
Both ATM and DNA-PK Are the Main Regulators of HIV-1 Post-Integrational DNA Repair

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
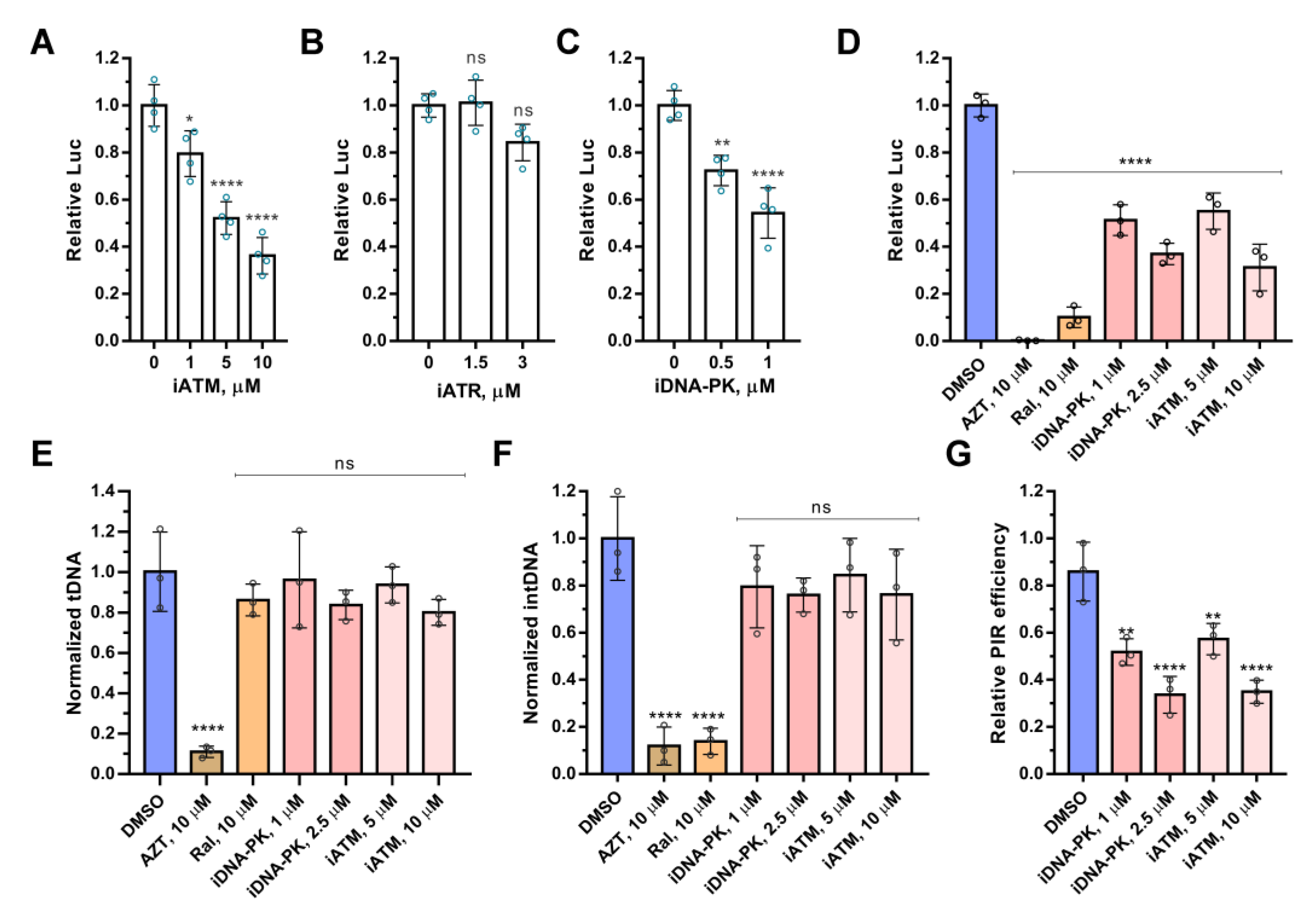
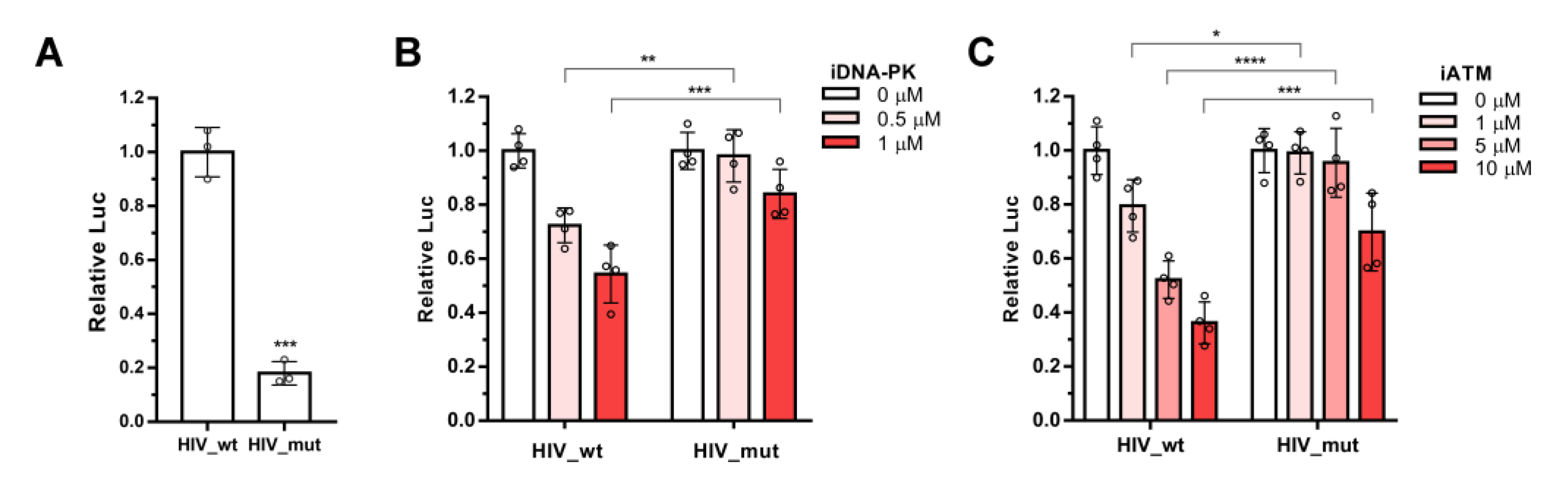
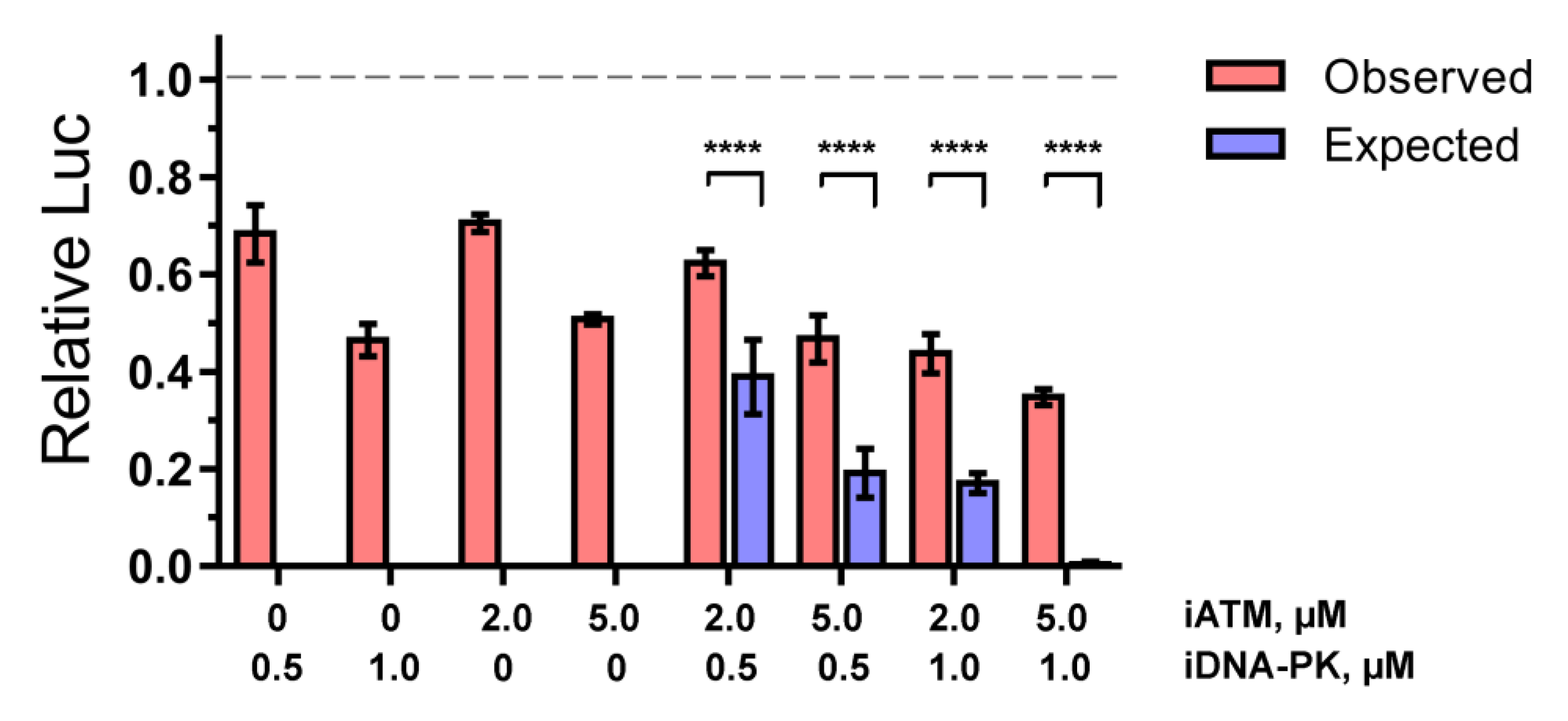
2.1. ATM and DNA-PKcs, but Not ATR, Are Essential for HIV-1 Post-Integrational Repair
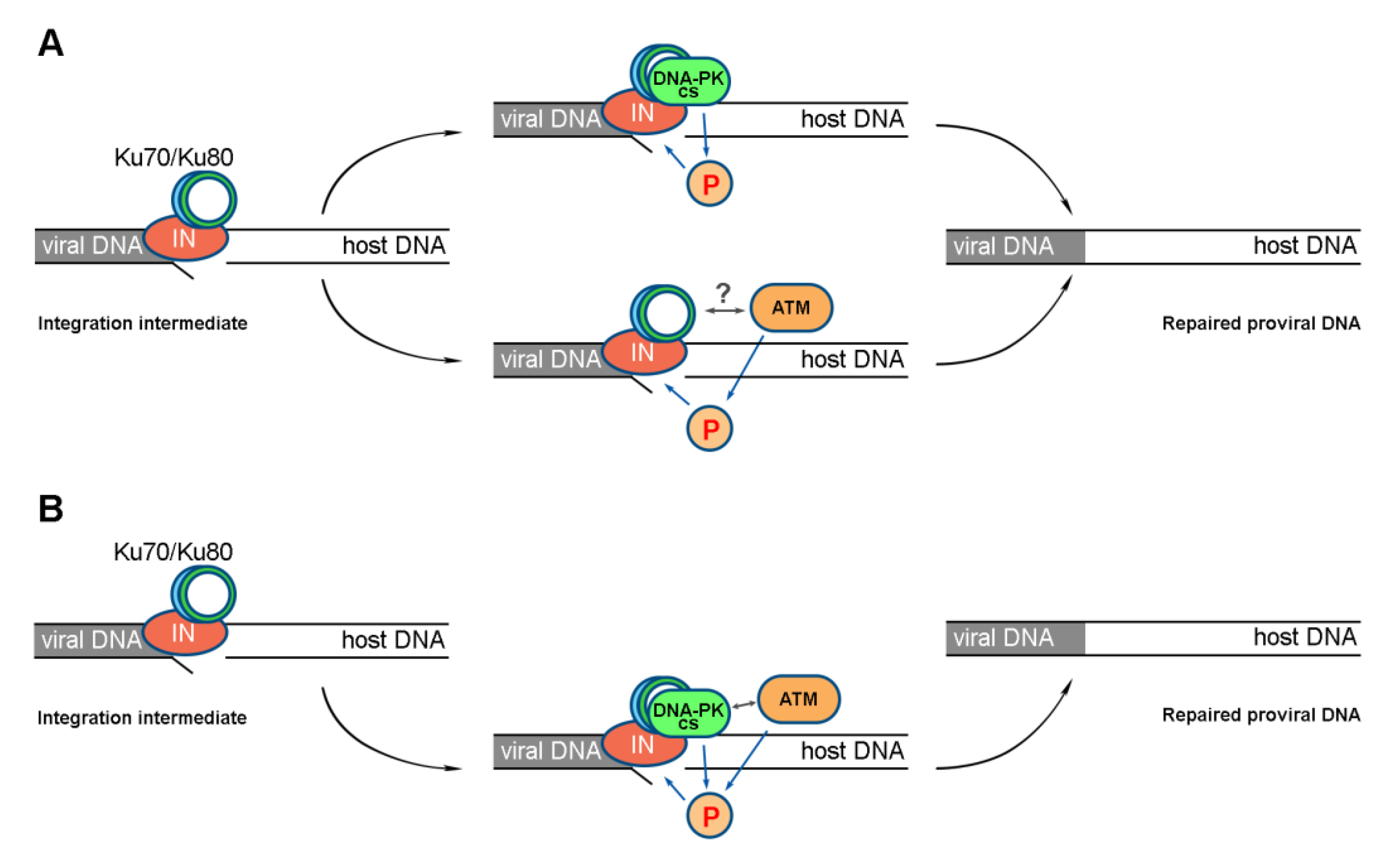
2.2. ATM Activation Depends on the Formation of a Complex between HIV-1 Integrase and the Cellular Ku70 Protein
2.3. Integration of Viral DNA Is a Signal for ATM and DNA-PKcs Activation
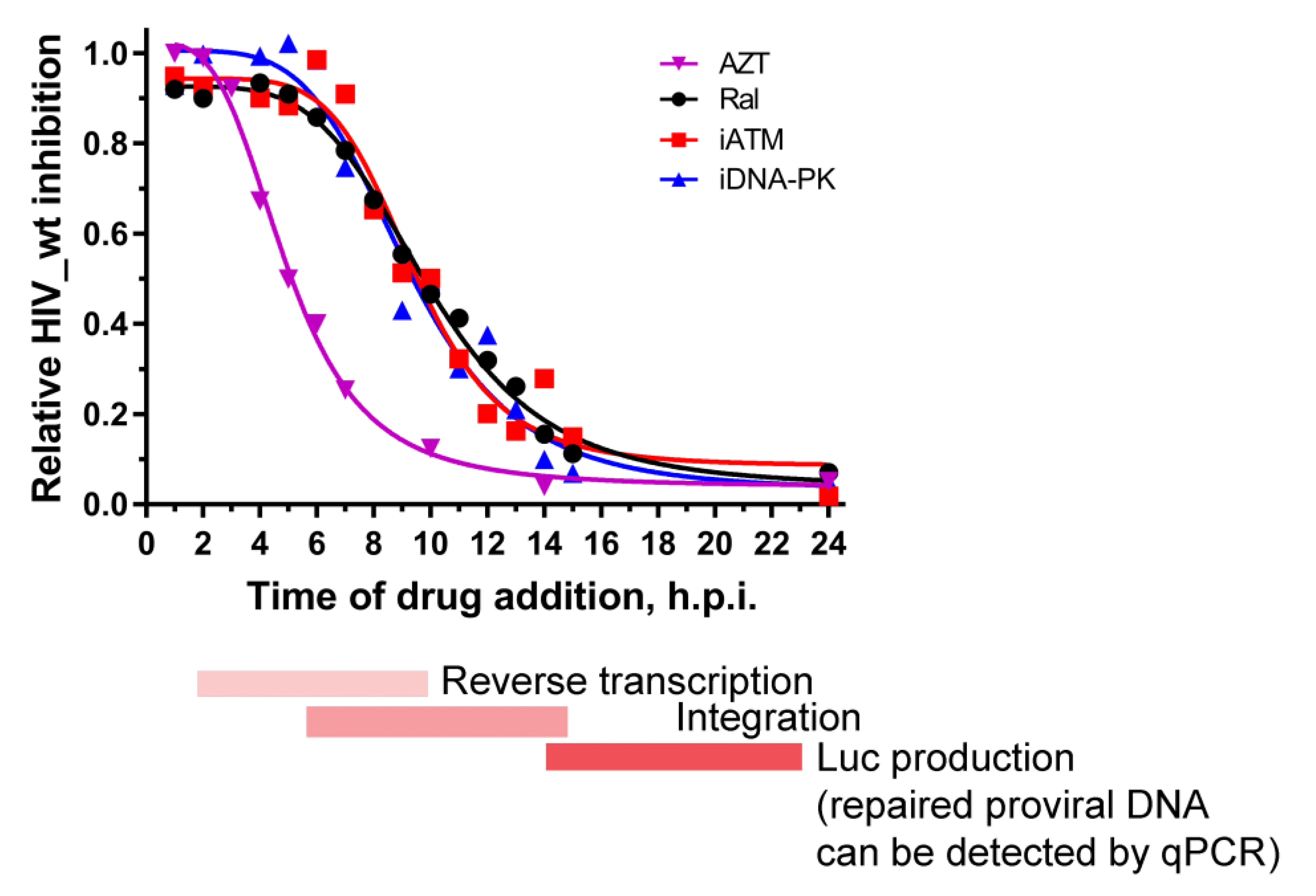
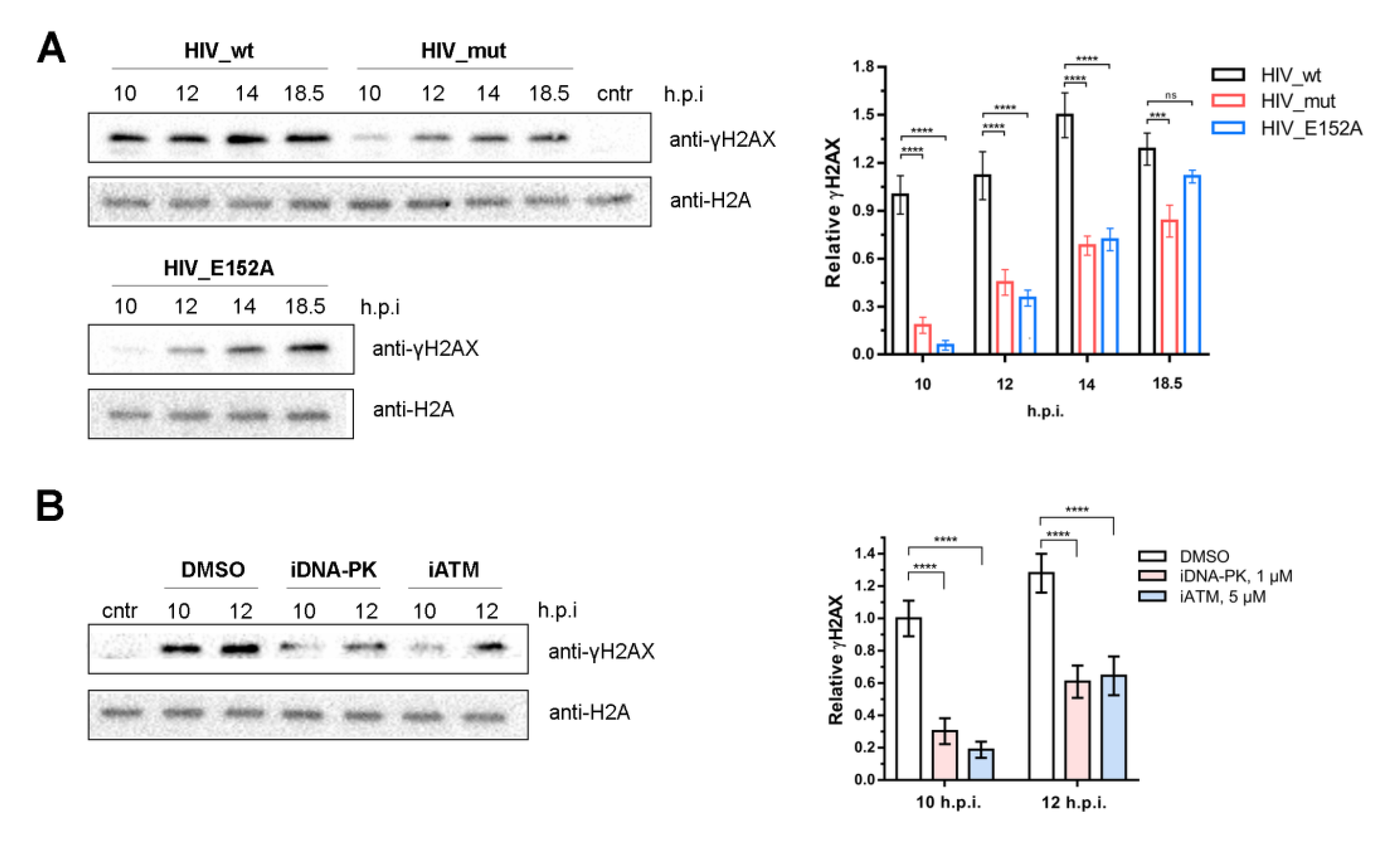
2.4. Activation of DNA-PKcs and ATM Is a Sequential Process
2.5. ATM and DNA-PKcs Activation Leads to γH2AX Histone Accumulation during PIR
3. Discussion
4. Materials and Methods
4.1. Cell Culture and Work with Lentiviral Vectors
4.2. HIV-Based Vectors Preparation
4.3. Cells Transduction
4.4. qPCR
4.5. Time-of-Addition
4.6. Western Blot
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Vannucci, L.; Lai, M.; Chiuppesi, F.; Ceccherini-Nelli, L.; Pistello, M. Viral vectors: A look back and ahead on gene transfer technology. New Microbiol. 2013, 36, 1–22. [Google Scholar] [PubMed]
- Milone, M.C.; O’Doherty, U. Clinical use of lentiviral vectors. Leukemia 2018, 32, 1529. [Google Scholar] [CrossRef]
