J.; Bittl, V.; Kirchner, S.; Sachsenheimer, T.; Brunner, H.L.; Luchtenborg, C.; Ozbalci, C.; Wiedemann, H.; Wegehingel, S.; Nickel, W.; et al. Sphingosine-1-Phosphate Lyase Deficient Cells as a Tool to Study Protein Lipid Interactions. PLoS ONE 2016, 11, e0153009. [Google Scholar] [CrossRef] [PubMed]
- Newton, A.C. Protein kinase C: Perfectly balanced. Crit. Rev. Biochem. Mol. Biol. 2018, 53, 208–230. [Google Scholar] [CrossRef]
- Kajimoto, T.; Caliman, A.D.; Tobias, I.S.; Okada, T.; Pilo, C.A.; Van, A.N.; Andrew McCammon, J.; Nakamura, S.I.; Newton, A.C. Activation of atypical protein kinase C by sphingosine 1-phosphate revealed by an aPKC-specific activity reporter. Sci. Signal. 2019, 12, eaat6662. [Google Scholar] [CrossRef] [PubMed]
- Spohner, A.K.; Jakobi, K.; Trautmann, S.; Thomas, D.; Schumacher, F.; Kleuser, B.; Lütjohann, D.; El-Hindi, K.; Grösch, S.; Pfeilschifter, J.; et al. Mouse Liver Compensates Loss of Sgpl1 by Secretion of Sphingolipids into Blood and Bile. Int. J. Mol. Sci. 2021, 22, 10617. [Google Scholar] [CrossRef] [PubMed]
- Merrill, A.H., Jr.; Nimkar, S.; Menaldino, D.; Hannun, Y.A.; Loomis, C.; Bell, R.M.; Tyagi, S.R.; Lambeth, J.D.; Stevens, V.L.; Hunter, R.; et al. Structural requirements for long-chain (sphingoid) base inhibition of protein kinase C in vitro and for the cellular effects of these compounds. Biochemistry 1989, 28, 3138–3145. [Google Scholar] [CrossRef] [PubMed]
- Bell, R.M.; Loomis, C.R.; Hannun, Y.A. Regulation of Protein Kinase C by Sphingosine/Lysosphingolipids. Clin. Chim. Acta 1988, 185, 265–286. [Google Scholar] [CrossRef]
- Hamaguchi, A.; Suzuki, E.; Murayama, K.; Fujimura, T.; Hikita, T.; Iwabuchi, K.; Handa, K.; Withers, D.A.; Masters, S.C.; Fu, H.; et al. Sphingosine-dependent protein kinase-1, directed to 14-3-3, is identified as the kinase domain of protein kinase C delta. J. Biol. Chem. 2003, 278, 41557–41565. [Google Scholar] [CrossRef]
- Li, G.; Kidd, J.; Kaspar, C.; Dempsey, S.; Bhat, O.M.; Camus, S.; Ritter, J.K.; Gehr, T.W.B.; Gulbins, E.; Li, P.L. Podocytopathy and Nephrotic Syndrome in Mice with Podocyte-Specific Deletion of the Asah1 Gene: Role of Ceramide Accumulation in Glomeruli. Am. J. Pathol. 2020, 190, 1211–1223. [Google Scholar] [CrossRef] [PubMed]
- Pecoits-Filho, R.; Lindholm, B.; Axelsson, J.; Stenvinkel, P. Update on interleukin-6 and its role in chronic renal failure. Nephrol. Dial. Transplant. 2003, 18, 1042–1045. [Google Scholar] [CrossRef] [PubMed]
- Nagayama, Y.; Braun, G.S.; Jakobs, C.M.; Maruta, Y.; van Roeyen, C.R.; Klinkhammer, B.M.; Boor, P.; Villa, L.; Raffetseder, U.; Trautwein, C.; et al. Gp130-dependent signaling in the podocyte. Am. J. Physiol. Physiol. 2014, 307, F346–F355. [Google Scholar] [CrossRef] [Green Version]
- Horino, T.; Kashio, T.; Inotani, S.; Ishihara, M.; Ichii, O.D. Membranous Nephropathy Associated With Multicentric Castleman Disease—Efficacy of Interleukin 6 Antibody for Nephrotic Syndrome. Am. J. Clin. Oncol. 2021, 28, e1–e2. [Google Scholar] [CrossRef]
- He, F.; Bao, D.; Su, H.; Wang, Y.; Lei, C.; Zhang, C.; Ye, C.; Tang, H.; Wan, C.; You, C.; et al. IL-6 increases podocyte motility via MLC-mediated focal adhesion impairment and cytoskeleton disassembly. J. Cell. Physiol. 2018, 233, 7173–7181. [Google Scholar] [CrossRef]
