
Genetic Variant Overlap Analysis Identifies Established and Putative Genes Involved in Pulmonary Fibrosis
 ,
,
Abstract
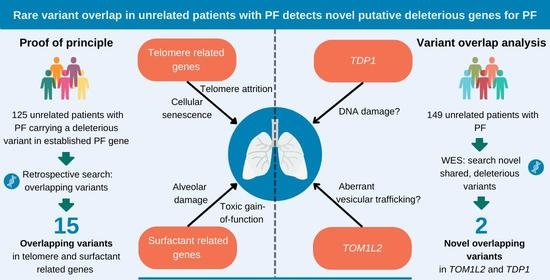
:
1. Introduction
2. Results
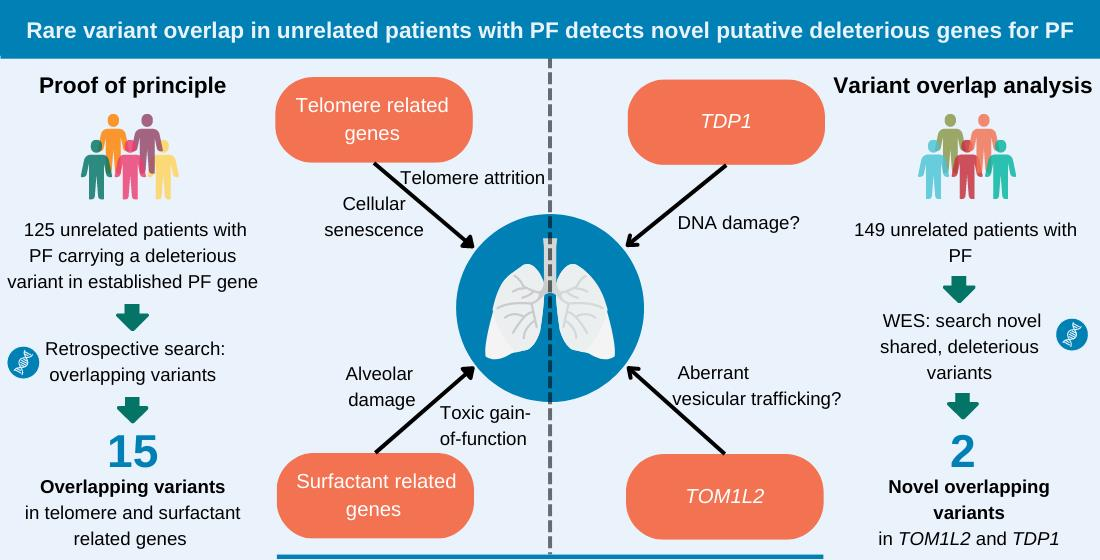
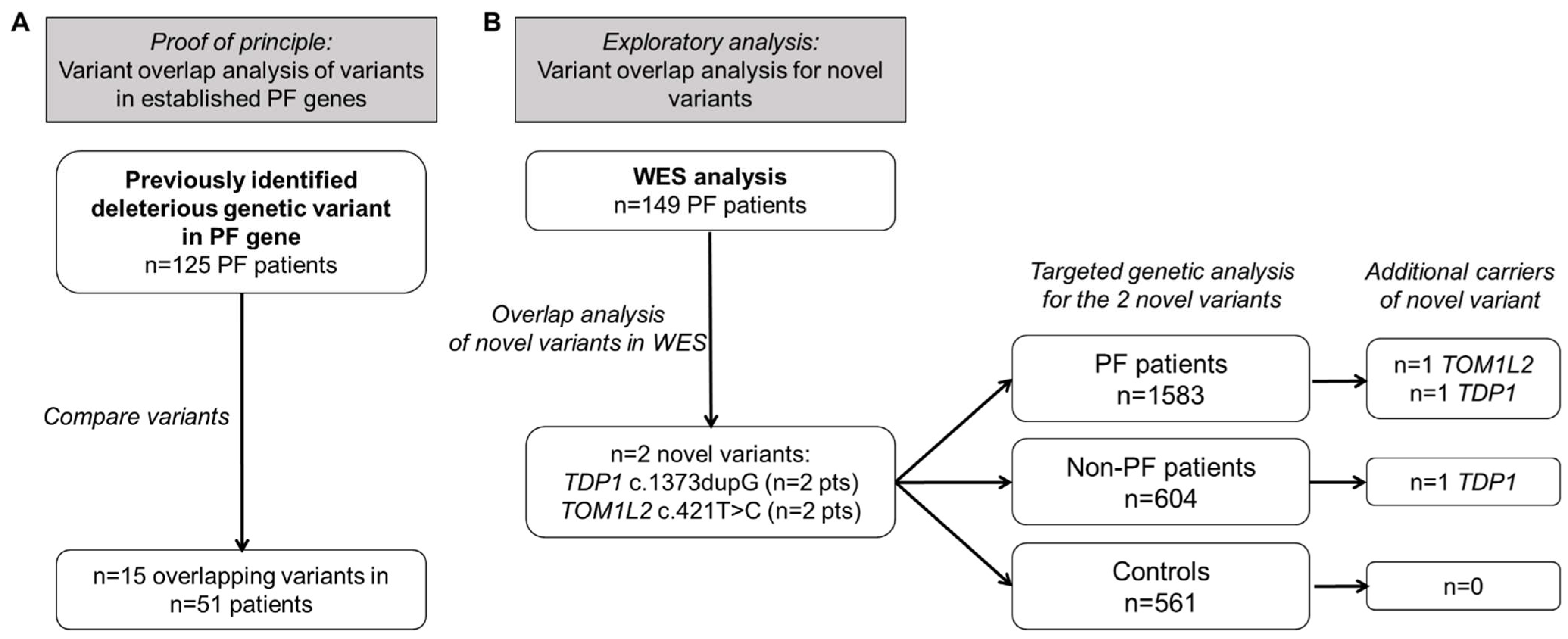
2.1. Overlap of Variants in Established PF Genes
2.2. Variant Overlap Analysis to Identify Novel Variants in PF
2.3. Novel Variant Patient Characteristics
2.4. TOM1L2 Presence in Lung Cells
2.5. TDP1 Expression in the Lung
2.6. Telomere Length
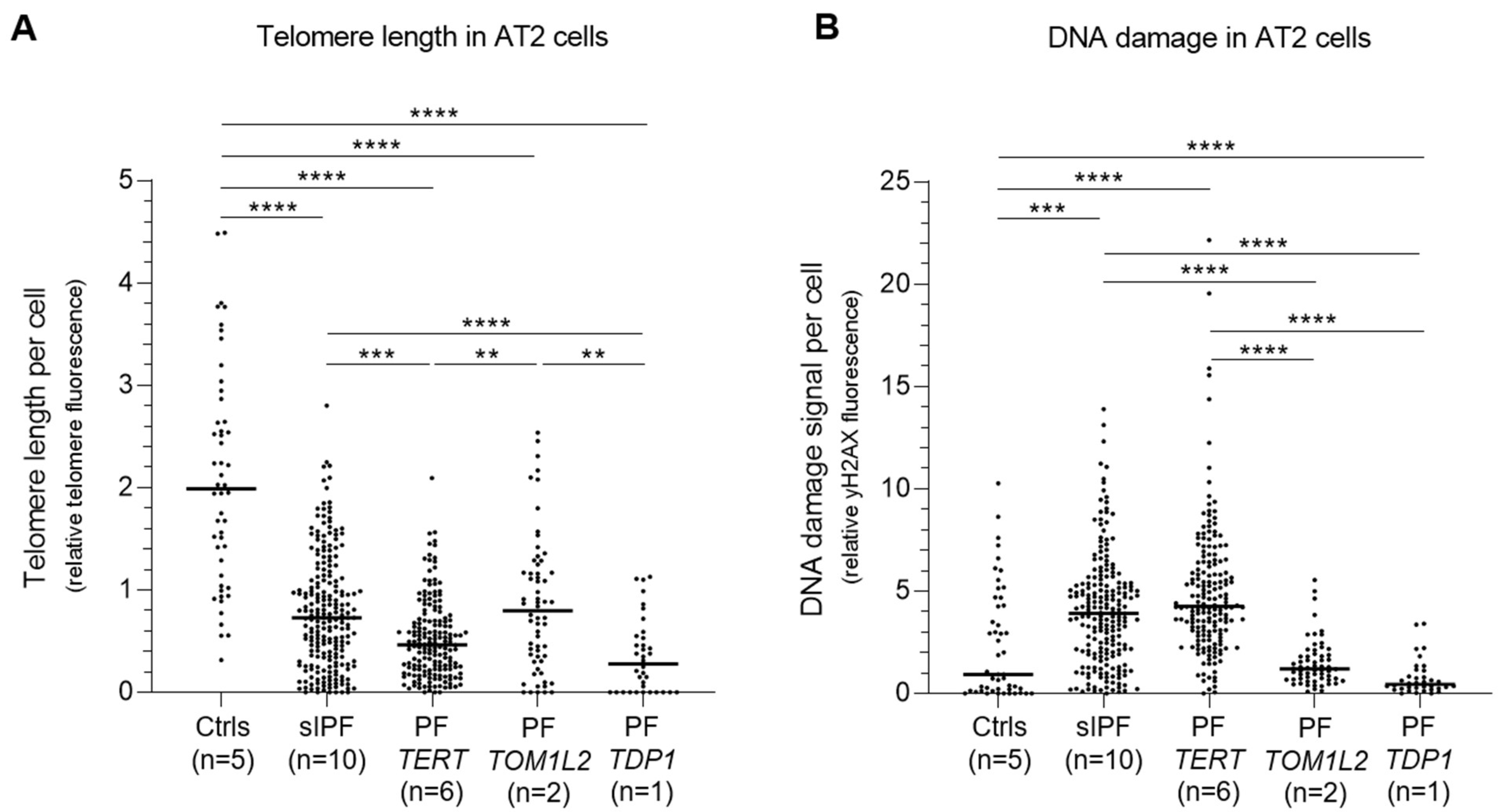
2.7. Telomere Length and DNA Damage in AT2 Cells of TOM1L2 and TDP1 Variant Carriers
3. Discussion
4. Methods and Materials
4.1. Patients
4.2. Samples
4.3. Whole Exome Sequencing
4.4. Variant Selection
4.5. Targeted Genetic Analysis
4.6. Immunofluorescent Staining of TOM1L2 and TDP1 in FFPE Diagnostic Lung Biopsies
4.7. Measurement of Average Telomere Length in Tissue and Blood Using MMqPCR
4.8. Quantification of TOM1L2 Expression with qPCR
4.9. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Martinez, F.J.; Collard, H.R.; Pardo, A.; Raghu, G.; Richeldi, L.; Selman, M.; Swigris, J.J.; Taniguchi, H.; Wells, A.U. Idiopathic Pulmonary Fibrosis. Nat. Rev. Dis. Prim. 2017, 3, 17074. [Google Scholar] [CrossRef] [PubMed]
- van Moorsel, C.H.M.; van der Vis, J.J.; Grutters, J.C. Genetic Disorders of the Surfactant System: Focus on Adult Disease. Eur. Respir. Rev. 2021, 30, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Borie, R.; Kannengiesser, C.; Sicre de Fontbrune, F.; Gouya, L.; Nathan, N.; Crestani, B. Management of Suspected Monogenic Lung Fibrosis in a Specialised Centre. Eur. Respir. Rev. 2017, 26, 160122. [Google Scholar] [CrossRef] [PubMed]
- Van Batenburg, A.A.; Kazemier, K.M.; Van Oosterhout, M.F.M.; Van Der Vis, J.J.; Van Es, H.W.; Grutters, J.C.; Goldschmeding, R.; Van Moorsel, C.H.M. From Organ to Cell: Multi-Level Telomere Length Assessment in Patients with Idiopathic Pulmonary Fibrosis. PLoS ONE 2020, 15, e0226785. [Google Scholar] [CrossRef]
- Newton, C.A.; Batra, K.; Torrealba, J.; Kozlitina, J.; Glazer, C.S.; Aravena, C.; Meyer, K.; Raghu, G.; Collard, H.R.; Garcia, C.K. Telomere-Related Lung Fibrosis Is Diagnostically Heterogeneous but Uniformly Progressive. Eur. Respir. J. 2016, 48, 1710–1720. [Google Scholar] [CrossRef] [PubMed]
- van Batenburg, A.A.; Kazemier, K.M.; van Oosterhout, M.F.M.; van der Vis, J.J.; Grutters, J.C.; Goldschmeding, R.; van Moorsel, C.H.M. Telomere Shortening and DNA Damage in Culprit Cells of Different Types of Progressive Fibrosing Interstitial Lung Disease. ERJ Open Res. 2021, 7, 00691–02020. [Google Scholar] [CrossRef] [PubMed]