- Arabi, F.; Mansouri, V.; Ahmadbeigi, N. Gene therapy clinical trials, where do we go? An overview. Biomed. Pharmacother. 2022, 153, 113324. [Google Scholar] [CrossRef]
- Chen, L.; Xie, T.; Wei, B.; Di, D.L. Current progress in CAR-T cell therapy for tumor treatment (Review). Oncol. Lett. 2022, 24, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Labbé, R.P.; Vessillier, S.; Rafiq, Q.A. Lentiviral Vectors for T Cell Engineering: Clinical Applications, Bioprocessing and Future Perspectives. Viruses 2021, 13, 1528. [Google Scholar] [CrossRef] [PubMed]
- Poorebrahim, M.; Sadeghi, S.; Fakhr, E.; Abazari, M.F.; Poortahmasebi, V.; Kheirollahi, A.; Askari, H.; Rajabzadeh, A.; Rastegarpanah, M.; Linē, A.; et al. Production of CAR T-cells by GMP-grade lentiviral vectors: Latest advances and future prospects. Crit. Rev. Clin. Lab. Sci. 2019, 56, 393–419. [Google Scholar] [CrossRef]
- Albinger, N.; Hartmann, J.; Ullrich, E. Current status and perspective of CAR-T and CAR-NK cell therapy trials in Germany. Gene Ther. 2021, 28, 513–527. [Google Scholar] [CrossRef] [PubMed]
- Qi, J.; Ding, C.; Jiang, X.; Gao, Y. Advances in Developing CAR T-Cell Therapy for HIV Cure. Front. Immunol. 2020, 11, 361. [Google Scholar] [CrossRef]
