
Cloning and Characterization of Trypanosoma congolense and T. vivax Nucleoside Transporters Reveal the Potential of P1-Type Carriers for the Discovery of Broad-Spectrum Nucleoside-Based Therapeutics against Animal African Trypanosomiasis

 , , ,
, , ,  , and
, and
Abstract
:1. Introduction
2. Results
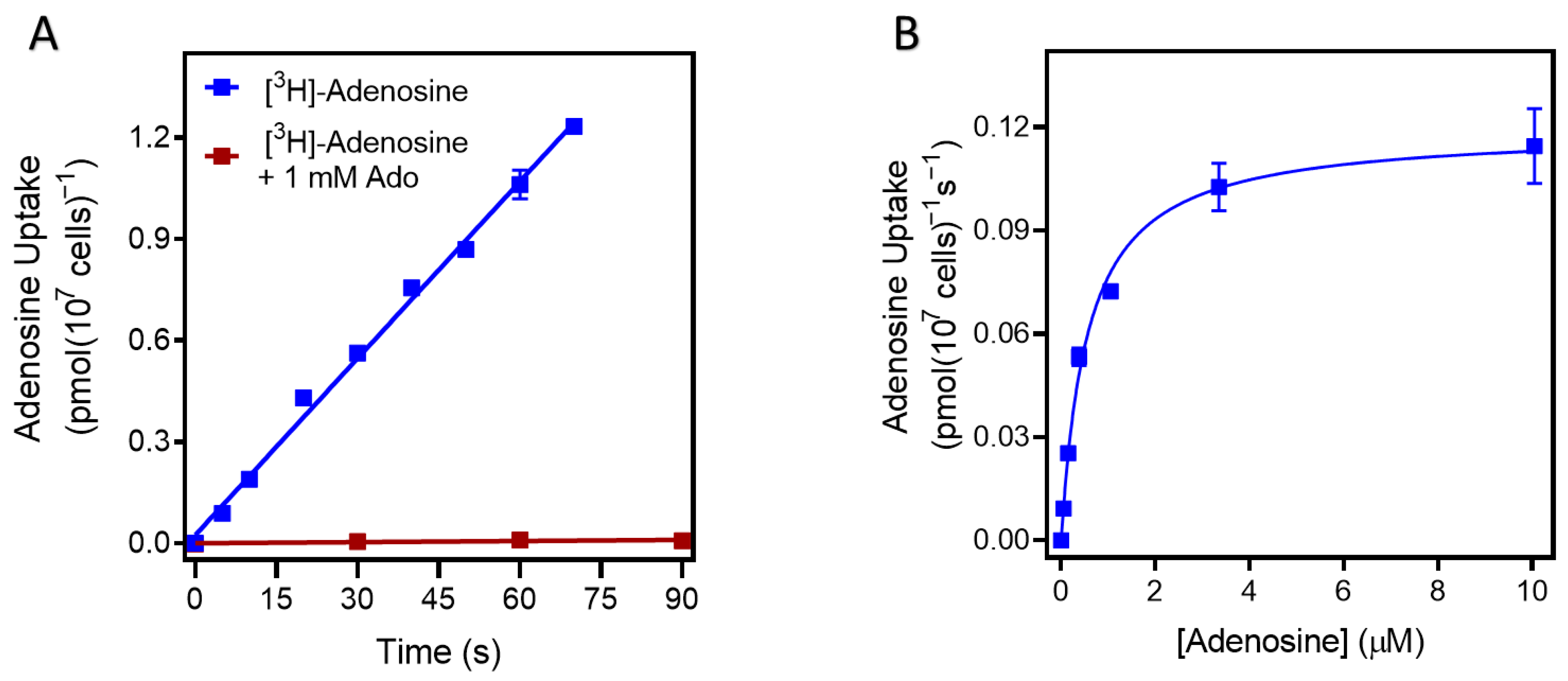
2.1. Nucleoside Transport Activity in T. congolense
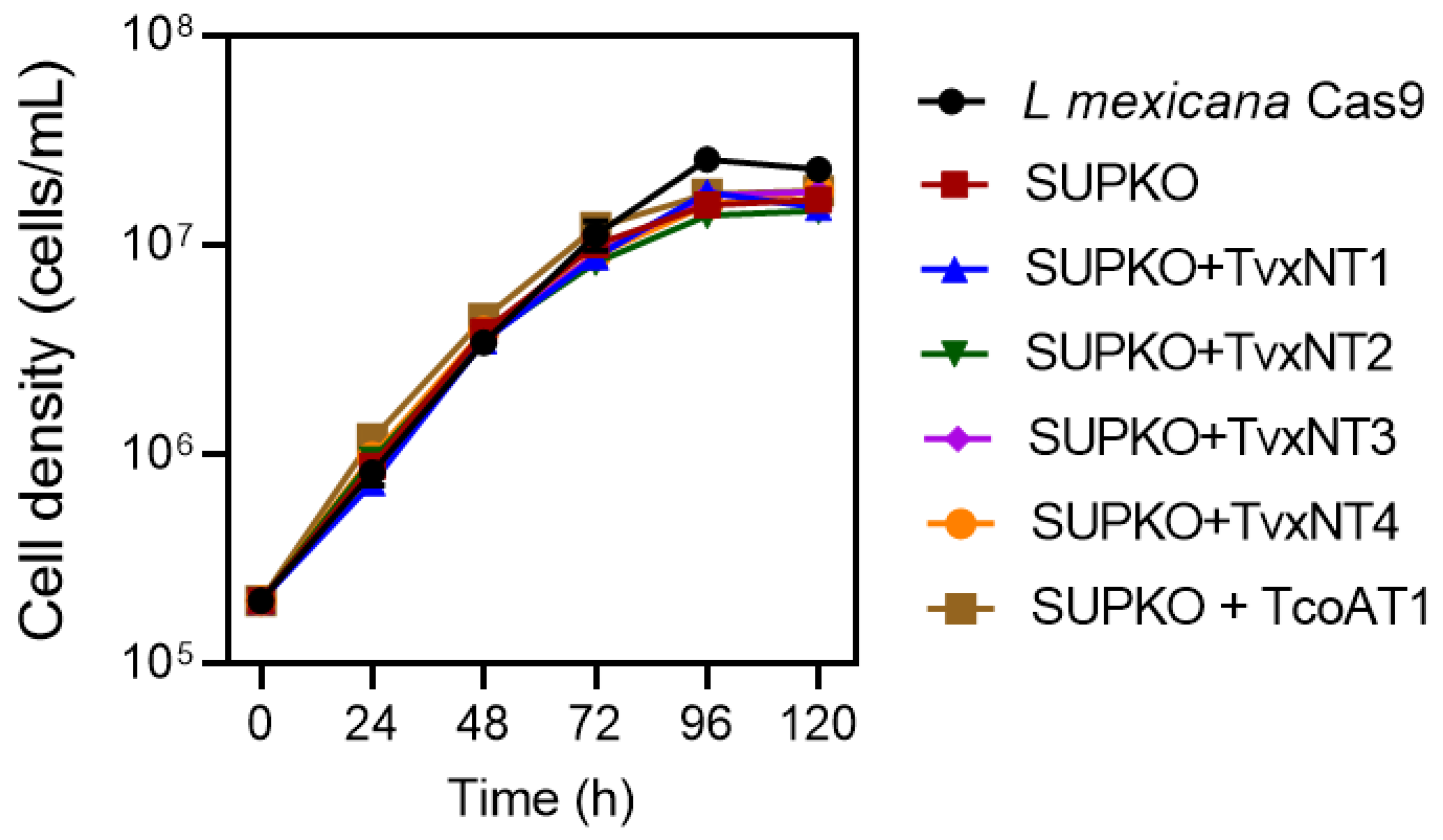
2.2. Expression of T. congolense and T. vivax Nucleoside Transporters in a L. mexicana NT-Null Cell Line
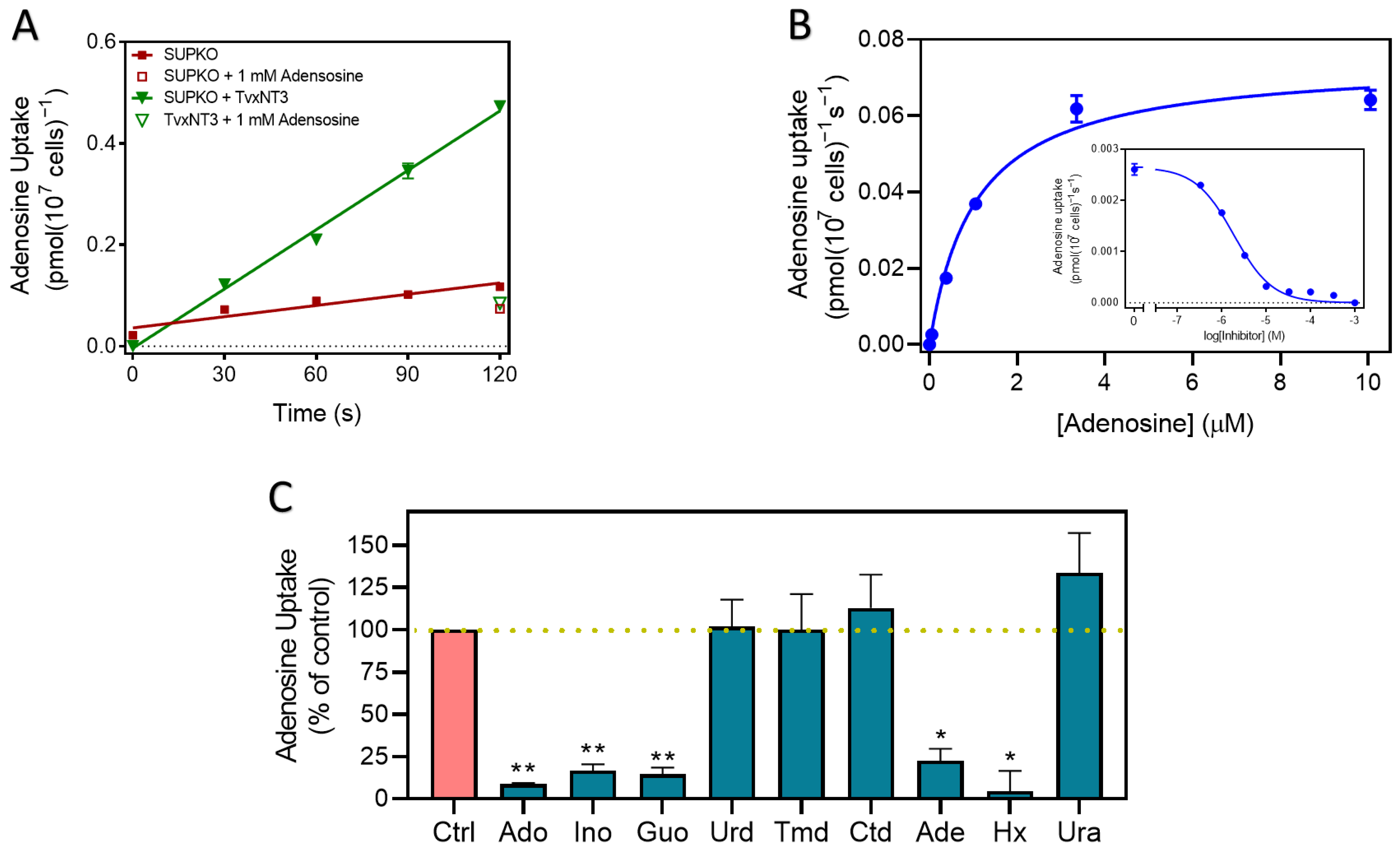
2.2.1. Nucleoside Transporters of T. vivax
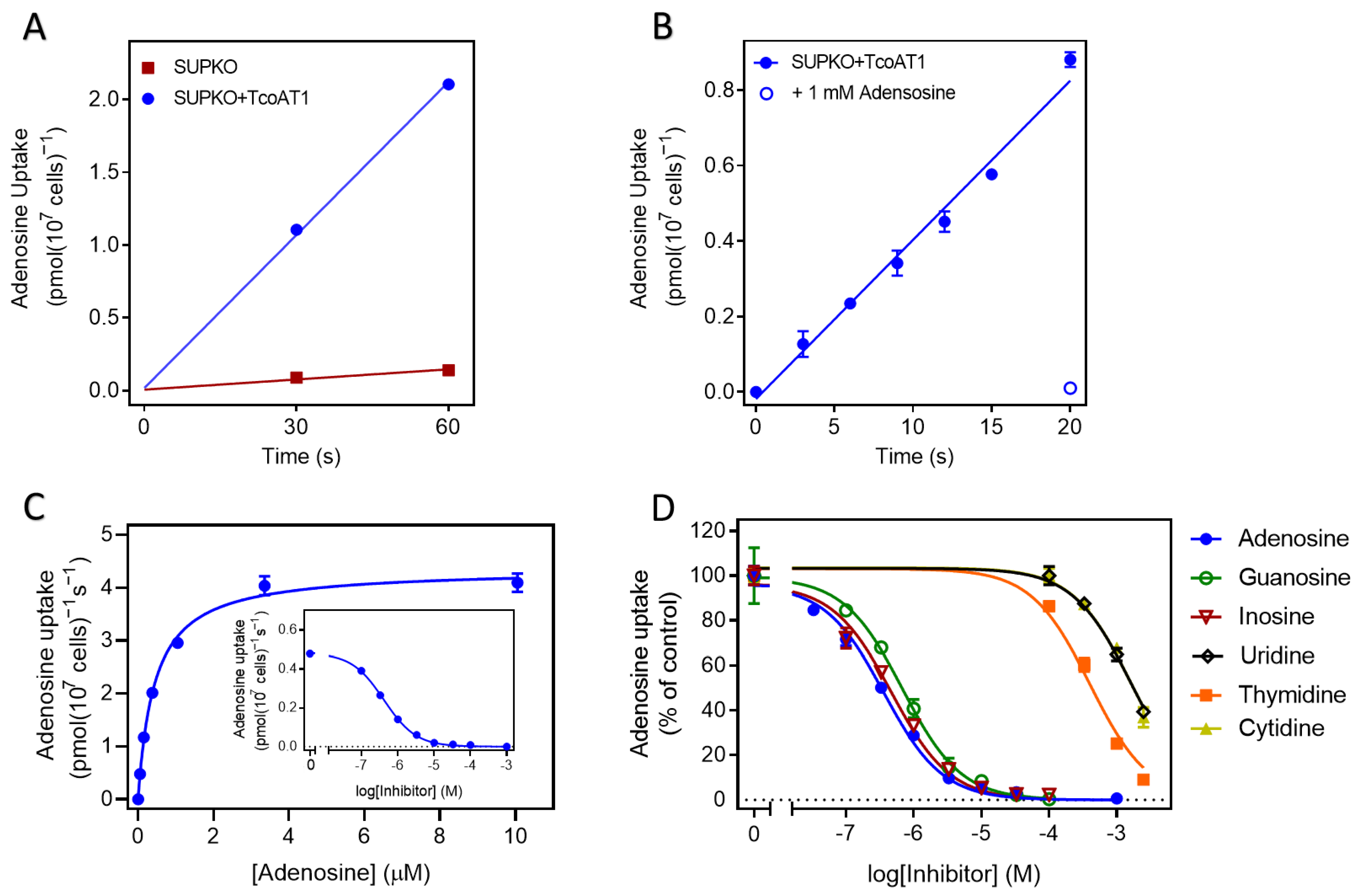
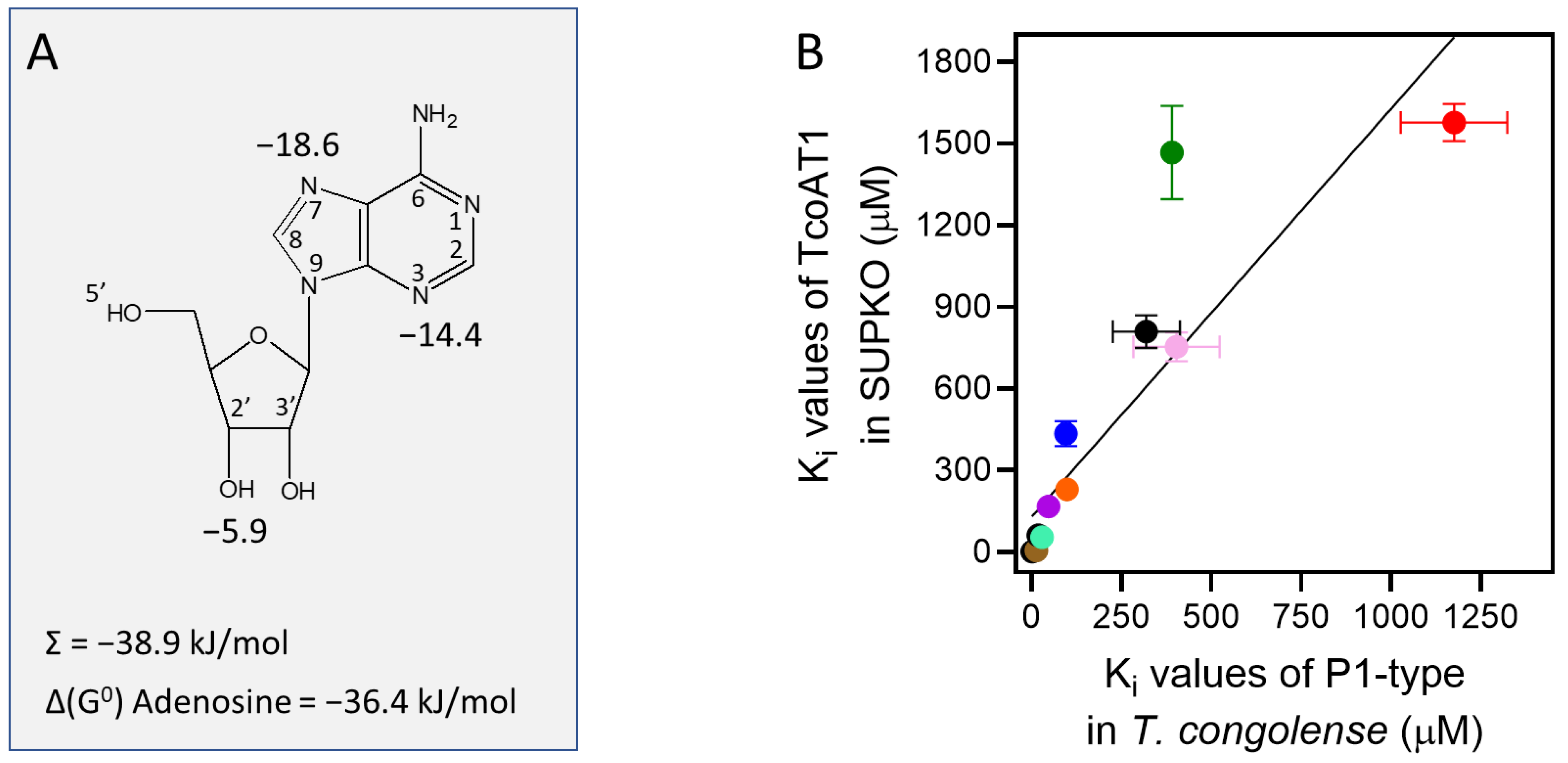
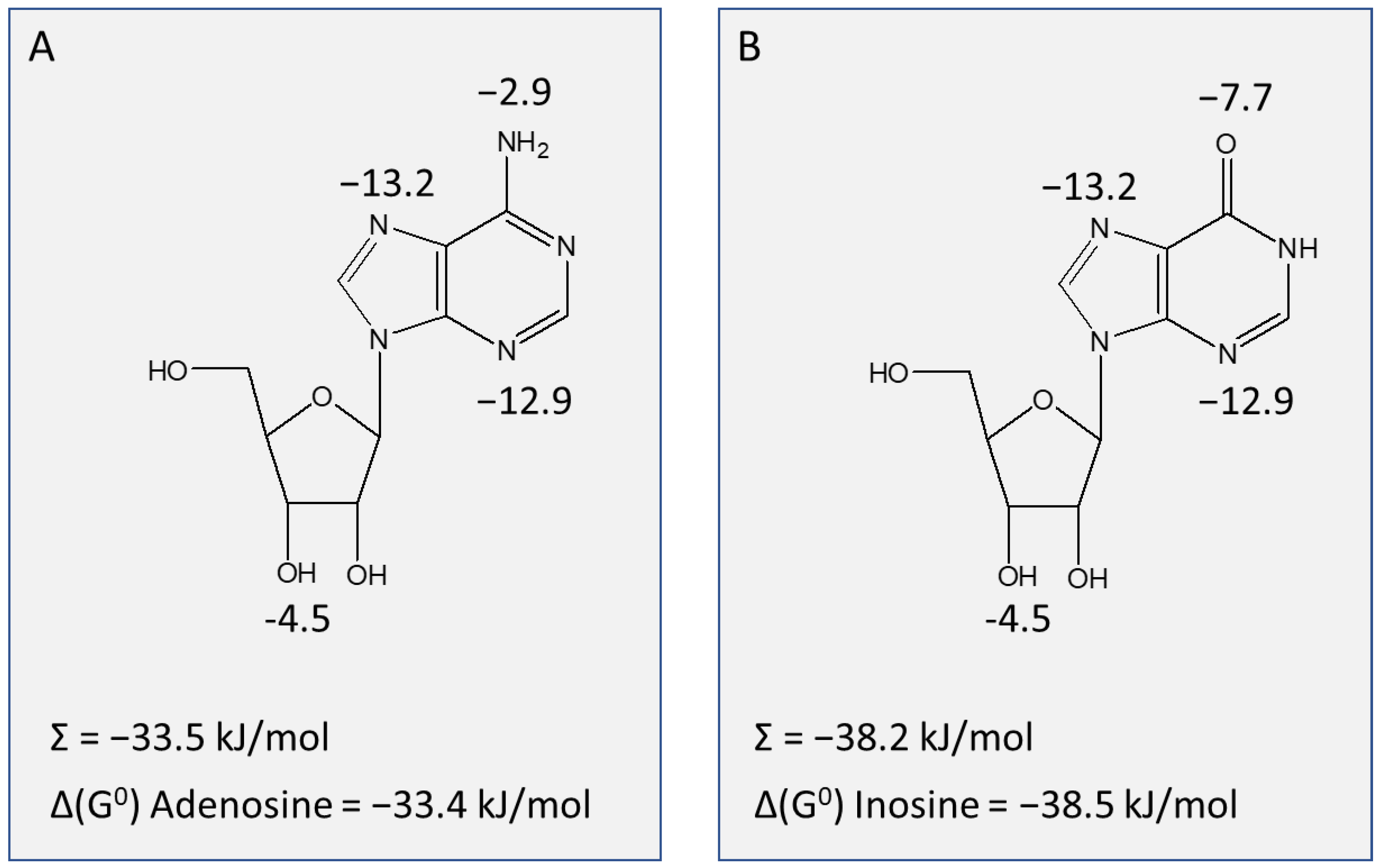
2.2.2. TcoAT1 Basic Characterization and Adenosine Binding Model
2.2.3. TvxNT3 Basic Characterization and Adenosine Binding Model
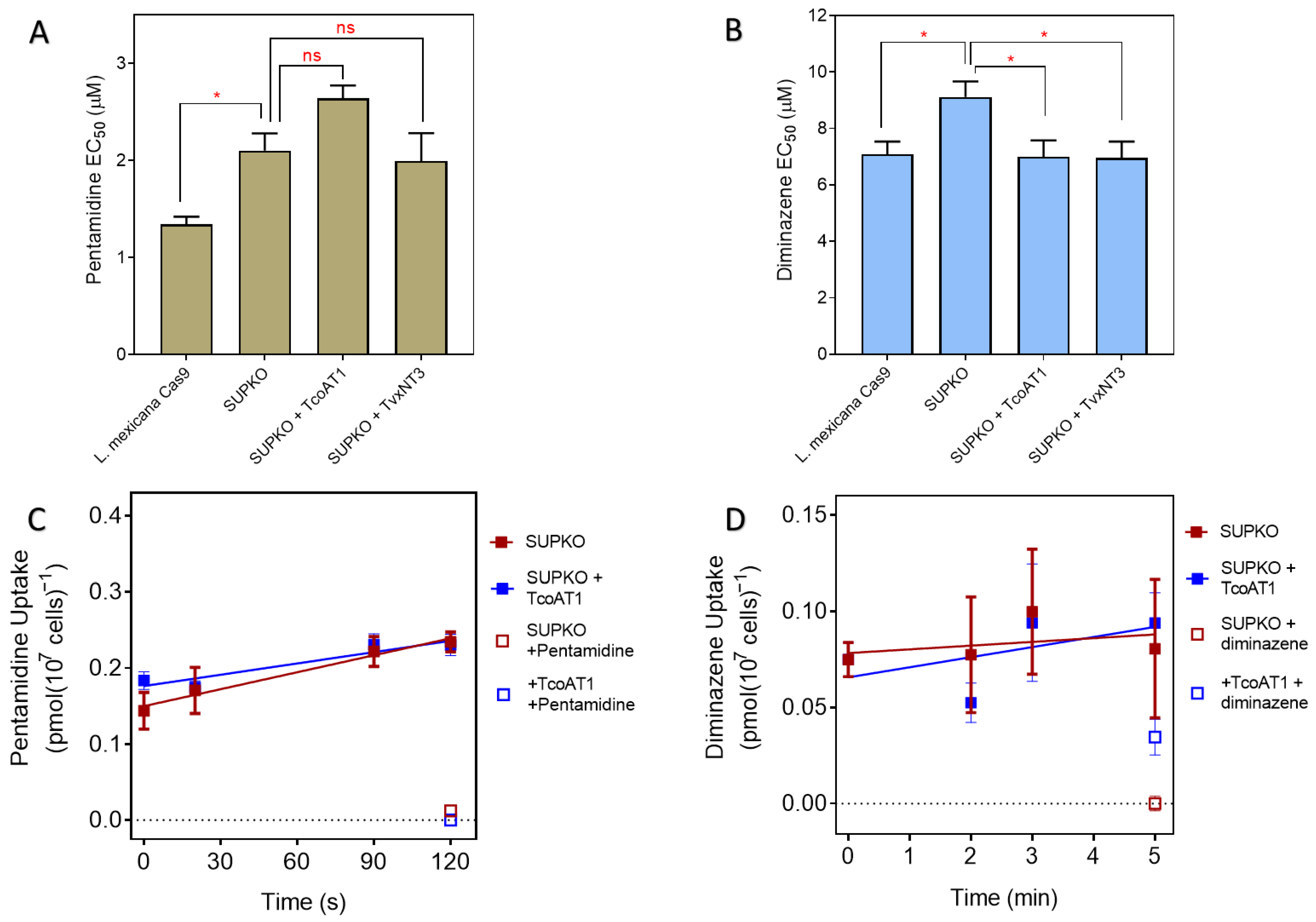
2.3. P1 Transporters Do Not Transport Any of the Trypanocides in Current Clinical Use
2.4. Trypanosomal P1 as Carrier of Novel Nucleoside Drugs
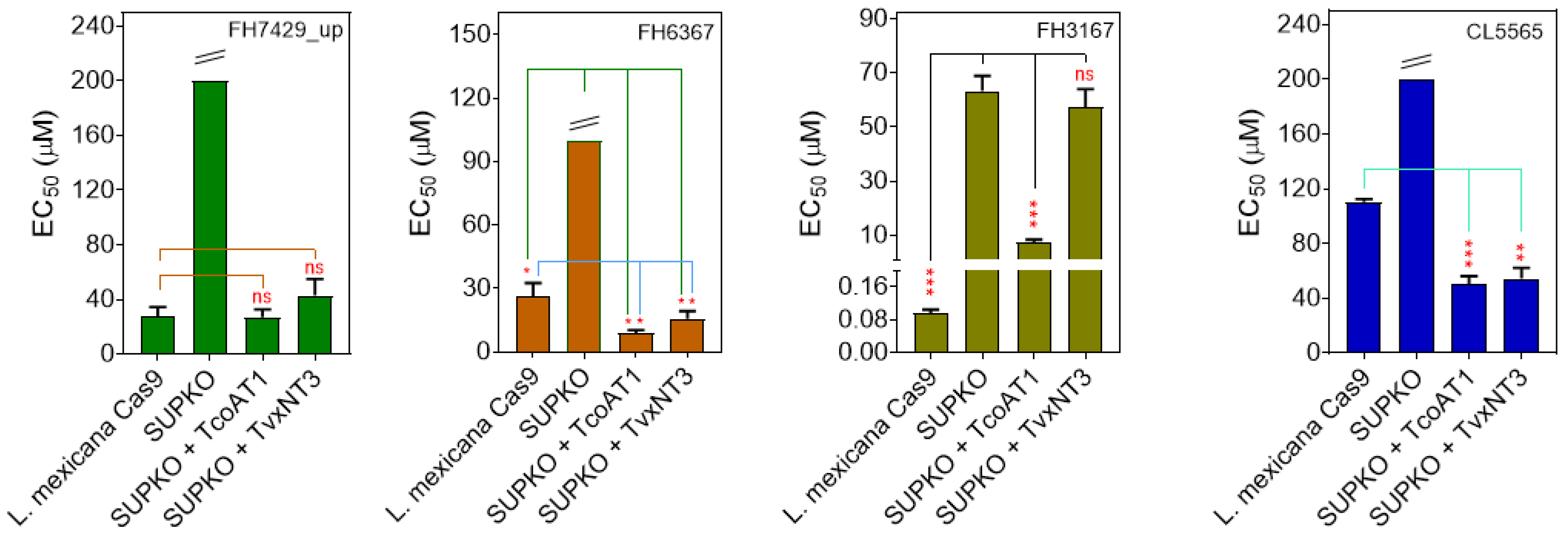
2.4.1. Affinity of Nucleoside Drugs to T. congolense Nucleoside Transporters
2.4.2. Affinity of Nucleoside Drugs to T. vivax Nucleoside Transporters
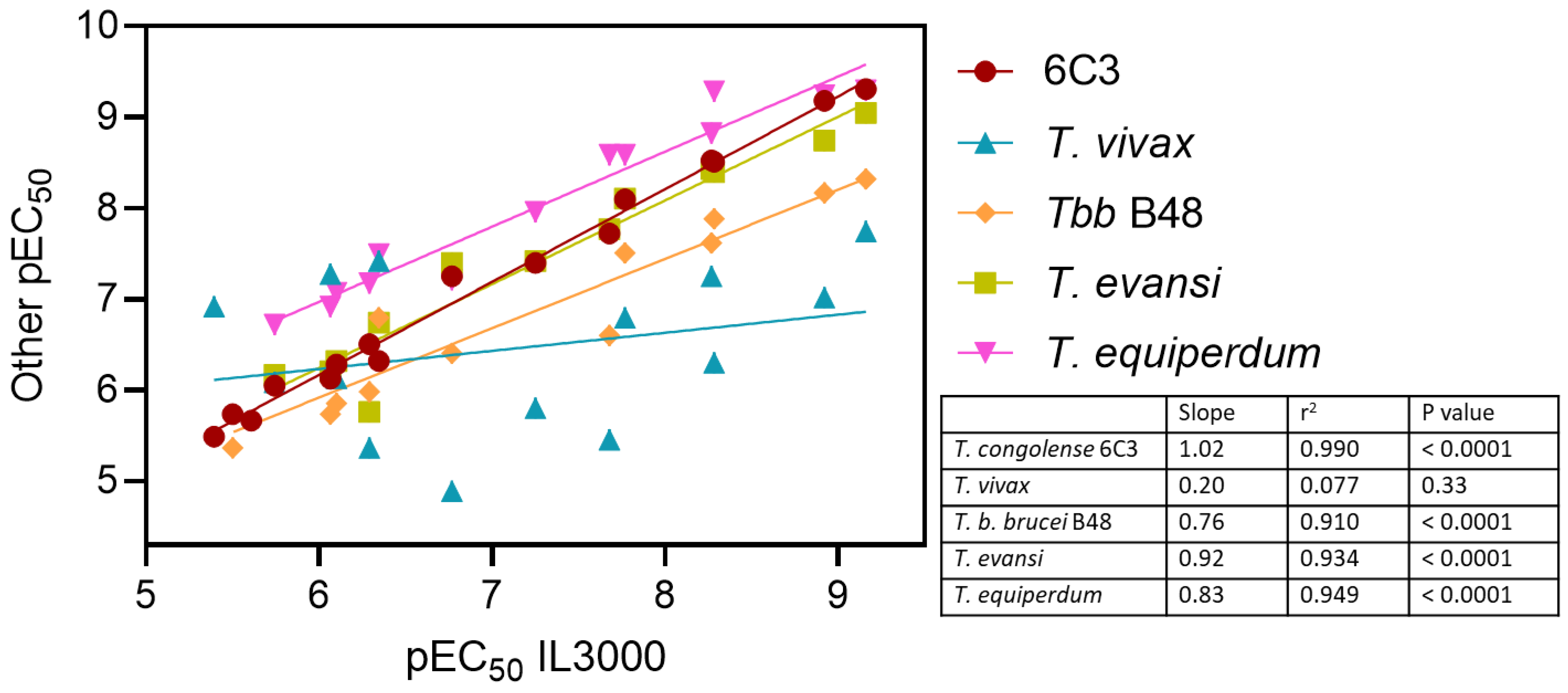
2.4.3. Antitrypanosomal Activity of Nucleoside Analogs In Vitro
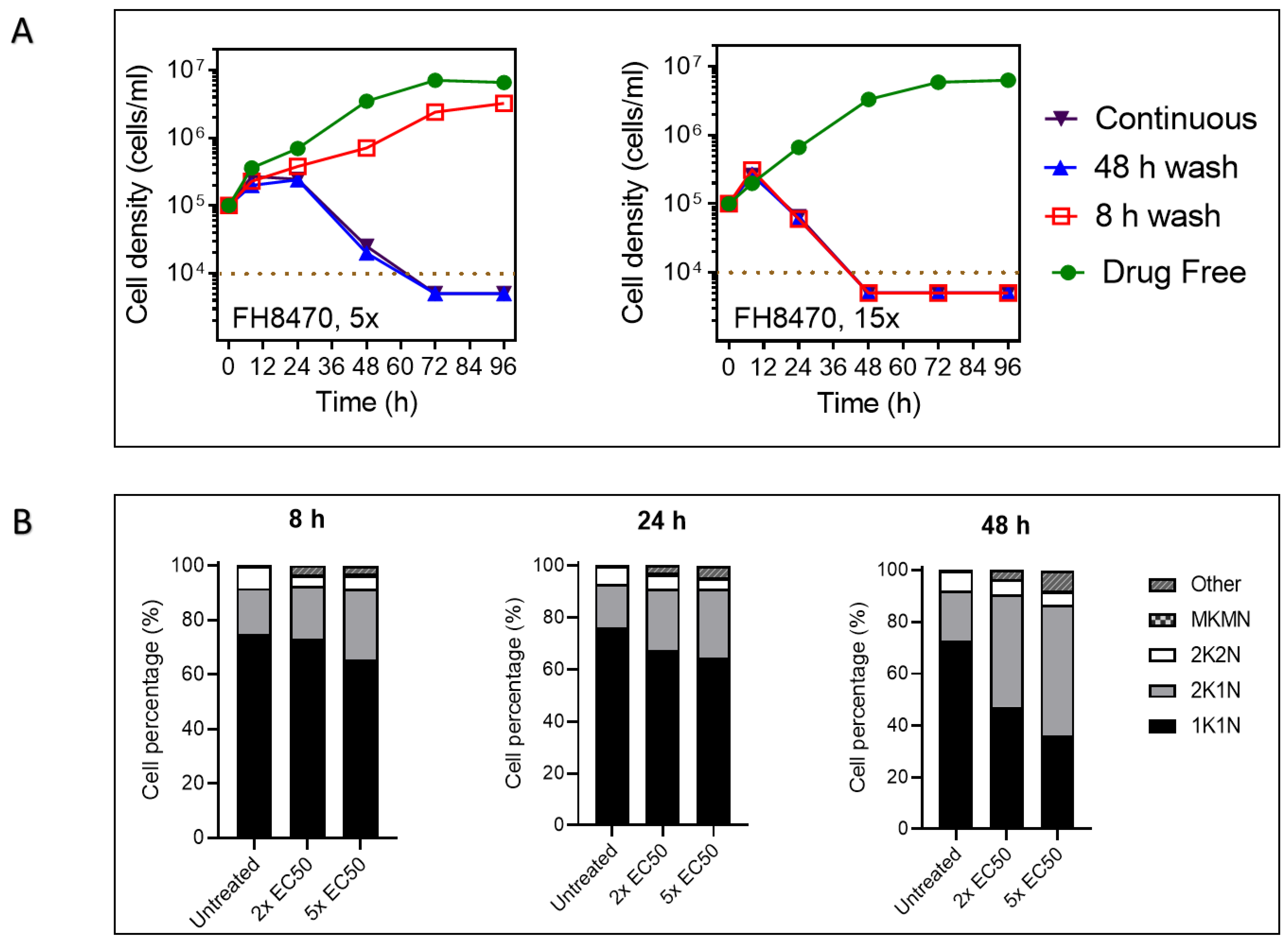
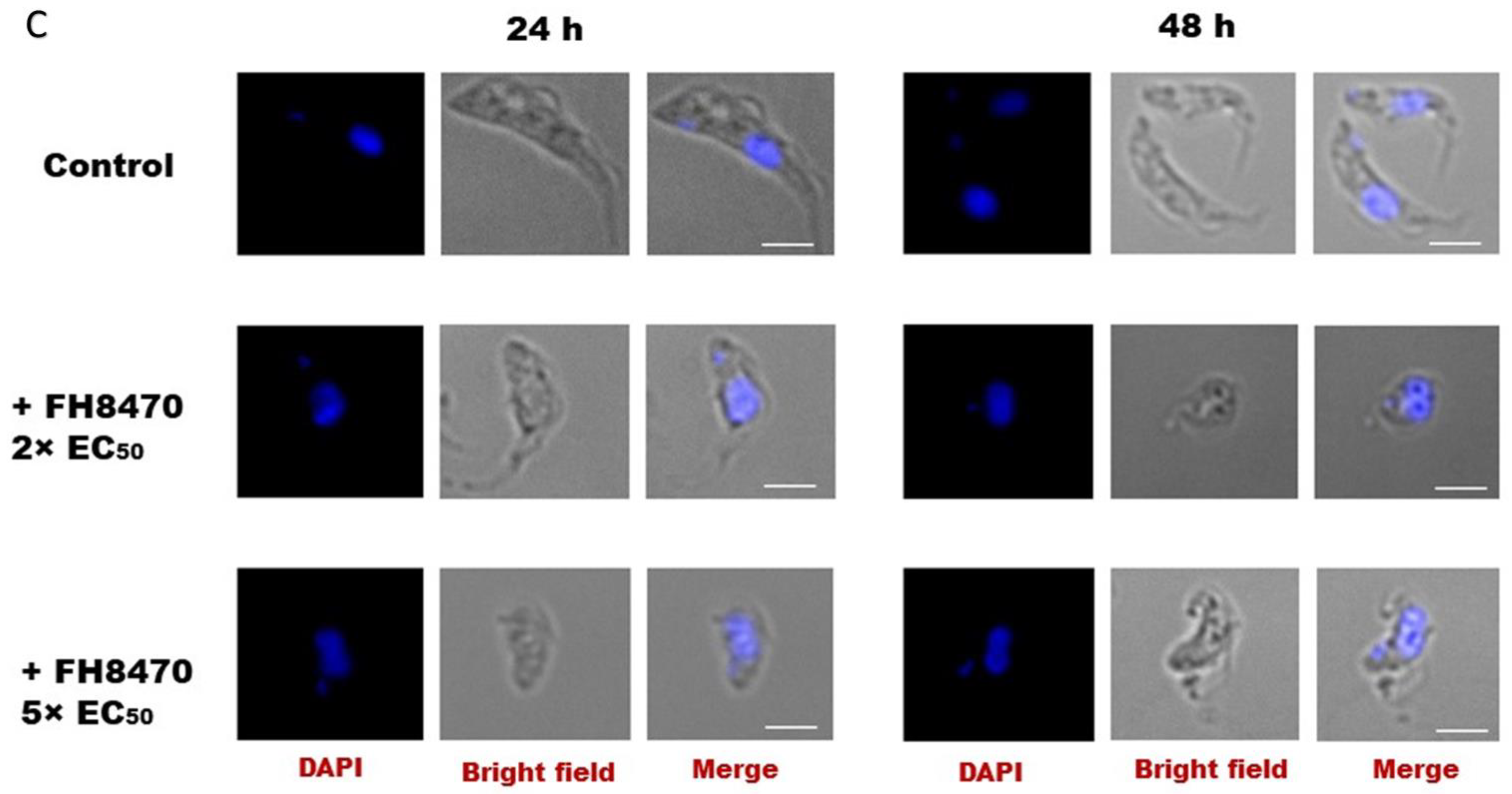
2.4.4. The Effect of Tubercidin Analogs on Growth, Cell Cycle and Morphology of T. congolense
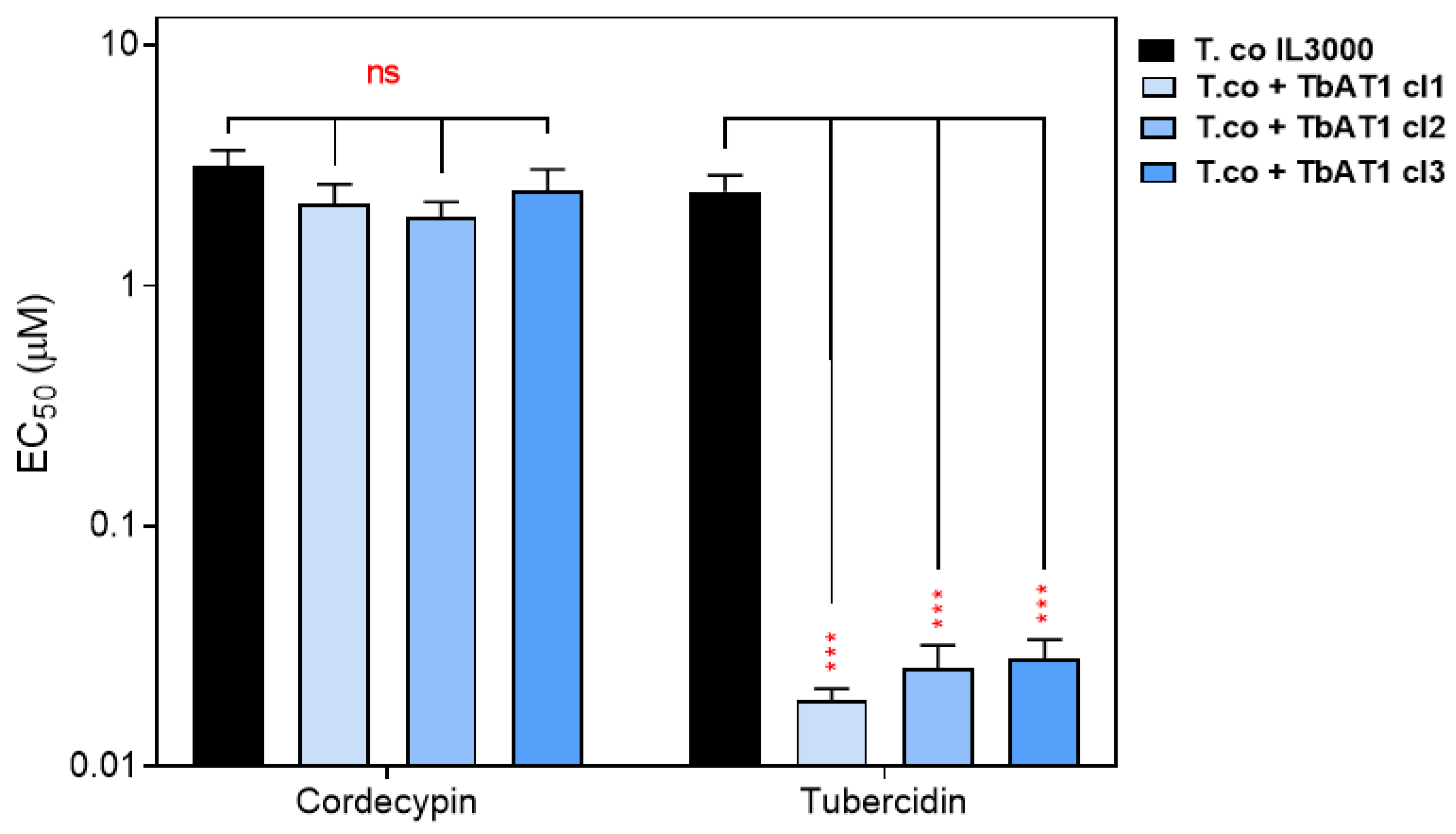
2.5. Toxicity of Tubercidin Analogs on Mammalian Cells
3. Discussion
4. Materials and Methods
4.1. Cell lines and Culture
4.2. Genetic Manipulation of L. mexicana Promastigotes
4.3. Drug Toxicity Assays
4.4. Radiolabel Drug and Nucleoside Uptake Assays
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Giordani, F.; Morrison, L.J.; Rowan, T.G.; De Koning, H.P.; Barrett, M.P. The animal trypanosomiases and their chemotherapy: A review. Parasitology 2016, 143, 1862–1889. [Google Scholar] [CrossRef]
- Franco, J.R.; Cecchi, G.; Priotto, G.; Paone, M.; Diarra, A.; Grout, L.; Simarro, P.P.; Zhao, W.; Argaw, D. Monitoring the elimination of human African trypanosomiasis at continental and country level: Update to 2018. PLoS Negl. Trop. Dis. 2020, 14, e0008261. [Google Scholar] [CrossRef]
- Autheman, D.; Crosnier, C.; Clare, S.; Goulding, D.A.; Brandt, C.; Harcourt, K.; Tolley, C.; Galaway, F.; Khushu, M.; Ong, H.; et al. An invariant Trypanosoma vivax vaccine antigen induces protective immunity. Nature 2021, 595, 96–100. [Google Scholar] [CrossRef] [PubMed]
- La Greca, F.; Magez, S. Vaccination against trypanosomiasis: Can it be done or is the trypanosome truly the ultimate immune destroyer and escape artist? Hum. Vaccin. 2011, 7, 1225–1233. [Google Scholar] [CrossRef] [PubMed]
- Magez, S.; Li, Z.; Nguyen, H.T.T.; Pinto Torres, J.E.; Van Wielendaele, P.; Radwanska, M.; Began, J.; Zoll, S.; Sterckx, Y.G. The history of anti-trypanosome vaccine development shows that highly immunogenic and exposed pathogen-derived antigens are not necessarily good target candidates: Enolase and ISG75 as examples. Pathogens 2021, 10, 1050. [Google Scholar] [CrossRef] [PubMed]
- Delespaux, V.; De Koning, H.P. Drugs and drug resistance in African trypanosomiasis. Drug Resist. Updat. 2007, 10, 30–50. [Google Scholar] [CrossRef]
- De Koning, H.P. The drugs of sleeping sickness: Their mechanisms of action and resistance, and a brief history. Trop. Med. Infect. Dis. 2020, 5, 14. [Google Scholar] [CrossRef] [PubMed]
- Desquesnes, M.; Dargantes, A.; Lai, D.H.; Lun, Z.R.; Holzmuller, P.; Jittapalapong, S. Trypanosoma evansi and surra: A review and perspectives on transmission, epidemiology and control, impact, and zoonotic aspects. Biomed. Res. Int. 2013, 2013, 321237. [Google Scholar] [CrossRef]
- Osório, A.L.; Madruga, C.R.; Desquesnes, M.; Soares, C.O.; Ribeiro, L.R.; Costa, S.C. Trypanosoma (Duttonella) vivax: Its biology, epidemiology, pathogenesis, and introduction in the New World–a review. Mem. Inst. Oswaldo Cruz 2008, 103, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Gizaw, Y.; Megersa, M.; Fayera, T. Dourine: A neglected disease of equids. Trop. Anim. Health Prod. 2017, 49, 887–897. [Google Scholar] [CrossRef] [PubMed]
- Anene, B.M.; Onah, D.N.; Nawa, Y. Drug resistance in pathogenic African trypanosomiasis: What hopes for the future? Vet. Parasitol. 2001, 96, 83–100. [Google Scholar] [CrossRef] [PubMed]
- Wilkinson, S.R.; Kelley, J.M. Trypanocidal drugs: Mechanisms, resistance and new targets. Expert Rev. Mol. Med. 2009, 11, e31. [Google Scholar] [CrossRef] [PubMed]
- Morrison, L.J.; Vezza, L.; Rowan, T.; Hope, J.C. Animal African trypanosomiasis: Time to increase focus on clinically relevant parasite and host species. Trends Parasitol. 2016, 32, 599–607. [Google Scholar] [CrossRef]
- Zweygarth, E.; Kaminsky, R. Evaluation of an arsenical compound (RM 110, mel Cy, Cymelarsan) against susceptible and drug-resistant Trypanosoma brucei brucei and T. b. evansi. Trop. Med. Parasitol. 1990, 41, 208–212. [Google Scholar] [PubMed]