- Hoffman, T.W.; Van Moorsel, C.H.M.; Borie, R.; Crestani, B. Pulmonary Phenotypes Associated with Genetic Variation in Telomere-Related Genes. Curr. Opin. Pulm. Med. 2018, 24, 269–280. [Google Scholar] [CrossRef]
- Nogee, L.M.; Dunbar, A.E.; Wert, S.E.; Askin, F.; Hamvas, A.; Whitsett, J.A. A Mutation in the Surfactant Protein C Gene Associated with Familial Interstitial Lung Disease. N. Engl. J. Med. 2001, 344, 573–579. [Google Scholar] [CrossRef]
- Armanios, M.Y.; Chen, J.J.-L.; Cogan, J.D.; Alder, J.K.; Ingersoll, R.G.; Markin, C.; Lawson, W.E.; Xie, M.; Vulto, I.; Phillips, J.A.; et al. Telomerase Mutations in Families with Idiopathic Pulmonary Fibrosis. N. Engl. J. Med. 2007, 356, 1317–1326. [Google Scholar] [CrossRef]
- Wang, Y.; Kuan, P.J.; Xing, C.; Cronkhite, J.T.; Torres, F.; Rosenblatt, R.L.; DiMaio, J.M.; Kinch, L.N.; Grishin, N.V.; Garcia, C.K. Genetic Defects in Surfactant Protein A2 Are Associated with Pulmonary Fibrosis and Lung Cancer. Am. J. Hum. Genet. 2009, 84, 52–59. [Google Scholar] [CrossRef] [Green Version]
- Cogan, J.D.; Kropski, J.A.; Zhao, M.; Mitchell, D.B.; Rives, L.; Markin, C.; Garnett, E.T.; Montgomery, K.H.; Mason, W.R.; McKean, D.F.; et al. Rare Variants in RTEL1 Are Associated with Familial Interstitial Pneumonia. Am. J. Respir. Crit. Care Med. 2015, 191, 646–655. [Google Scholar] [CrossRef]
- Gilissen, C.; Hoischen, A.; Brunner, H.G.; Veltman, J.A. Disease Gene Identification Strategies for Exome Sequencing. Eur. J. Hum. Genet. 2012, 20, 490–497. [Google Scholar] [CrossRef]
- Stuart, B.D.; Choi, J.; Zaidi, S.; Xing, C.; Holohan, B.; Chen, R.; Choi, M.; Dharwadkar, P.; Torres, F.; Girod, C.E.; et al. Exome Sequencing Links Mutations in PARN and RTEL1 with Familial Pulmonary Fibrosis and Telomere Shortening. Nat. Genet. 2015, 47, 512–517. [Google Scholar] [CrossRef]
- Hong, D.; Dai, D.; Liu, J.; Zhang, C.; Jin, T.; Shi, Y.; Jiang, G.; Mei, M.; Wang, L.; Qian, L. Clinical and Genetic Spectrum of Interstitial Lung Disease in Chinese Children Associated with Surfactant Protein C Mutations. Ital. J. Pediatr. 2019, 45, 117. [Google Scholar] [CrossRef]
- Guillot, L.; Epaud, R.; Thouvenin, G.; Jonard, L.; Mohsni, A.; Couderc, R.; Counil, F.; de Blic, J.; Taam, R.A.; Le Bourgeois, M.; et al. New Surfactant Protein C Gene Mutations Associated with Diffuse Lung Disease. J. Med. Genet. 2009, 46, 490–494. [Google Scholar] [CrossRef]