- Jiang, Z.; Liang, H.; Pan, H.; Liang, Y.; Wang, H.; Yang, X.; Lu, P.; Zhang, X.; Yang, J.; Zhang, D.; et al. HIV-1-Specific CAR-T Cells with Cell-Intrinsic PD-1 Checkpoint Blockade Enhance Anti-HIV Efficacy in vivo. Front. Microbiol. 2021, 12, 1891. [Google Scholar] [CrossRef]
- Hale, M.; Mesojednik, T.; Romano Ibarra, G.S.; Sahni, J.; Bernard, A.; Sommer, K.; Scharenberg, A.M.; Rawlings, D.J.; Wagner, T.A. Engineering HIV-Resistant, Anti-HIV Chimeric Antigen Receptor T Cells. Mol. Ther. 2017, 25, 570–579. [Google Scholar] [CrossRef] [Green Version]
- Hu, W.S.; Hughes, S.H. HIV-1 Reverse Transcription. Cold Spring Harb. Perspect. Med. 2012, 2, a006882. [Google Scholar] [CrossRef] [PubMed]
- Rozina, A.; Anisenko, A.; Kikhai, T.; Silkina, M.; Gottikh, M. Complex Relationships between HIV-1 Integrase and Its Cellular Partners. Int. J. Mol. Sci. 2022, 23, 12341. [Google Scholar] [CrossRef] [PubMed]
- Lesbats, P.; Engelman, A.N.; Cherepanov, P. Retroviral DNA Integration. Chem. Rev. 2016, 116, 12730–12757. [Google Scholar] [CrossRef]
- Skalka, A.M.; Katz, R.A. Retroviral DNA integration and the DNA damage response. Cell Death Differ. 2005, 12, 971–978. [Google Scholar] [CrossRef]
- Knyazhanskaya, E.S.; Shadrina, O.A.; Anisenko, A.N.; Gottikh, M.B. Role of DNA-dependent protein kinase in the HIV-1 replication cycle. Mol. Biol. 2016, 50, 639–654. [Google Scholar] [CrossRef]