- Kinabo, L.D.B. Pharmacology of existing drugs for animal trypanosomiasis. Acta Trop. 1993, 54, 169–183. [Google Scholar] [CrossRef]
- Geerts, S.; Holmes, P.H.; Eisler, M.C.; Diall, O. African bovine trypanosomiasis: The problem of drug resistance. Trends Parasitol. 2001, 17, 25–28. [Google Scholar] [CrossRef] [PubMed]
- Munday, J.C.; Settimo, L.; De Koning, H.P. Transport proteins determine drug sensitivity and resistance in a protozoan parasite, Trypanosoma brucei. Front. Pharmacol. 2015, 6, 32. [Google Scholar] [CrossRef]
- Ungogo, M.A.; Campagnaro, G.D.; Alghamdi, A.H.; Natto, M.J.; De Koning, H.P. Differences in transporters rather than drug targets are the principal determinants of the different innate sensitivities of Trypanosoma congolense and Trypanozoon subgenus trypanosomes to diamidines and melaminophenyl arsenicals. Int. J. Mol. Sci. 2022, 23, 2844. [Google Scholar] [CrossRef] [PubMed]
- Vincent, I.M.; Creek, D.; Watson, D.G.; Kamleh, M.A.; Woods, D.J.; Wong, P.E.; Burchmore, R.J.; Barrett, M.P. A molecular mechanism for eflornithine resistance in African trypanosomes. PLoS Pathog. 2010, 6, e1001204. [Google Scholar] [CrossRef] [PubMed]
- Stewart, M.L.; Burchmore, R.J.S.; Clucas, C.; Hertz-Fowler, C.; Brook, K.; Tait, A.; McLeod, A.; Turner, C.M.R.; De Koning, H.P.; Wong, P.E.; et al. Multiple genetic mechanisms lead to the loss of functional TbAT1 expression in drug resistant trypanosomes. Eukaryot. Cell 2010, 9, 336–343. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baker, N.; De Koning, H.P.; Mäser, P.; Horn, D. Drug resistance in African trypanosomiasis: The melarsoprol and pentamidine story. Trends Parasitol. 2013, 29, 110–118. [Google Scholar] [CrossRef] [PubMed]
- Alghamdi, A.H.; Munday, J.C.; Campagnaro, G.D.; Gurvic, D.; Svensson, F.; Okpara, C.E.; Kumar, A.; Quintana, J.; Martin Abril, M.E.; Milić, P.; et al. Positively selected modifications in the pore of TbAQP2 allow pentamidine to enter Trypanosoma brucei. eLife 2020, 9, e56416. [Google Scholar] [CrossRef] [PubMed]
- Kell, D.B. The transporter-mediated cellular uptake and efflux of pharmaceutical drugs and biotechnology products: How and why phospholipid bilayer transport Is negligible in real biomembranes. Molecules 2021, 26, 5629. [Google Scholar] [CrossRef]
- Hassan, H.F.; Coombs, G.H. Purine and pyrimidine metabolism in parasitic protozoa. FEMS Microbiol. Rev. 1988, 4, 47–83. [Google Scholar]
- De Koning, H.P.; Bridges, D.J.; Burchmore, R. Purine and pyrimidine transport in protozoa: From biology to therapy. FEMS Microbiol. Rev. 2005, 29, 987–1020. [Google Scholar] [CrossRef] [PubMed]
- Campagnaro, G.D.; De Koning, H.P. Purine and pyrimidine transporters of pathogenic protozoa-conduits for therapeutic agents. Med. Res. Rev. 2020, 40, 1679–1714. [Google Scholar] [CrossRef] [PubMed]
- Campagnaro, G.D.; de Freitas Nascimento, J.; Girard, R.B.M.; Silber, A.M.; De Koning, H.P. Cloning and characterisation of the Equilibrative Nucleoside Transporter family of Trypanosoma cruzi: Ultra-high affinity and selectivity to survive in the intracellular niche. Biochim. Biophys. Acta Gen. Subj. 2018, 1862, 2750–2763. [Google Scholar] [CrossRef] [PubMed]
- Campagnaro, G.D.; Elati, H.A.A.; Balaska, S.; Martin Abril, M.E.; Natto, M.J.; Hulpia, F.; Lee, K.; Sheiner, L.; Van Calenbergh, S.; De Koning, H.P. A Toxoplasma gondii oxopurine transporter binds nucleobases and nucleosides using different binding modes. Int. J. Mol. Sci. 2022, 23, 710. [Google Scholar] [CrossRef]
- Aldfer, M.M.; AlSiari, T.A.; Elati, H.A.A.; Natto, M.J.; Alfayez, I.A.; Campagnaro, G.D.; Sani, B.; Burchmore, R.J.S.; Diallinas, G.; De Koning, H.P. Nucleoside transport and nucleobase uptake null mutants in Leishmania mexicana for the routine expression and characterisation of purine and pyrimidine transporters. Int. J. Mol. Sci. 2022, 23, 8139. [Google Scholar] [CrossRef]
- Aldfer, M.M.; Alfayez, I.A.; Campagnaro, G.D.; Gayen, N.; Elmahallawy, E.K.; Murillo, A.M.; Marsiccobetre, S.; Van Calenbergh, S.; Silber, A.M.; De Koning, H.P. TcrNT2 is a conduit for the uptake of 5-F-2’deoxyuridine and tubercidin analogues in Trypanosoma cruzi. Molecules 2022, 27, 8045. [Google Scholar] [CrossRef]
- Campagnaro, G.D.; Alzahrani, K.J.H.; Munday, J.C.; De Koning, H.P. Trypanosoma brucei bloodstream forms express highly specific and separate transporters for adenine and hypoxanthine; evidence for a new protozoan purine transporter family? Mol. Biochem. Parasitol. 2018, 220, 46–56. [Google Scholar] [CrossRef] [PubMed]
- De Koning, H.P. Pyrimidine transporters of protozoa–A class apart? Trends Parasitol. 2007, 23, 190. [Google Scholar] [CrossRef]
- De Koning, H.P.; Jarvis, S.M. Hypoxanthine uptake through a purine-selective nucleobase transporter in Trypanosoma brucei brucei procyclics is driven by protonmotive force. Eur. J. Biochem. 1997, 247, 1102–1110. [Google Scholar] [CrossRef] [PubMed]
- De Koning, H.P.; Watson, C.J.; Jarvis, S.M. Characterisation of a nucleoside/proton symporter in procyclic Trypanosoma brucei brucei. J. Biol. Chem. 1998, 273, 9486–9494. [Google Scholar] [CrossRef] [PubMed]