- Tredano, M.; Griese, M.; Brasch, F.; Schumacher, S.; de Blic, J.; Marque, S.; Houdayer, C.; Elion, J.; Couderc, R.; Bahuau, M. Mutation of SFTPC in Infantile Pulmonary Alveolar Proteinosis with or without Fibrosing Lung Disease. Am. J. Med. Genet. 2004, 126A, 18–26. [Google Scholar] [CrossRef]
- Coghlan, M.A.; Shifren, A.; Huang, H.J.; Russell, T.D.; Mitra, R.D.; Zhang, Q.; Wegner, D.J.; Cole, F.S.; Hamvas, A. Sequencing of Idiopathic Pulmonary Fibrosis-Related Genes Reveals Independent Single Gene Associations. BMJ Open Respir. Res. 2014, 1, e000057. [Google Scholar] [CrossRef]
- Cameron, H.S.; Somaschini, M.; Carrera, P.; Hamvas, A.; Whitsett, J.A.; Wert, S.E.; Deutsch, G.; Nogee, L.M. A Common Mutation in the Surfactant Protein C Gene Associated with Lung Disease. J. Pediatr. 2005, 146, 370–375. [Google Scholar] [CrossRef]
- van der Vis, J.J.; van der Smagt, J.J.; Hennekam, F.A.M.; Grutters, J.C.; van Moorsel, C.H.M. Pulmonary Fibrosis and a TERT Founder Mutation With a Latency Period of 300 Years. Chest 2020, 158, 612–619. [Google Scholar] [CrossRef]
- Kelich, J.; Aramburu, T.; van der Vis, J.J.; Showe, L.; Kossenkov, A.; van der Smagt, J.; Massink, M.; Schoemaker, A.; Hennekam, E.; Veltkamp, M.; et al. Telomere Dysfunction Implicates POT1 in Patients with Idiopathic Pulmonary Fibrosis. J. Exp. Med. 2022, 219, e2021168. [Google Scholar] [CrossRef]
- van Batenburg, A.A.; Kazemier, K.M.; Peeters, T.; van Oosterhout, M.F.M.; van der Vis, J.J.; Grutters, J.C.; Goldschmeding, R.; van Moorsel, C.H.M. Cell Type–Specific Quantification of Telomere Length and DNA Double-Strand Breaks in Individual Lung Cells by Fluorescence In Situ Hybridization and Fluorescent Immunohistochemistry. J. Histochem. Cytochem. 2018, 66, 485–495. [Google Scholar] [CrossRef] [PubMed]
- Justet, A.; Klay, D.; Porcher, R.; Cottin, V.; Ahmad, K.; Molina, M.M.; Nunes, H.; Reynaud-Gaubert, M.; Naccache, J.M.; Manali, E.; et al. Safety and Efficacy of Pirfenidone and Nintedanib in Patients with Idiopathic Pulmonary Fibrosis and Carrying a Telomere-Related Gene Mutation. Eur. Respir. J. 2021, 57, 4–7. [Google Scholar] [CrossRef] [PubMed]
- Dressen, A.; Abbas, A.R.; Cabanski, C.; Reeder, J.; Ramalingam, T.R.; Neighbors, M.; Bhangale, T.R.; Brauer, M.J.; Hunkapiller, J.; Reeder, J.; et al. Analysis of Protein-Altering Variants in Telomerase Genes and Their Association with MUC5B Common Variant Status in Patients with Idiopathic Pulmonary Fibrosis: A Candidate Gene Sequencing Study. Lancet Respir. Med. 2018, 6, 603–614. [Google Scholar] [CrossRef] [PubMed]