- Brass, A.L.; Dykxhoorn, D.M.; Benita, Y.; Yan, N.; Engelman, A.; Xavier, R.J.; Lieberman, J.; Elledge, S.J. Identification of host proteins required for HIV infection through a functional genomic screen. Science 2008, 319, 921–926. [Google Scholar] [CrossRef]
- Zhou, H.; Xu, M.; Huang, Q.; Gates, A.T.; Zhang, X.D.; Castle, J.C.; Stec, E.; Ferrer, M.; Strulovici, B.; Hazuda, D.J.; et al. Genome-Scale RNAi Screen for Host Factors Required for HIV Replication. Cell Host Microbe 2008, 4, 495–504. [Google Scholar] [CrossRef]
- Espeseth, A.S.; Fishel, R.; Hazuda, D.; Huang, Q.; Xu, M.; Yoder, K.; Zhou, H. Sirna screening of a targeted library of DNA repair factors in HIV infection reveals a role for base excision repair in HIV integration. PLoS ONE 2011, 6, e17612. [Google Scholar] [CrossRef]
- König, R.; Zhou, Y.; Elleder, D.; Diamond, T.L.; Bonamy, G.M.C.; Irelan, J.T.; Chiang, C.y.; Tu, B.P.; De Jesus, P.D.; Lilley, C.E.; et al. Global analysis of host-pathogen interactions that regulate early-stage HIV-1 replication. Cell 2008, 135, 49–60. [Google Scholar] [CrossRef]
- Anisenko, A.N.; Knyazhanskaya, E.S.; Isaguliants, M.G.; Gottikh, M.B. A qPCR assay for measuring the post-integrational DNA repair in HIV-1 replication. J. Virol. Methods 2018, 262, 12–19. [Google Scholar] [CrossRef]