- Stein, A.; Vaseduvan, G.; Carter, N.S.; Ullman, B.; Landfear, S.M.; Kavanaugh, M.P. Equilibrative nucleoside transporter family members from Leishmania donovani are electrogenic proton symporters. J. Biol. Chem. 2003, 278, 35127–35134. [Google Scholar] [CrossRef] [PubMed]
- Young, J.D.; Yao, S.Y.; Baldwin, J.M.; Cass, C.E.; Baldwin, S.A. The human concentrative and equilibrative nucleoside transporter families, SLC28 and SLC29. Mol. Aspects Med. 2013, 34, 529–547. [Google Scholar] [CrossRef] [PubMed]
- Boswell-Casteel, R.C.; Hays, F.A. Equilibrative nucleoside transporters–a review. Nucleosides Nucleotides Nucleic Acids 2017, 36, 7–30. [Google Scholar] [CrossRef] [PubMed]
- Hulpia, F.; Mabille, D.; Campagnaro, G.D.; Schumann, G.; Maes, L.; Roditi, I.; Hofer, A.; De Koning, H.P.; Galjon, G.; Van Calenbergh, S. Combining tubercidin and cordycepin scaffolds results in highly active candidates to treat late-stage sleeping sickness. Nat. Commun. 2019, 10, 5564. [Google Scholar] [CrossRef] [PubMed]
- Sanchez, M.A.; Tryon, R.; Green, J.; Boor, I.; Landfear, S.M. Six related nucleoside/nucleobase transporters from Trypanosoma brucei exhibit distinct biochemical functions. J. Biol. Chem. 2002, 277, 21499–21504. [Google Scholar] [CrossRef]
- Al-Salabi, M.I.; Wallace, L.J.M.; Lüscher, A.; Mäser, P.; Candlish, D.; Rodenko, B.; Gould, M.K.; Jabeen, I.; Ajith, S.N.; De Koning, H.P. Molecular interactions underlying the unusually high adenosine affinity of a novel Trypanosoma brucei nucleoside transporter. Mol. Pharmacol. 2007, 71, 921–929. [Google Scholar] [CrossRef] [PubMed]
- Carter, N.S.; Fairlamb, A.H. Arsenical-resistant trypanosomes lack an unusual adenosine transporter. Nature 1993, 361, 173–176. [Google Scholar] [CrossRef] [PubMed]
- Mäser, P.; Sütterlin, C.; Kralli, A.; Kaminsky, R. A nucleoside transporter from Trypanosoma brucei involved in drug resistance. Science 1999, 285, 242–244. [Google Scholar] [CrossRef]
- Carter, N.S.; Barrett, M.P.; De Koning, H.P. A drug resistance determinant from Trypanosoma brucei. Trends Microbiol. 1999, 7, 469–471. [Google Scholar] [CrossRef]
- De Koning, H.P.; Jarvis, S.M. Adenosine transporters in bloodstream forms of T. b. brucei: Substrate recognition motifs and affinity for trypanocidal drugs. Mol. Pharmacol. 1999, 56, 1162–1170. [Google Scholar] [CrossRef]
- Munday, J.C.; Tagoe, D.N.A.; Eze, A.A.; Krezdorn, J.A.; Rojas López, K.E.; Alkhaldi, A.A.M.; McDonald, F.; Still, J.; Alzahrani, K.J.; Settimo, L.; et al. Functional analysis of drug resistance-associated mutations in the Trypanosoma brucei adenosine transporter 1 (TbAT1) and the proposal of a structural model for the protein. Mol. Microbiol. 2015, 96, 887–900. [Google Scholar] [CrossRef] [PubMed]
- Geiser, F.; Lüscher, A.; De Koning, H.P.; Seebeck, T.; Mäser, P. Molecular pharmacology of adenosine transport in Trypanosoma brucei: P1/P2 revisited. Mol. Pharmacol. 2005, 68, 589–595. [Google Scholar] [CrossRef]
- Vodnala, S.K.; Lundbäck, T.; Yeheskieli, E.; Sjöberg, B.; Gustavsson, A.L.; Svensson, R.; Olivera, G.C.; Eze, A.A.; De Koning, H.P.; Hammarström, L.G.J.; et al. Structure-activity relationships of synthetic cordycepin analogues as experimental therapeutics for African trypanosomiasis. J. Med. Chem. 2013, 56, 9861–9873. [Google Scholar] [CrossRef] [PubMed]
- Hulpia, F.; Campagnaro, G.D.; Scortichini, M.; Van Hecke, K.; Maes, L.; De Koning, H.P.; Caljon, G.; Van Calenbergh, S. Revisiting tubercidin against kinetoplastid parasites: Aromatic substitutions at position 7 improve activity and reduce toxicity. Eur. J. Med. Chem. 2019, 164, 689–705. [Google Scholar] [CrossRef] [PubMed]
- Carter, N.S.; Berger, B.J.; Fairlamb, A.H. Uptake of diamidine drugs by the P2 nucleoside transporter in melarsen-sensitive and -resistant Trypanosoma brucei brucei. J. Biol. Chem. 1995, 270, 28153–28157. [Google Scholar] [CrossRef]
- De Koning, H.P.; MacLeod, A.; Barrett, M.P.; Cover, B.; Jarvis, S.M. Further evidence for a link between melarsoprol resistance and P2 transporter function in African trypanosomes. Mol. Biochem. Parasitol. 2000, 106, 181–185. [Google Scholar] [CrossRef] [PubMed]
- Witola, W.H.; Inoue, N.; Ohashi, K.; Onuma, M. RNA-interference silencing of the adenosine transporter-1 gene in Trypanosoma evansi confers resistance to diminazene aceturate. Exp. Parasitol. 2004, 107, 47–57. [Google Scholar] [CrossRef]
- Munday, J.C.; Rojas López, K.E.; Eze, A.A.; Delespaux, V.; Van Den Abbeele, J.; Rowan, T.; Barrett, M.P.; Morrison, L.J.; De Koning, H.P. Functional expression of TcoAT1 reveals it to be a P1-type nucleoside transporter with no capacity for diminazene uptake. Int J. Parasitol. Drugs Drug Resist. 2013, 3, 69–76. [Google Scholar] [CrossRef]
- Jackson, A.P.; Allison, H.C.; Barry, J.D.; Field, M.C.; Hertz-Fowler, C.; Berriman, M. A cell-surface phylome for African trypanosomes. PLoS Negl. Trop. Dis. 2013, 7, e2121. [Google Scholar] [CrossRef]
- Delespaux, V.; Chitanga, S.; Geysen, D.; Goethals, A.; Van den Bossche, P.; Geerts, S. SSCP analysis of the P2 purine transporter TcoAT1 gene of Trypanosoma congolense leads to a simple PCR-RFLP test allowing the rapid identification of diminazene resistant stocks. Acta Trop. 2006, 100, 96–102. [Google Scholar] [CrossRef] [PubMed]