- Moriya, K.; Niizuma, H.; Rikiishi, T.; Yamaguchi, H.; Sasahara, Y.; Kure, S. Novel Compound Heterozygous RTEL1 Gene Mutations in a Patient With Hoyeraal-Hreidarsson Syndrome. Pediatr. Blood Cancer 2016, 63, 1683–1684. [Google Scholar] [CrossRef]
- Borie, R.; Bouvry, D.; Cottin, V.; Gauvain, C.; Cazes, A.; Debray, M.-P.; Cadranel, J.; Dieude, P.; Degot, T.; Dominique, S.; et al. Regulator of Telomere Length 1 (RTEL1) Mutations Are Associated with Heterogeneous Pulmonary and Extra-Pulmonary Phenotypes. Eur. Respir. J. 2019, 53, 1800508. [Google Scholar] [CrossRef]
- van Moorsel, C.H.M.; ten Klooster, L.; van Oosterhout, M.F.M.; de Jong, P.A.; Adams, H.; Wouter van Es, H.; Ruven, H.J.T.; van der Vis, J.J.; Grutters, J.C. SFTPA2 Mutations in Familial and Sporadic Idiopathic Interstitial Pneumonia. Am. J. Respir. Crit. Care Med. 2015, 192, 1249–1252. [Google Scholar] [CrossRef]
- Nogee, L.M.; Dunbar, A.E.; Wert, S.; Askin, F.; Hamvas, A.; Whitsett, J.A. Mutations in the Surfactant Protein C Gene Associated With Interstitial Lung Disease. Chest 2002, 121, 20S–21S. [Google Scholar] [CrossRef]
- Van Moorsel, C.H.M.; Van Oosterhout, M.F.M.; Barlo, N.P.; De Jong, P.A.; Van Der Vis, J.J.; Ruven, H.J.T.; Van Es, H.W.; Van Den Bosch, J.M.M.; Grutters, J.C. Surfactant Protein C Mutations Are the Basis of a Significant Portion of Adult Familial Pulmonary Fibrosis in a Dutch Cohort. Am. J. Respir. Crit. Care Med. 2010, 182, 1419–1425. [Google Scholar] [CrossRef]
- Tsakiri, K.D.; Cronkhite, J.T.; Kuan, P.J.; Xing, C.; Raghu, G.; Weissler, J.C.; Rosenblatt, R.L.; Shay, J.W.; Garcia, C.K. Adult-Onset Pulmonary Fibrosis Caused by Mutations in Telomerase. Proc. Natl. Acad. Sci. 2007, 104, 7552–7557. [Google Scholar] [CrossRef]
- Diaz de Leon, A.; Cronkhite, J.T.; Katzenstein, A.L.A.; Godwin, J.D.; Raghu, G.; Glazer, C.S.; Rosenblatt, R.L.; Girod, C.E.; Garrity, E.R.; Xing, C.; et al. Telomere Lengths, Pulmonary Fibrosis and Telomerase (TERT) Mutations. PLoS ONE 2010, 5, e10680. [Google Scholar] [CrossRef] [Green Version]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and Guidelines for the Interpretation of Sequence Variants: A Joint Consensus Recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–423. [Google Scholar] [CrossRef]
- Kumar, P.; Henikoff, S.; Ng, P.C. Predicting the Effects of Coding Non-Synonymous Variants on Protein Function Using the SIFT Algorithm. Nat. Protoc. 2009, 4, 1073–1081. [Google Scholar] [CrossRef]