- Knyazhanskaya, E.; Anisenko, A.; Shadrina, O.; Kalinina, A.; Zatsepin, T.; Zalevsky, A.; Mazurov, D.; Gottikh, M. NHEJ pathway is involved in post-integrational DNA repair due to Ku70 binding to HIV-1 integrase. Retrovirology 2019, 16, 30. [Google Scholar] [CrossRef]
- Blackford, A.N.; Jackson, S.P. ATM, ATR, and DNA-PK: The Trinity at the Heart of the DNA Damage Response. Mol. Cell 2017, 66, 801–817. [Google Scholar] [CrossRef]
- Daniel, R.; Katz, R.A.; Skalka, A.M. A role for DNA-PK in retroviral DNA integration. Science 1999, 284, 644–647. [Google Scholar] [CrossRef] [PubMed]
- Daniel, R.; Katz, R.A.; Merkel, G.; Hittle, J.C.; Yen, T.J.; Skalka, A.M. Wortmannin Potentiates Integrase-Mediated Killing of Lymphocytes and Reduces the Efficiency of Stable Transduction by Retroviruses. Mol. Cell. Biol. 2001, 21, 1164–1172. [Google Scholar] [CrossRef] [PubMed]
- Sakurai, Y.; Komatsu, K.; Agematsu, K.; Matsuoka, M. DNA double strand break repair enzymes function at multiple steps in retroviral infection. Retrovirology 2009, 6, 114. [Google Scholar] [CrossRef]
- Lau, A.; Swinbank, K.M.; Ahmed, P.S.; Taylor, D.L.; Jackson, S.P.; Smith, G.C.M.; O’Connor, M.J. Suppression of HIV-1 infection by a small molecule inhibitor of the ATM kinase. Nat. Cell Biol. 2005, 7, 493–500. [Google Scholar] [CrossRef]
- Daelemans, D.; Pauwels, R.; De Clercq, E.; Pannecouque, C. A time-of-drug addition approach to target identification of antiviral compounds. Nat. Protoc. 2011, 6, 925–933. [Google Scholar] [CrossRef] [PubMed]