- Vitouley, H.S.; Mungube, E.O.; Allegye-Cudjoe, E.; Diall, O.; Bocoum, Z.; Diarra, B.; Randolph, T.F.; Bauer, B.; Clausen, P.H.; Geysen, D.; et al. Improved PCR-RFLP for the detection of diminazene resistance in Trypanosoma congolense under field conditions using filter papers for sample storage. PLoS Negl. Trop. Dis. 2011, 5, e1223. [Google Scholar] [CrossRef] [PubMed]
- Mamoudou, A.; Delespaux, V.; Chepnda, V.; Hachimou, Z.; Andrikaye, J.P.; Zoli, A.; Geerts, S. Assessment of the occurrence of trypanocidal drug resistance in trypanosomes of naturally infected cattle in the Adamaoua region of Cameroon using the standard mouse test and molecular tools. Acta Trop. 2008, 106, 115–118. [Google Scholar] [CrossRef]
- Mewamba, E.M.; Farikou, O.; Kamga, R.M.N.; Magang, M.E.K.; Tume, C.; Tiofack, A.A.Z.; Ravel, S.; Simo, G. Molecular identification of diminazene aceturate-resistant strains of Trypanosoma congolense in naturally infected domestic animals of Yoko in the centre region of Cameroon. Vet. Parasitol. Reg. Stud. Rep. 2020, 20, 100405. [Google Scholar] [CrossRef]
- Simo, G.; Magang, E.M.K.; Mewamba, E.M.; Farikou, O.; Kamga, R.M.N.; Tume, C.; Solano, P.; Ravel, S. Molecular identification of diminazene aceturate resistant trypanosomes in tsetse flies from Yoko in the Centre region of Cameroon and its epidemiological implications. Parasite Epidemiol. Control 2020, 9, e00135. [Google Scholar] [CrossRef]
- Bridges, D.; Gould, M.K.; Nerima, B.; Mäser, P.; Burchmore, R.J.S.; De Koning, H.P. Loss of the High Affinity Pentamidine Transporter is responsible for high levels of cross-resistance between arsenical and diamidine drugs in African trypanosomes. Mol. Pharmacol. 2007, 71, 1098–1108. [Google Scholar] [CrossRef] [PubMed]
- Carruthers, L.V.; Munday, J.C.; Ebiloma, G.U.; Steketee, P.; Jayaraman, S.; Campagnaro, G.D.; Ungogo, M.A.; Donnachie, A.; Lemgruber, L.; Rowan, T.G.; et al. Diminazene resistance in Trypanosoma congolense is not caused by reduced transport capacity but associated with reduced mitochondrial membrane potential. Mol. Microbiol. 2021, 116, 564–588. [Google Scholar] [CrossRef] [PubMed]
- Alzahrani, K.J.H.; Ali, J.A.M.; Eze, A.A.; Looi, W.L.; Tagoe, D.N.A.; Creek, D.J.; Barrett, M.P.; De Koning, H.P. Functional and genetic evidence that nucleoside transport is highly conserved in Leishmania species: Implications for pyrimidine-based chemotherapy. Int. J. Parasitol. Drugs Drug Resist. 2017, 7, 206–226. [Google Scholar] [CrossRef]
- Vasudevan, G.; Carter, N.S.; Drew, M.E.; Beverley, S.M.; Sanchez, M.A.; Seyfang, A.; Ullman, B.; Landfear, S.M. Cloning of Leishmania nucleoside transporter genes by rescue of a transport-deficient mutant. Proc. Natl. Acad. Sci. USA 1998, 95, 9873–9878. [Google Scholar] [CrossRef]
- Carter, N.S.; Drew, M.E.; Sanchez, M.; Vasudevan, G.; Landfear, S.M.; Ullman, B. Cloning of a novel inosine-guanosine transporter gene from Leishmania donovani by functional rescue of a transport-deficient mutant. J. Biol. Chem. 2000, 275, 20935–20941. [Google Scholar] [CrossRef]
- Beneke, T.; Madden, R.; Makin, L.; Valli, J.; Sunter, J.; Gluenz, E. A CRISPR Cas9 high-throughput genome editing toolkit for kinetoplastids. R. Soc. Open Sci. 2017, 4, 170095. [Google Scholar] [CrossRef]
- Hulpia, F.; Campagnaro, G.D.; Alzahrani, K.J.; Alfayez, I.A.; Ungogo, M.A.; Mabille, D.; Maes, L.; De Koning, H.P.; Caljon, G.; Van Calenbergh, S. Structure-activity relationship exploration of 3′-deoxy-7-deazapurine nucleoside analogues as anti-Trypanosoma brucei agents. ACS Infect. Dis. 2020, 6, 2045–2056. [Google Scholar] [CrossRef]
- Hulpia, F.; Bouton, J.; Campagnaro, G.D.; Alfayez, I.A.; Mabille, D.; Maes, L.; De Koning, H.P.; Caljon, G.; Van Calenbergh, S. C6-O-alkylated 7-deazainosine nucleoside analogues: Discovery of potent and selective anti-sleeping sickness agents. Eur. J. Med. Chem. 2020, 188, 112018. [Google Scholar] [CrossRef]
- De Almeida Fiuza, L.F.; Batista, D.G.J.; Girão, R.D.; Hulpia, F.; Finamore, P.; Aldfer, M.M.; Elmahallawy, E.K.; De Koning, H.P.; Moreira, O.C.; Van Calenbergh, S.; et al. Phenotypic evaluation of nucleoside analogues against Trypanosoma cruzi infection: In vitro and in vivo approaches. Molecules 2022, 27, 8087. [Google Scholar] [CrossRef] [PubMed]
- Natto, M.J.; Hulpia, F.; Kalkman, E.R.; Baillie, S.; Alhejeli, A.; Miyamoto, Y.; Eckmann, L.; Van Calenbergh, S.; De Koning, H.P. Deazapurine nucleoside analogues for the treatment of Trichomonas vaginalis. ACS Infect. Dis. 2021, 7, 1752–1764. [Google Scholar] [CrossRef] [PubMed]
- Mabille, D.; Ilbeigi, K.; Hendrickx, S.; Ungogo, M.A.; Hulpia, F.; Lin, C.; Maes, L.; De Koning, H.P.; Van Calenbergh, S.; Caljon, G. Nucleoside analogues for the treatment of animal African trypanosomiasis. Int. J. Parasitol. Drugs Drug Resist. 2022, 19, 21–30. [Google Scholar] [CrossRef] [PubMed]
- Burchmore, R.; Wallace, L.J.M.; Candlish, D.; Al-Salabi, M.I.; Beal, P.; Barrett, M.P.; Baldwin, S.A.; De Koning, H.P. Cloning, heterologous expression, and in situ characterization of the first high affinity nucleobase transporter from a protozoan. J. Biol. Chem. 2003, 278, 23502–23507. [Google Scholar] [CrossRef]