- Adzhubei, I.; Jordan, D.M.; Sunyaev, S.R. Predicting Functional Effect of Human Missense Mutations Using PolyPhen-2. Curr. Protoc. Hum. Genet. 2013, 76, 7–20. [Google Scholar] [CrossRef]
- Steinhaus, R.; Proft, S.; Schuelke, M.; Cooper, D.N.; Schwarz, J.M.; Seelow, D. MutationTaster2021. Nucleic Acids Res. 2021, 49, W446–W451. [Google Scholar] [CrossRef]
- Alder, J.K.; Cogan, J.D.; Brown, A.F.; Anderson, C.J.; Lawson, W.E.; Lansdorp, P.M.; Phillips, J.A.; Loyd, J.E.; Chen, J.J.-L.; Armanios, M. Ancestral Mutation in Telomerase Causes Defects in Repeat Addition Processivity and Manifests As Familial Pulmonary Fibrosis. PLoS Genet. 2011, 7, e1001352. [Google Scholar] [CrossRef]
- Wang, T.; Liu, N.S.; Seet, L.F.; Hong, W. The Emerging Role of VHS Domain-Containing Tom1, Tom1L1 and Tom1L2 in Membrane Trafficking. Traffic 2010, 11, 1119–1128. [Google Scholar] [CrossRef]
- Katoh, Y.; Imakagura, H.; Futatsumori, M.; Nakayama, K. Recruitment of Clathrin onto Endosomes by the Tom1-Tollip Complex. Biochem. Biophys. Res. Commun. 2006, 341, 143–149. [Google Scholar] [CrossRef]
- Noth, I.; Zhang, Y.; Ma, S.-F.; Flores, C.; Barber, M.; Huang, Y.; Broderick, S.M.; Wade, M.S.; Hysi, P.; Scuirba, J.; et al. Genetic Variants Associated with Idiopathic Pulmonary Fibrosis Susceptibility and Mortality: A Genome-Wide Association Study. Lancet Respir. Med. 2013, 1, 309–317. [Google Scholar] [CrossRef]
- Bonella, F.; Campo, I.; Zorzetto, M.; Boerner, E.; Ohshimo, S.; Theegarten, D.; Taube, C.; Costabel, U. Potential Clinical Utility of MUC5B Und TOLLIP Single Nucleotide Polymorphisms (SNPs) in the Management of Patients with IPF. Orphanet J. Rare Dis. 2021, 16, 111. [Google Scholar] [CrossRef]
- Girirajan, S.; Hauck, P.M.; Williams, S.; Vlangos, C.N.; Szomju, B.B.; Solaymani-Kohal, S.; Mosier, P.D.; White, K.L.; McCoy, K.; Elsea, S.H. Tom1l2 Hypomorphic Mice Exhibit Increased Incidence of Infections and Tumors and Abnormal Immunologic Response. Mamm. Genome 2008, 19, 246–262. [Google Scholar] [CrossRef]
- Franco, M.; Furstoss, O.; Simon, V.; Benistant, C.; Hong, W.J.; Roche, S. The Adaptor Protein Tom1L1 Is a Negative Regulator of Src Mitogenic Signaling Induced by Growth Factors. Mol. Cell. Biol. 2006, 26, 1932–1947. [Google Scholar] [CrossRef] [PubMed]
- Tumbarello, D.A.; Waxse, B.J.; Arden, S.D.; Bright, N.A.; Kendrick-Jones, J.; Buss, F. Autophagy Receptors Link Myosin VI to Autophagosomes to Mediate Tom1-Dependent Autophagosome Maturation and Fusion with the Lysosome. Nat. Cell Biol. 2012, 14, 1024–1035. [Google Scholar] [CrossRef] [PubMed]