- Mah, L.J.; El-Osta, A.; Karagiannis, T.C. γH2AX: A sensitive molecular marker of DNA damage and repair. Leukemia 2010, 24, 679–686. [Google Scholar] [CrossRef]
- Daniel, R.; Ramcharan, J.; Rogakou, E.; Taganov, K.D.; Greger, J.G.; Bonner, W.; Nussenzweig, A.; Katz, R.A.; Skalka, A.M. Histone H2AX is phosphorylated at sites of retroviral DNA integration but is dispensable for postintegration repair. J. Biol. Chem. 2004, 279, 45810–45814. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Yu, Y.; Hamrick, H.E.; Duerksen-Hughes, P.J. ATM, ATR and DNA-PK: Initiators of the cellular genotoxic stress responses. Carcinogenesis 2003, 24, 1571–1580. [Google Scholar] [CrossRef] [Green Version]
- Ryan, E.L.; Hollingworth, R.; Grand, R.J. Activation of the DNA Damage Response by RNA Viruses. Biomolecules 2016, 6, 2. [Google Scholar] [CrossRef]
- Yoder, K.E.; Bushman, F.D. Repair of gaps in retroviral DNA integration intermediates. J. Virol. 2000, 74, 11191–11200. [Google Scholar] [CrossRef] [PubMed]
- Yoder, K.E.; Espeseth, A.; Wang, X.h.; Fang, Q.; Russo, M.T.; Lloyd, R.S.; Hazuda, D.; Sobol, R.W.; Fishel, R. The Base Excision Repair Pathway Is Required for Efficient Lentivirus Integration. PLoS ONE 2011, 6, e17862. [Google Scholar] [CrossRef]