- Henriques, C.; Sanchez, M.A.; Tryon, R.; Landfear, S.M. Molecular and functional characterization of the first nucleobase transporter gene from African trypanosomes. Mol. Biochem. Parasitol. 2003, 130, 101–110. [Google Scholar] [CrossRef]
- Tetaud, E.; Lecuix, I.; Sheldrake, T.; Baltz, T.; Fairlamb, A.H. A new expression vector for Crithidia fasciculata and Leishmania. Mol. Biochem. Parasitol. 2002, 120, 195–204. [Google Scholar] [CrossRef] [PubMed]
- Gudin, S.; Quashie, N.B.; Candlish, D.; Al-Salabi, M.I.; Jarvis, S.M.; Ranford-Cartwright, L.C.; De Koning, H.P. Trypanosoma brucei: A survey of pyrimidine transport activities. Exp. Parasitol. 2006, 114, 103–108. [Google Scholar] [CrossRef] [PubMed]
- Ali, J.A.M.; Creek, D.J.; Burgess, K.; Allison, H.C.; Field, M.C.; Mäser, P.; De Koning, H.P. Pyrimidine salvage in Trypanosoma brucei bloodstream forms and the trypanocidal action of halogenated pyrimidines. Mol. Pharmacol. 2013, 83, 439–453. [Google Scholar] [CrossRef]
- Munday, J.C.; Eze, A.A.; Baker, N.; Glover, L.; Clucas, C.; Aguinaga Andrés, D.; Natto, M.J.; Teka, I.A.; McDonald, J.; Lee, R.S.; et al. Trypanosoma brucei Aquaglyceroporin 2 is a high affinity transporter for pentamidine and melaminophenyl arsenic drugs and is the main genetic determinant of resistance to these drugs. J. Antimicrob. Chemother. 2014, 69, 651–663. [Google Scholar] [CrossRef]
- Matovu, E.; Stewart, M.; Geiser, F.; Brun, R.; Mäser, P.; Wallace, L.J.M.; Burchmore, R.J.; Enyaru, J.C.K.; Barrett, M.P.; Kaminsky, R.; et al. Mechanisms of Arsenical and Diamidine Uptake and Resistance in Trypanosoma brucei. Eukaryot. Cell 2003, 2, 1003–1008. [Google Scholar] [CrossRef] [PubMed]
- Graf, F.E.; Ludin, P.; Wenzler, T.; Kaiser, M.; Brun, R.; Pati Pyana, P.; Büscher, P.; De Koning, H.P.; Horn, D.; Mäser, P. Aquaporin 2 mutations in Trypanosoma brucei gambiense field isolates correlate with decreased susceptibility to pentamidine and melarsoprol. PLoS Negl. Trop. Dis. 2013, 7, e2475. [Google Scholar] [CrossRef]
- Hammarton, T.C. Cell cycle regulation in Trypanosoma brucei. Mol. Biochem. Parasitol. 2007, 153, 1–8. [Google Scholar] [CrossRef]
- Thomas, J.A.; Baker, N.; Hutchinson, S.; Dominicus, C.; Trenaman, A.; Glover, L.; Alsford, S.; Horn, D. Insights into antitrypanosomal drug mode-of-action from cytology-based profiling. PLoS Negl. Trop. Dis. 2018, 12, e0006980. [Google Scholar] [CrossRef]
- Ibrahim, H.M.; Al-Salabi, M.I.; El Sabbagh, N.; Quashie, N.B.; Alkhaldi, A.A.; Escale, R.; Smith, T.K.; Vial, H.J.; De Koning, H.P. Symmetrical choline-derived dications display strong anti-kinetoplastid activity. J. Antimicrob. Chemother. 2011, 66, 111–125. [Google Scholar] [CrossRef] [PubMed]
- Giordani, F.; Khalaf, A.I.; Gillingwater, K.; Munday, J.C.; De Koning, H.P.; Suckling, C.J.; Barrett, M.P.; Scott, F.J. Novel Minor Groove Binders cure animal African trypanosomiasis in an in vivo mouse model. J. Med. Chem. 2019, 62, 3021–3035. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Koning, H.P.; Gould, M.K.; Sterk, G.J.; Tenor, H.; Kunz, S.; Luginbuehl, E.; Seebeck, T. Pharmacological validation of Trypanosoma brucei phosphodiesterases as novel drug targets. J. Infect. Dis. 2012, 206, 229–237. [Google Scholar] [CrossRef]
- Nvau, J.B.; Alenezi, S.; Ungogo, M.A.; Alfayez, I.A.M.; Natto, M.J.; Igoli, J.O.; Gray, A.I.; Ferro, V.A.; Watson, D.G.; De Koning, H.P. Antiparasitic and cytotoxic activity of Bokkosin, a novel diterpene substituted chromanyl benzoquinone from Calliandra portoricensis. Front. Chem. 2020, 8, 574103. [Google Scholar] [CrossRef] [PubMed]
- Spector, T.; Jones, T.E.; LaFon, S.W.; Nelson, D.J.; Berens, R.L.; Marr, J.J. Monophosphates of formycin B and allopurinol riboside: Interactions with leishmanial and mammalian succino-AMP synthetase and GMP reductase. Biochem. Pharmacol. 1984, 33, 1611–1617. [Google Scholar] [CrossRef]
- Steketee, P.C.; Dickie, E.A.; Iremonger, J.; Crouch, K.; Paxton, E.; Jayaraman, S.; Alfituri, O.A.; Awuah-Mensah, G.; Ritchie, R.; Schnaufer, A.; et al. Divergent metabolism between Trypanosoma congolense and Trypanosoma brucei results in differential sensitivity to metabolic inhibition. PloS Path. 2021, 17, e1009734. [Google Scholar] [CrossRef]
- Coustou, V.; Guegan, F.; Plazolles, N.; Baltz, T. Complete in vitro life cycle of Trypanosoma congolense: Development of genetic tools. PLoS Negl. Trop. Dis. 2010, 4, e618. [Google Scholar] [CrossRef] [PubMed]
- Awuah-Mensah, G.; McDonald, J.; Steketee, P.C.; Autheman, D.; Whipple, S.; D’Archivio, S.; Brandt, C.; Clare, S.; Harcourt, K.; Wright, G.J.; et al. Reliable, scalable functional genetics in bloodstream-form Trypanosoma congolense in vitro and in vivo. PLoS Path. 2021, 17, e1009224. [Google Scholar] [CrossRef]
- Chamond, N.; Cosson, A.; Blom-Potar, M.C.; Jouvion, G.; d’Archivio, S.; Medina, M.; Droin-Bergère, S.; Huerre, M.; Goyard, S.; Minoprio, P. Trypanosoma vivax infections: Pushing ahead with mouse models for the study of Nagana. I. Parasitological, hematological and pathological parameters. PLoS Negl. Trop. Dis. 2010, 4, e792. [Google Scholar] [CrossRef] [PubMed]