- Jain, N.; Zhang, T.; Kee, W.H.; Li, W.; Cao, X. Protein kinase C delta associates with and phosphorylates Stat3 in an inter-leukin-6-dependent manner. J. Biol. Chem. 1999, 274, 24392–24400. [Google Scholar] [CrossRef]
- Schuringa, J.J.; Dekker, L.V.; Vellenga, E.; Kruijer, W. Sequential activation of Rac-1, SEK-1/MKK-4, and protein kinase Cdelta is required for interleukin-6-induced STAT3 Ser-727 phosphorylation and transactivation. J. Biol. Chem. 2001, 276, 27709–27715. [Google Scholar] [CrossRef]
- Schaefer, L.; Ren, S.; Schaefer, R.M.; Mihalik, D.; Babelova, A.; Huwiler, A.; Pfeilschifter, J. Nephrin expression is increased in anti-Thy1.1-induced glomerulonephritis in rats. Biochem. Biophys. Res. Commun. 2004, 324, 247–254. [Google Scholar] [CrossRef]
- Ren, S.Y.; Xin, C.Y.; Beck, K.F.; Saleem, M.A.; Mathieson, P.; Pavenstadt, H.; Pfeilschifter, J.; Huwiler, A. PPAR alpha ac-tivation upregulates nephrin expression in human embryonic kidney epithelial cells and podocytes by a dual mechanism. Biochem. Biophys. Res. Commun. 2005, 338, 1818–1824. [Google Scholar] [CrossRef] [PubMed]
- Pavenstädt, H.; Späth, M.; Schlunck, G.; Nauck, M.; Fischer, R.; Wanner, C.; Schollmeyer, P. Effect of nucleotides on the cytosolic free calcium activity and inositol phosphate formation in human glomerular epithelial cells. Br. J. Pharmacol. 1992, 107, 189–195. [Google Scholar] [CrossRef]
- Saleem, M.A.; O’Hare, M.J.; Reiser, J.; Coward, R.J.; Inward, C.D.; Farren, T.; Xing, C.Y.; Ni, L.; Mathieson, P.W.; Mundel, P. A conditionally immortalized human podocyte cell line demonstrating nephrin and podocin expression. J. Am. Soc. Nephrol. 2002, 13, 630–638. [Google Scholar] [CrossRef]
- Stepanovska Tanturovska, B.; Zivkovic, A.; Imeri, F.; Homann, T.; Kleuser, B.; Stark, H.; Huwiler, A. ST-2191, an Anellated Bismorpholino Derivative of Oxy-Fingolimod, Shows Selective S1P(1) Agonist and Functional Antagonist Potency In Vitro and In Vivo. Molecules 2021, 26, 5134. [Google Scholar] [CrossRef]
- Schmidt, H.; Schmidt, R.; Geisslinger, G. LC–MS/MS-analysis of sphingosine-1-phosphate and related compounds in plasma samples. Prostaglandins Other Lipid Mediat. 2006, 81, 162–170. [Google Scholar] [CrossRef]






Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Imeri, F.; Stepanovska Tanturovska, B.; Manaila, R.; Pavenstädt, H.; Pfeilschifter, J.; Huwiler, A. Loss of S1P Lyase Expression in Human Podocytes Causes a Reduction in Nephrin Expression That Involves PKCδ Activation. Int. J. Mol. Sci. 2023, 24, 3267. https://doi.org/10.3390/ijms24043267
Imeri F, Stepanovska Tanturovska B, Manaila R, Pavenstädt H, Pfeilschifter J, Huwiler A. Loss of S1P Lyase Expression in Human Podocytes Causes a Reduction in Nephrin Expression That Involves PKCδ Activation. International Journal of Molecular Sciences. 2023; 24(4):3267. https://doi.org/10.3390/ijms24043267
Chicago/Turabian StyleImeri, Faik, Bisera Stepanovska Tanturovska, Roxana Manaila, Hermann Pavenstädt, Josef Pfeilschifter, and Andrea Huwiler. 2023. "Loss of S1P Lyase Expression in Human Podocytes Causes a Reduction in Nephrin Expression That Involves PKCδ Activation" International Journal of Molecular Sciences 24, no. 4: 3267. https://doi.org/10.3390/ijms24043267