- Bennett, G.R.; Peters, R.; Wang, X.H.; Hanne, J.; Sobol, R.W.; Bundschuh, R.; Fishel, R.; Yoder, K.E. Repair of oxidative DNA base damage in the host genome influences the HIV integration site sequence preference. PLoS ONE 2014, 9, e103164. [Google Scholar] [CrossRef]
- Fu, S.; Phan, A.T.; Mao, D.; Wang, X.; Gao, G.; Goff, S.P.; Zhu, Y. HIV-1 exploits the Fanconi anemia pathway for viral DNA integration. Cell Rep. 2022, 39, 110840. [Google Scholar] [CrossRef]
- Cooper, A.; García, M.; Petrovas, C.; Yamamoto, T.; Koup, R.A.; Nabel, G.J. HIV-1 causes CD4 cell death through DNA-dependent protein kinase during viral integration. Nature 2013, 498, 376–379. [Google Scholar] [CrossRef] [PubMed]
- Smith, J.A.; Wang, F.X.; Zhang, H.; Wu, K.J.; Williams, K.J.; Daniel, R. Evidence that the Nijmegen breakage syndrome protein, an early sensor of double-strand DNA breaks (DSB), is involved in HIV-1 post-integration repair by recruiting the ataxia telangiectasia-mutated kinase in a process similar to, but distinct from, cellul. Virol. J. 2008, 5, 11. [Google Scholar] [CrossRef]
- Mazurov, D.; Ilinskaya, A.; Heidecker, G.; Lloyd, P.; Derse, D. Quantitative Comparison of HTLV-1 and HIV-1 Cell-to-Cell Infection with New Replication Dependent Vectors. PLoS Pathog. 2010, 6, e1000788. [Google Scholar] [CrossRef] [PubMed]