- d’Archivio, S.; Medina, M.; Cosson, A.; Chamond, N.; Rotureau, B.; Minoprio, P.; Goyard, S. Genetic engineering of Trypanosoma (Dutonella) vivax and in vitro differentiation under axenic conditions. PLoS Negl. Trop. Dis. 2011, 5, e1461. [Google Scholar] [CrossRef]
- D’Archivio, S.; Cosson, A.; Medina, M.; Lang, T.; Minoprio, P.; Goyard, S. Non-invasive in vivo study of the Trypanosoma vivax infectious process consolidates the brain commitment in late infections. PLoS Negl. Trop. Dis. 2013, 7, e1976. [Google Scholar] [CrossRef] [PubMed]
- Sanchez, M.A.; Ullman, B.; Landfear, S.M.; Carter, N.S. Cloning and functional expression of a gene encoding a P1 type nucleoside transporter from Trypanosoma brucei. J. Biol. Chem. 1999, 274, 30244–30249. [Google Scholar] [CrossRef] [Green Version]
- De Koning, H.P. Transporters in African trypanosomes: Role in drug action and resistance. Int. J. Parasitol. 2001, 31, 512–522. [Google Scholar] [CrossRef] [PubMed]
- Lüscher, A.; De Koning, H.P.; Mäser, P. Chemotherapeutic strategies against Trypanosoma brucei: Drug targets vs. drug targeting. Curr. Pharm. Des. 2007, 13, 555–567. [Google Scholar] [CrossRef] [PubMed]
- Ogbunude, P.O.J.; Ikediobi, C.O.; Ukoha, A.I. Adenosine cycle in African trypanosomes. Ann. Trop. Med. Parasitol. 1985, 79, 7–11. [Google Scholar] [CrossRef] [PubMed]
- James, D.M.; Born, G.V.R. Uptake of purine bases and nucleosides in African trypanosomes. Parasitology 1980, 81, 383–393. [Google Scholar] [CrossRef]
- Ortiz, D.; Sanchez, M.A.; Quecke, P.; Landfear, S.M. Two novel nucleobase/pentamidine transporters from Trypanosoma brucei. Mol. Biochem. Parasitol. 2009, 163, 67–76. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.; Ferreira de Almeida Fiuza, L.; Cardoso Santos, C.; Ferreira Nunes, D.; Cruz Moreira, O.; Bouton, J.; Karalic, I.; Maes, L.; Caljon, G.; Hulpia, F.; et al. 6-Methyl-7-aryl-7-deazapurine nucleosides as anti-Trypanosoma cruzi agents: Structure-activity relationship and in vivo efficacy. ChemMedChem 2021, 16, 2231–2253. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.; Hulpia, F.; Karalic, I.; De Schepper, L.; Maes, L.; Caljon, G.; Van Calenbergh, S. 6-Methyl-7-deazapurine nucleoside analogues as broad-spectrum antikinetoplastid agents. Int. J. Parasitol. Drugs Drug Resist. 2021, 17, 57–66. [Google Scholar] [CrossRef]
- Richards, S.; Morrison, L.J.; Torr, S.J.; Barrett, M.P.; Manangwa, O.; Mramba, F.; Auty, H. Pharma to farmer: Field challenges of optimizing trypanocide use in African animal trypanosomiasis. Trends Parasitol. 2021, 37, 831–843. [Google Scholar] [CrossRef] [PubMed]
- Fidalgo, L.M.; Gille, L. Mitochondria and trypanosomatids: Targets and drugs. Pharm. Res. 2011, 28, 2758–2770. [Google Scholar] [CrossRef]
- Eze, A.A.; Gould, M.K.; Munday, J.C.; Tagoe, D.N.A.; Stelmanis, V.; Schnaufer, A.; De Koning, H.P. Loss of mitochondrial membrane potential is a late adaptation of Trypanosoma brucei brucei to isometamidium preceded by mutations in the γ subunit of the F1F0-ATPase. PLoS Negl. Trop. Dis. 2016, 10, e0004791. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Drew, M.E.; Morris, J.C.; Wang, Z.; Wells, L.; Sanchez, M.; Landfear, S.M.; Englund, P.T. The adenosine analog tubercidin inhibits glycolysis in Trypanosoma brucei as revealed by an RNA interference library. J. Biol. Chem. 2003, 278, 46596–46600. [Google Scholar] [CrossRef] [PubMed]
- Anyam, J.V.; Daikwo, P.E.; Ungogo, M.A.; Nweze, N.E.; Igoli, N.P.; Gray, A.I.; De Koning, H.P.; Igoli, J.O. Two New Antiprotozoal Diterpenes from the Roots of Acacia nilotica. Front. Chem. 2021, 9, 624741. [Google Scholar] [CrossRef] [PubMed]
- Wallace, L.J.M.; Candlish, D.; De Koning, H.P. Different substrate recognition motifs of human and trypanosome nucleobase transporters: Selective uptake of purine antimetabolites. J. Biol. Chem. 2002, 277, 26149–26156. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Y.; Prusoff, W.H. Relationship between the inhibition constant (Ki) and the concentration of inhibitor which causes 50 per cent inhibition (I50) of an enzymatic reaction. Biochem. Pharmacol. 1973, 22, 3099–3108. [Google Scholar] [PubMed]













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Tc Km or Ki (µM) | ΔG0 (kJ/mol) | n | Tbb P1 1 (µM) | ΔG0 (kJ/mol) |
|---|---|---|---|---|---|
| Adenosine | 0.48 ± 0.03 | −36.0 | 3 | 0.36 ± 0.05 | −36.8 |
| Tubercidin | 403 ± 120 | −19.3 | 3 | 78 ± 6.4 | −23.4 |
| Cordycepin | 12.0 ± 2 | −28.0 | 3 | 210 ± 48 | −21.0 |
| Inosine | 0.27 ± 0.02 | −37.5 | 3 | 0.44 ± 0.10 | −36.3 |
| Guanosine | 1.06 ± 0.07 | −34.1 | 3 | 1.8 ± 0.3 | −32.7 |
| Uridine | 390 ± 9.1 | −19.4 | 3 | 830 ± 86 | −18.7 |
| Thymidine | 94.6 ± 11.5 | −23.0 | 3 | 44 ± 10 | −11.4 |
| Cytidine | 1176 ± 148 | −16.7 | 3 | >250 |
| Gene ID | Given Name | Number of Amino Acids | Number of TMDs | Closest Related T. brucei Transporter | Remarks |
|---|---|---|---|---|---|
| Nucleoside transporters | |||||
| TcIL3000_9_2500 | TcoAT1/TcoNT10 | 472 | 10 | TbNT10 | P1-type |
| TvY486_0202110 | TvxNT1 | 472 | 11 | TbNT4 | P1-type |
| TvY486_1112030 | TvxNT2 | 470 | 11 | TbNT12 | P1-type |
| TvY486_0043680 | TvxNT3 | 464 | 10 | TbNT6 | P1-type |