- Nathan, J.A.; Tae Kim, H.; Ting, L.; Gygi, S.P.; Goldberg, A.L. Why Do Cellular Proteins Linked to K63-Polyubiquitin Chains Not Associate with Proteasomes? EMBO J. 2013, 32, 552–565. [Google Scholar] [CrossRef] [PubMed]
- Beers, M.F.; Hawkins, A.; Maguire, J.A.; Kotorashvili, A.; Zhao, M.; Newitt, J.L.; Ding, W.; Russo, S.; Guttentag, S.; Gonzales, L.; et al. A Nonaggregating Surfactant Protein C Mutant Is Misdirected to Early Endosomes and Disrupts Phospholipid Recycling. Traffic 2011, 12, 1196–1210. [Google Scholar] [CrossRef]
- Hawkins, A.; Guttentag, S.H.; Deterding, R.; Funkhouser, W.K.; Goralski, J.L.; Chatterjee, S.; Mulugeta, S.; Beers, M.F. A Non-BRICHOS SFTPC Mutant (SP-C I73T ) Linked to Interstitial Lung Disease Promotes a Late Block in Macroautophagy Disrupting Cellular Proteostasis and Mitophagy. Am. J. Physiol. Cell. Mol. Physiol. 2014, 308, L33–L47. [Google Scholar] [CrossRef]
- Dickens, J.A.; Rutherford, E.N.; Abreu, S.; Chambers, J.E.; Ellis, M.O.; van Schadewijk, A.; Hiemstra, P.S.; Marciniak, S.J. Novel Insights into Surfactant Protein C Trafficking Revealed through the Study of a Pathogenic Mutant. Eur. Respir. J. 2022, 59. [Google Scholar] [CrossRef]
- Collaco, A.; Jakab, R.; Hegan, P.; Mooseker, M.; Ameen, N. α-AP-2 Directs Myosin VI-Dependent Endocytosis of Cystic Fibrosis Transmembrane Conductance Regulator Chloride Channels in the Intestine. J. Biol. Chem. 2010, 285, 17177–17187. [Google Scholar] [CrossRef]
- Gooptu, B. Surfactant Protein C Mutations and Familial Pulmonary Fibrosis: Stuck in a Loop on the Scenic Route. Eur. Respir. J. 2022, 59, 2102147. [Google Scholar] [CrossRef]
- Fam, H.K.; Chowdhury, M.K.; Walton, C.; Choi, K.; Boerkoel, C.F.; Hendson, G. Expression Profile and Mitochondrial Colocalization of Tdp1 in Peripheral Human Tissues. J. Mol. Histol. 2013, 44, 481–494. [Google Scholar] [CrossRef]
- Das, B.B.; Antony, S.; Gupta, S.; Dexheimer, T.S.; Redon, C.E.; Garfield, S.; Shiloh, Y.; Pommier, Y. Optimal Function of the DNA Repair Enzyme TDP1 Requires Its Phosphorylation by ATM and/or DNA-PK. EMBO J. 2009, 28, 3667–3680. [Google Scholar] [CrossRef] [Green Version]
- Borda, M.A.; Palmitelli, M.; Verón, G.; González-Cid, M.; de Campos Nebel, M. Tyrosyl-DNA-Phosphodiesterase I (TDP1) Participates in the Removal and Repair of Stabilized-Top2α Cleavage Complexes in Human Cells. Mutat. Res.-Fundam. Mol. Mech. Mutagen. 2015, 781, 37–48. [Google Scholar] [CrossRef]