- Yung, E.; Sorin, M.; Pal, A.; Craig, E.; Morozov, A.; Delattre, O.; Kappes, J.; Ott, D.; Kalpana, G.V. Inhibition of HIV-1 virion production by a transdominant mutant of integrase interactor 1. Nat. Med. 2001, 7, 920–926. [Google Scholar] [CrossRef]
- Vandergeeten, C.; Fromentin, R.; Merlini, E.; Lawani, M.B.; DaFonseca, S.; Bakeman, W.; McNulty, A.; Ramgopal, M.; Michael, N.; Kim, J.H.; et al. Cross-clade ultrasensitive PCR-based assays to measure HIV persistence in large-cohort studies. J. Virol. 2014, 88, 12385–12396. [Google Scholar] [CrossRef] [Green Version]






Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Anisenko, A.; Nefedova, A.; Agapkina, Y.; Gottikh, M. Both ATM and DNA-PK Are the Main Regulators of HIV-1 Post-Integrational DNA Repair. Int. J. Mol. Sci. 2023, 24, 2797. https://doi.org/10.3390/ijms24032797
Anisenko A, Nefedova A, Agapkina Y, Gottikh M. Both ATM and DNA-PK Are the Main Regulators of HIV-1 Post-Integrational DNA Repair. International Journal of Molecular Sciences. 2023; 24(3):2797. https://doi.org/10.3390/ijms24032797
Chicago/Turabian StyleAnisenko, Andrey, Anastasiia Nefedova, Yulia Agapkina, and Marina Gottikh. 2023. "Both ATM and DNA-PK Are the Main Regulators of HIV-1 Post-Integrational DNA Repair" International Journal of Molecular Sciences 24, no. 3: 2797. https://doi.org/10.3390/ijms24032797