| TvY486_0014570 | TvxNT4 | 373 | 7 | TbNT10 | P1-type |
| Nucleobase transporters | |||||
| TvY486_0011610 | (fragment) | 284 | 6 | NT8 | ----- |
| TvY486_1103750 | (fragment) | 150 | 4 | NT8 | ----- |
| TvY486_1103760 | TvxNT5 | 385 | 9 | NT8 | NBT |
| TvY486_0041960 | TvxNT6 | 437 | 10 | NT8 | NBT |
| TvY486_1103740 | TvxNT7 | 436 | 10 | NT8 | NBT |
| Purine | Km or Ki (μM) | ΔG0 | δ(ΔG0) | n |
|---|---|---|---|---|
| Adenosine | 0.42 ± 0.03 | −36.4 | 3 | |
| Inosine | 0.55 ± 0.09 | −35.7 | 0.68 | 3 |
| Guanosine | 0.80 ± 0.09 | −34.8 | 1.62 | 3 |
| 2′-deoxyadenosine | 0.50 ± 0.07 | −36.0 | 0.44 | 3 |
| 5′-deoxyadenosine | 0.45 ± 0.08 | −36.2 | 0.18 | 3 |
| Cordycepin | 4.59 ± 0.63 | −30.5 | 5.94 | 3 |
| 2′,3′-dideoxyinosine | 85.4 ± 7.1 | −23.2 | 12.51 | 3 |
| 2′,3′-dideoxyadenosine | 51.9 ± 2.6 | −24.4 | 11.95 | 3 |
| 1-deazaadenosine | 1.03 ± 0.34 | −34.2 | 2.24 | 4 |
| 3-deazaadenosine | 138 ± 37 | −22.0 | 14.37 | 3 |
| Tubercidin | 754 ± 54.3 | −17.8 | 18.58 | 3 |
| Uridine | 1467 ± 172 | −16.2 | 20.23 | 3 |
| Thymidine | 434 ± 46.4 | −19.2 | 17.21 | 4 |
| Cytidine | 1577 ± 69.2 | −16.0 | 20.41 | 3 |
| Adenine | 761± 103 | −17.8 | 18.60 | 5 |
| Hypoxanthine | 61.2 ± 3.26 | −24.0 | 11.68 | 3 |
| Ki | |||||
|---|---|---|---|---|---|
| AVG | SEM | n | ΔG0 | δ(ΔG0) | |
| Adenosine | 1.41 | 0.35 | 3 | −33.38 | |
| Guanosine | 1.37 | 0.13 | 3 | −33.45 | 0.07 |
| Inosine | 0.18 | 0.05 | 3 | −38.46 | 5.08 |
| 2′-deoxyadenosine | 1.06 | 0.29 | 3 | −34.1 | 0.70 |
| 5′-deoxyadenosine | 1.10 | 0.12 | 3 | −34.0 | 0.63 |
| Cordycepin | 8.6 | 1.4 | 3 | −28.91 | −4.47 |
| Tubercidin | 287 | 55 | 3 | −20.21 | −13.17 |
| Nebularine | 4.6 | 1.1 | 4 | −30.4 | −2.94 |
| 6-thioinosine | 4.03 | 0.78 | 4 | −30.8 | −7.7 |
| 3-deazainosine | 33.0 | 17.4 | 4 | −25.6 | −12.9 |
| Adenine | 2.43 | 0.14 | 2 | −32.04 | −1.34 |
| Hypoxanthine | 1.75 | 0.17 | 3 | −32.85 | −7.45 |
| Uridine | 720 | 110 | 3 | −17.93 | −15.45 |
| Thymidine | 603 | 51 | 3 | −18.37 | −15.01 |
| Compound | 3′ | 6-Subst | 7-Subst | LmexCas9 | SUPKO | SUPKO+TcoAT1 | SUPKO+TvxNT3 | |||
|---|---|---|---|---|---|---|---|---|---|---|
| EC50 (µM) | EC50 (µM) | RF (Cas9) | EC50 (µM) | RF (SUPKO) | EC50 (µM) | RF (SUPKO) | ||||
| Tubercidin | OH | NH2 | 0.44 ± 0.06 | >100 | >230 | >100 | - | >100 | - | |
| Cordycepin | NH2 | 91.6 ± 12.9 | >100 | >1.09 | >100 | - | >100 | - | ||
| FH7429_u | NH2 | Br | 27.3 ± 7.0 | >200 | >3.6 | 27.0 ± 5.8 | >7.4 | 42.9 ± 12.0 | >2.33 | |
| FH7429_d | NH2 | 15.1 ± 1.7 | >100 | >6.63 | 65.3 ± 7.6 | >1.53 | >100 | - | ||
| FH6367 | OH | NH2 | CF3 | 26.1 ± 6.4 | >100 | >3.83 | 3.79 ± 0.34 | >26.4 | 7.4 ± 1.4 | >13.5 |
| FH3169 | OH | NH2 | Cl | 0.23 ± 0.005 | 15.4 ± 2.7 | 67.7 | 9.41 ± 0.56 | 1.64 | 15.9 ± 2.5 | 0.97 |
| TH1008 | OH | NH2 | Pyridin-2-yl | 11.7 ± 0.5 | >100 | >8.5 | >100 | - | >100 | - |
| TH1003 | OH | NH2 | Br | 2.43 ± 0.14 | >100 | >41.2 | 71.1 ± 5.0 | >1.4 | >100 | - |
| FH3167 | OH | NH2 | F | 0.096 ± 0.009 | 63.4 ± 5.5 | 660 | 7.5 ± 0.9 | 8.48 | 57.2 ± 6.8 | 1.1 |
| CL4510 | OH | OCH3 | 0.39 ± 0.05 | 2.28 ± 0.21 | 5.84 | 2.2 ± 0.03 | 1.03 | 2.05 ± 0.04 | 1.1 | |
| FH8517 | NH2 | F | 3.09 ± 0.04 | 9.96 ± 2.04 | 3.22 | 6.1 ± 0.7 | 1.64 | 9.42 ± 1.41 | 1.06 | |
| FH8505 | NH2 | ethynyl | 92.7 ± 4.3 | >100 | >1.08 | 89.9 ± 12.5 | >1.1 | 80.0 ± 7.7 | >1.25 | |
| FH8496 | NH2 | I | 51.8 ± 10.3 | 52.6 ± 0.0 | 1.01 | 23.9 ± 5.6 | >2.2 | 40.0 ± 9.2 | >1.2 | |
| FH9531 | OH | O-iPr | Cl | >100 | >100 | - | >100 | - | >100 | - |
| FH10659 | NH2 | -vinyl | >100 | >100 | - | >100 | - | >100 | - | |
| Pentamidine | 1.43 ± 0.05 | 1.83 ± 0.29 | 1.28 | 1.73 ± 0.20 | 1.1 | 2.31 ± 0.18 | 0.77 | |||
| TcoAT1 in L. mexicana SUPKO | T. congolense IL300 | |||||
|---|---|---|---|---|---|---|
| Km or Ki (μM) | ΔG0 (kJ/mol) | n | Km or Ki (μM) | ΔG0 (kJ/mol) | n | |
| Adenosine | 0.42 ± 0.03 | −36.4 | 3 | 0.48 ± 0.03 | −36.0 | 3 |
| Tubercidin | 693 ± 25 | −17.8 | 3 | 403 ± 120 | −19.4 | 3 |
| Cordycepin | 4.6 ± 0.6 | −30.5 | 3 | 12.0 ± 2.0 | −28.1 | 3 |
| FH7429_u | 259 ± 11 | −20.5 | 3 | |||
| FH7429_d | 810 ± 60 | −18.0 | 3 | 319 ± 93 | −19.9 | 3 |
| FH8496 | 135 ± 11 | −20.6 | 3 | 46.2 ± 1.7 | −24.7 | 3 |
| FH8470 | 60.6 ± 10.7 | −24.1 | 3 | 17.5 ± 3.4 | −27.1 | 4 |
| FH8517 | 229 ± 15 | −20.8 | 3 | 98.1 ± 2.3 | −22.9 | 3 |
| FH10659 | 59.3 ± 4.3 | −17.7 | 4 | 28.5 ± 8.1 | −25.9 | 6 |
| TH1012 | 146 ± 58 | −21.9 | 3 | |||
| CL4510 | 226 ± 26 | −20.8 | 3 | |||
| Km or Ki (µM) | ΔG0 (kJ/mol) | n | |
|---|---|---|---|
| Adenosine | 1.41 ± 0.35 | −33.4 | 3 |
| Tubercidin | 287 ± 55 | −20.2 | 3 |
| Cordycepin | 8.57 ± 1.45 | −28.9 | 3 |
| FH 7429_u | 169 ± 4 | −21.5 | 3 |
| FH 7429_d | 388 ± 13 | −19.5 | 3 |
| CL4510 | 16.1 ± 2.8 | −27.4 | 3 |
| CL5565 | 32.0 ± 9.2 | −25.7 | 3 |
| FH15963 | 225 ± 42 | −20.8 | 3 |
| FH15967 | 109 ± 30 | −22.6 | 3 |
| T. congolense IL3000 | T. congolense 6C3 | T. vivax ** | T.b.b. B48 | T. evansi | T. equiperdum | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Compound | C7 | 2′-OH | 3′-OH | 6-Position | EC50 (µM) | EC50 (µM) | RF † | EC50 (µM) | EC50 (µM) | EC50 (µM) | EC50 (µM) |
| Tubercidin | None | + | + | NH2 | 3.16 ± 0.50 | 1.83 ± 0.39 | 0.58 | 4.30 ± 1.30 | |||