- Kosmider, B.; Lin, C.R.; Karim, L.; Tomar, D.; Vlasenko, L.; Marchetti, N.; Bolla, S.; Madesh, M.; Criner, G.J.; Bahmed, K. Mitochondrial Dysfunction in Human Primary Alveolar Type II Cells in Emphysema. EBioMedicine 2019, 46, 305–316. [Google Scholar] [CrossRef]
- Borie, R.; Kannengiesser, C.; Antoniou, K.; Bonella, F.; Crestani, B.; Fabre, A.; Froidure, A.; Galvin, L.; Griese, M.; Grutters, J.C.; et al. European Respiratory Society Statement on Familial Pulmonary Fibrosis. Eur. Respir. J. 2022. [Google Scholar] [CrossRef]
- Pontén, F.; Jirström, K.; Uhlen, M. The Human Protein Atlas—A Tool for Pathology. J. Pathol. 2008, 216, 387–393. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Number of Unrelated Patients | Transcript | Nucleotide Change | Amino Acid Change | ACMG Classification | Publication * |
|---|---|---|---|---|---|---|
| PARN | 2 | NM_001134477.2 | c.1214G > C | p.(Ser405Thr) | VUS | |
| PARN | 4 | NM_001134477.2 | c.98C > T | p.(Pro33Leu) | VUS | Dressen et al. (2018); Justet et al. (2021); Van Batenburg et al. (2018) [21,22,23] |
| POT1 | 2 | NM_015450.2 | c.776T > C | p.(Leu259Ser) | LP | Kelich et al. (2022) [20] |
| RTEL1 | 3 | NM_001283009.1 | c.1972_1974del | p.(Leu658del) | LP | Justet et al. (2021) [22] |
| RTEL1 | 2 | NM_001283009.1 | c.3811C > T | p.(Arg1271Trp) | VUS | |
| RTEL1 | 5 | NM_001283009.1 | c.2956C > T | p.(Arg986 *) | P | Borie et al. (2019); Moriya et al. (2016); Van Batenburg et al. (2020) [4,24,25] |
| SFTPA2 | 2 | NM_001098668.2 | c.629A > C | p.(Asn210Thr) | LP | Van Moorsel et al. (2015) [26] |
| SFTPC | 3 | NM_003018.3 | c.218T > C | p.(Ile73Thr) | P | Nogee et al. (2002); Van Moorsel et al. (2010) and many others [27,28] |
| TERT | 2 | NM_198253.2 | c.2701C > T | p.(Arg901Trp) | P | |
| TERT | 2 | NM_198253.2 | c.1726C > T | p.(Tyr576His) | VUS | |
| TERT | 3 | NM_198253.2 | c.1584T > G | p.(Cys528Trp) | LP | Justet et al. (2021) [22] |
| TERT | 4 | NM_198253.2 | c.2312C > T | p.(Pro771Leu) | LP | |
| TERT | 2 | NM_198253.2 | c.2594G > A | p.(Arg865His) | P | Diaz de Leon et al. (2010); Justet et al. (2021); Newton et al. (2016); Tsakiri et al. (2007) [5,22,29,30] |
| TERT | 2 | NM_198253.2 | c.299G > A | p.(Gly100Asp) | VUS | Justet et al. (2021) [22] |
| TERT | 13 | NM_198253.2 | c.2005C > T | p.(Arg669Trp) | LP | Dressen et al. (2018); Justet et al. (2021); Newton et al. (2016); van der Vis et al. (2020) # [5,19,22,23] |
| Gene | Transcript | Nucleotide Change | Amino Acid Change | Translation Impact | SIFT | PolyPhen-2 | CADD | Mutation Taster |
|---|---|---|---|---|---|---|---|---|
| TDP1 | NM_018319.4 | c.1373dupG | p.(Ser459fs*5) | frameshift | - | - | 35.0 | Deleterious |
| TOM1L2 | NM_001288786.2 | c.421T > C | p.(Tyr141His) | missense | Damaging | Probably Damaging | 28.1 | Benign (deleterious for all other transcipts) |
| ID | Variant | Familial Disease | Sex | Diagnosis | Age at Diagnosis (Death) | Age LTx | HRCT Pattern | Histology (Tissue) | TL < 10th Percentile for Age |
|---|---|---|---|---|---|---|---|---|---|
| Patient 1 | TOM1L2 c.421T > C | Yes | Male | fHP + secondary UIP characteristics | 52 | 60 (b) | probable UIP | UIP characteristics (explant) | No |
| Patient 2 # | TOM1L2 c.421T > C | No | Male | IPF | 47 (55) | 51 (u) | UIP | UIP + DIP component (explant) | Yes |
| Patient 3 # | TOM1L2 c.421T > C | No | Male | NCIP | 55 (59) | NA | NSIP | UIP (VATS biopsy) | No |
| Patient 4 | TDP1 c.1373dupG | Yes | Male | IPF | 62 (63) | NA | UIP with DAD | UIP (VATS biopsy) | Yes |
| Patient 5 # | TDP1 c.1373dupG | No | Male | IPF | 50 (61) | 52 (b) | UIP, paraseptal emphysema | UIP (explant) | No |
| Patient 6 | TDP1 c.1373dupG | No | Female | LAM | 46 | NA | multiple thin-walled cysts | NA | No |
| Patient 7 | TDP1 c.1373dupG | No | Male | pretransplant COPD/emphysema; posttransplant progressive pulmonary fibrosis in right lung with UIP characteristics | 56 (62) | 57 (u) | emphysema, UIP | emphysema, presence of fibroblast foci (explant) | Yes |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Groen, K.; van der Vis, J.J.; van Batenburg, A.A.; Kazemier, K.M.; Grutters, J.C.; van Moorsel, C.H.M. Genetic Variant Overlap Analysis Identifies Established and Putative Genes Involved in Pulmonary Fibrosis. Int. J. Mol. Sci. 2023, 24, 2790. https://doi.org/10.3390/ijms24032790
Groen K, van der Vis JJ, van Batenburg AA, Kazemier KM, Grutters JC, van Moorsel CHM. Genetic Variant Overlap Analysis Identifies Established and Putative Genes Involved in Pulmonary Fibrosis. International Journal of Molecular Sciences. 2023; 24(3):2790. https://doi.org/10.3390/ijms24032790
Chicago/Turabian StyleGroen, Karlijn, Joanne J. van der Vis, Aernoud A. van Batenburg, Karin M. Kazemier, Jan C. Grutters, and Coline H. M. van Moorsel. 2023. "Genetic Variant Overlap Analysis Identifies Established and Putative Genes Involved in Pulmonary Fibrosis" International Journal of Molecular Sciences 24, no. 3: 2790. https://doi.org/10.3390/ijms24032790