| Cordycepin | None | + | − | NH2 | 2.46 ± 0.42 | 2.16 ± 0.44 | 0.88 | 0.12 ± 0.02 | |||
| FH7429_d * | None | + | − | NH2 | 0.021 ± 0.001 * | 0.019 ± 0.002 * | 0.91 | 3.48 ± 0.09 | 0.25 ± 0.02 | 0.017 ± 0.002 | 0.0026 ± 0.0006 |
| FH10677 | None | − | + | NH2 | 43.3 ± 14.5 | 10.9 ± 2.3 | 0.25 | >64.0 | 38.0 ± 2.3 | >100 | 17.4 ± 2.3 |
| FH7429_u | Br | + | − | NH2 | 0.00069 ± 0.00007 | 0.00049 ± 0.00009 | 0.71 | 0.018 ± 0.003 | 0.0048 ± 0.0005 | 0.00090 ± 0.00022 | 0.00051 ± 0.00007 |
| FH8496 | I | + | − | NH2 | 0.0054 ± 0.0010 | 0.0030 ± 0.0002 | 0.56 | 0.056 ± 0.001 | 0.024 ± 0.002 | 0.0037 ± 0.0012 | 0.0015 ± 0.0005 |
| FH8470 * | Cl | + | − | NH2 | 0.0012 ± 0.0002 | 0.00066 ± 0.00003 | 0.53 a | 0.096 ± 0.082 | 0.0068 ± 0.0009 | 0.0018 ± 0.0002 | 0.00057 ± 0.00008 |
| FH8517 | F | + | − | NH2 | 0.0052 ± 0.0003 | 0.0031 ± 0.0003 | 0.60 | 0.50 ± 0.01 | 0.013 ± 0.002 | 0.0040 ± 0.0006 | 0.00052 ± 0.00011 |
| FH10679 | Br | − | + | NH2 | 0.51 ± 0.09 | 0.31 ± 0.07 | 0.60 | 4.30 ± 0.90 | 1.04 ± 0.19 | 1.74 ± 0.53 | 0.067 ± 0.013 |
| TH1003 | Br | + | + | NH2 | 0.79 ± 0.14 | 0.52 ± 0.031 | 0.66 | 0.74 ± 0.04 | 1.40 ± 0.09 | 0.48 ± 0.06 | 0.085 ± 0.026 |
| FH3169 | Cl | + | + | NH2 | 0.86 ± 0.12 | 0.75 ± 0.14 | 0.87 | 0.053 ± 0.007 | 1.84 ± 0.33 | 0.62 ± 0.06 | 0.12 ± 0.018 |
| FH3167 | F | + | + | NH2 | 0.45 ± 0.62 | 0.48 ± 0.10 | 1.06 | 0.038 ± 0.006 | 0.16 ± 0.03 | 0.18 ± 0.023 | 0.032 ± 0.007 |
| FH6367 | CF3 | + | + | NH2 | 1.80 ± 0.44 | 0.89 ± 0.17 | 0.50 | 0.82 ± 0.09 | 0.90 ± 0.13 | 0.68 ± 0.11 | 0.19 ± 0.03 |
| FH9531 | Cl | + | + | O-iPr | 4.04 ± 0.36 * | 3.23 ± 0.36 * | 0.80 | 0.12 ± 0.01 | |||
| FH8505 | ethynyl | + | − | NH2 | 0.017 ± 0.002 | 0.0080 ± 0.002 | 0.46 a | 0.16 ± 0.01 | 0.031 ± 0.004 | 0.0078 ± 0.0020 | 0.0026 ± 0.0008 |
| FH10659 | vinyl | + | − | NH2 | 0.056 ± 0.004 | 0.040 ± 0.003 | 0.70 | 1.57 ± 0.43 | 0.038 ± 0.0015 | 0.011 ± 0.001 | |
| TH1008 * | Pyridin-2-yl | + | + | NH2 | 0.17 ± 0.05 | 0.056 ± 0.013 | 0.33 | 12.9 ± 4.4 | 0.39 ± 0.03 | 0.040 ± 0.005 | 0.061 ± 0.005 |
| Diminazene | 0.23 ± 0.04 | 1.32 ± 0.43 | 5.7 a | 0.34 ± 0.13 | |||||||
| Pentamidine | 0.55 ± 0.11 | 0.44 ± 0.15 | 0.79 | 0.34 ± 0.06 | |||||||
| HEK EC50 (µM) | SI † T. evansi | SI † T. equiperdum | SI † T. congolense | |
|---|---|---|---|---|
| FH7429_u | 2.18 ± 0.41 | 2430 | 4271 | 3158 |
| FH7429_d | >200 | >11,800 | >76,000 | 9700 |
| FH6367 | 0.49 ± 0.01 | 0.7 | 2.6 | 0.3 |
| FH3169 | 4.55 ± 0.19 | 7.4 | 37 | 5.3 |
| TH1008 | 18.5 ± 1.5 | 466 | 303 | 110 |
| TH1003 | 2.37 ± 0.12 | 4.9 | 28 | 3.0 |
| FH8470 | 12.0 ± 0.32 | 6830 | 21,100 | 9800 |
| FH3167 | 0.49 ± 0.05 | 2.7 | 15 | 1.1 |
| FH8517 | 1.71 ± 0.11 | 431 | 3310 | 330 |
| FH8505 | 1.17 ± 0.06 | 151 | 446 | 68 |
| FH8496 | 4.91 ± 0.03 | 1340 | 3260 | 902 |
| FH10679 | >200 | >115 | >3000 | >390 |
| FH10677 | >200 | >12 | >4.6 |
| Transporter | Substrate | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| ADE | ADO | GUA | GUO | HYP | INO | CTD | TMD | URD | |
| TbP2/AT1 | 0.38 a; 0.30 c | 0.59 a; 0.92 c | NE 3,c | >500 c | NE 4,c | NE 3,c | NE 2,c | NE 2,c | |
| TbP1/NT2 | NE 3,c | 0.15 a; 0.26 b; 0.38 c | 1.8 c | NE 1,c | 0.44 c | NE 3,c | 44 c | 830 c | |
| TbNT5 d | 2 | 2.2 | 49.4 | ||||||
| TbNT6 d | 1.4 | 4.3 | |||||||
| TbNT7 d | 0.3 | 1.8 | |||||||
| TbNT9 e | 148 | 0.068 | 6.2 | 320 | 2.75 | 510 | 235 | ||
| TbNT10 e | 0.41 | 0.53 | |||||||
| TbNT11 f | 2.7; 8 | 266 | 651 | 141 | |||||
| TbNT8/H4 g | 2.6 | 860 | 2.6 | 4.7 | 0.55 | 20 | 95 | ||
| TcoAT1 | 786 | 0.42 | 0.80 | 61.2 | 0.55 | 1630 | 49.6 | 147 | |
| TvxNT3 | 2.43 | 1.41 | 1.37 | 1.75 | 0.086 | >100 | 603 | 720 | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ungogo, M.A.; Aldfer, M.M.; Natto, M.J.; Zhuang, H.; Chisholm, R.; Walsh, K.; McGee, M.; Ilbeigi, K.; Asseri, J.I.; Burchmore, R.J.S.; et al. Cloning and Characterization of Trypanosoma congolense and T. vivax Nucleoside Transporters Reveal the Potential of P1-Type Carriers for the Discovery of Broad-Spectrum Nucleoside-Based Therapeutics against Animal African Trypanosomiasis. Int. J. Mol. Sci. 2023, 24, 3144. https://doi.org/10.3390/ijms24043144
Ungogo MA, Aldfer MM, Natto MJ, Zhuang H, Chisholm R, Walsh K, McGee M, Ilbeigi K, Asseri JI, Burchmore RJS, et al. Cloning and Characterization of Trypanosoma congolense and T. vivax Nucleoside Transporters Reveal the Potential of P1-Type Carriers for the Discovery of Broad-Spectrum Nucleoside-Based Therapeutics against Animal African Trypanosomiasis. International Journal of Molecular Sciences. 2023; 24(4):3144. https://doi.org/10.3390/ijms24043144
Chicago/Turabian StyleUngogo, Marzuq A., Mustafa M. Aldfer, Manal J. Natto, Hainan Zhuang, Robyn Chisholm, Katy Walsh, MarieClaire McGee, Kayhan Ilbeigi, Jamal Ibrahim Asseri, Richard J. S. Burchmore, and et al. 2023. "Cloning and Characterization of Trypanosoma congolense and T. vivax Nucleoside Transporters Reveal the Potential of P1-Type Carriers for the Discovery of Broad-Spectrum Nucleoside-Based Therapeutics against Animal African Trypanosomiasis" International Journal of Molecular Sciences 24, no. 4: 3144. https://doi.org/10.3390/ijms24043144