
CD4 T-Cell Subsets and the Pathophysiology of Inflammatory Bowel Disease
 , , ,
, , , 
{kind=link}
{kind=link}
Abstract
:1. Introduction
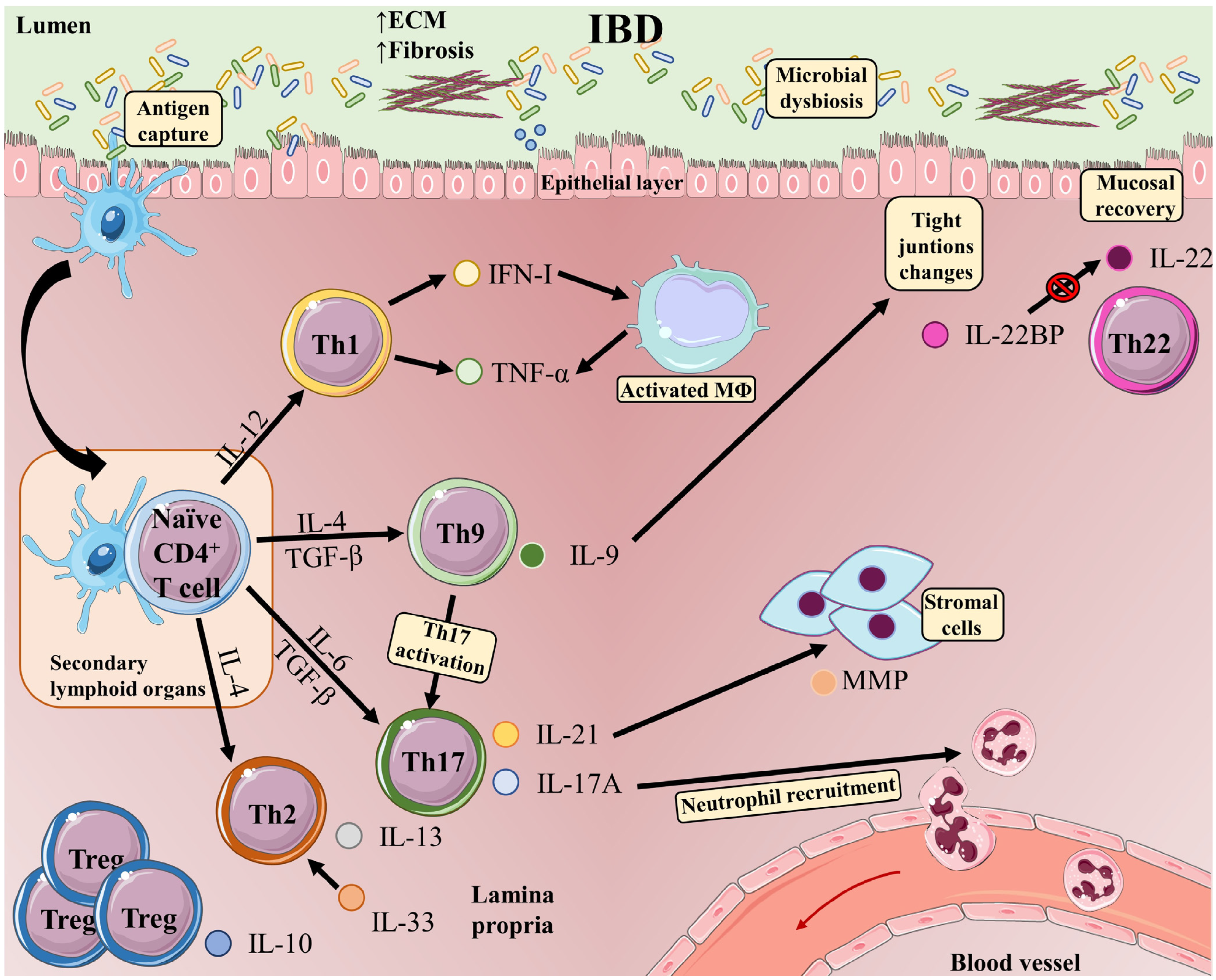
2. T-Cells in IBD
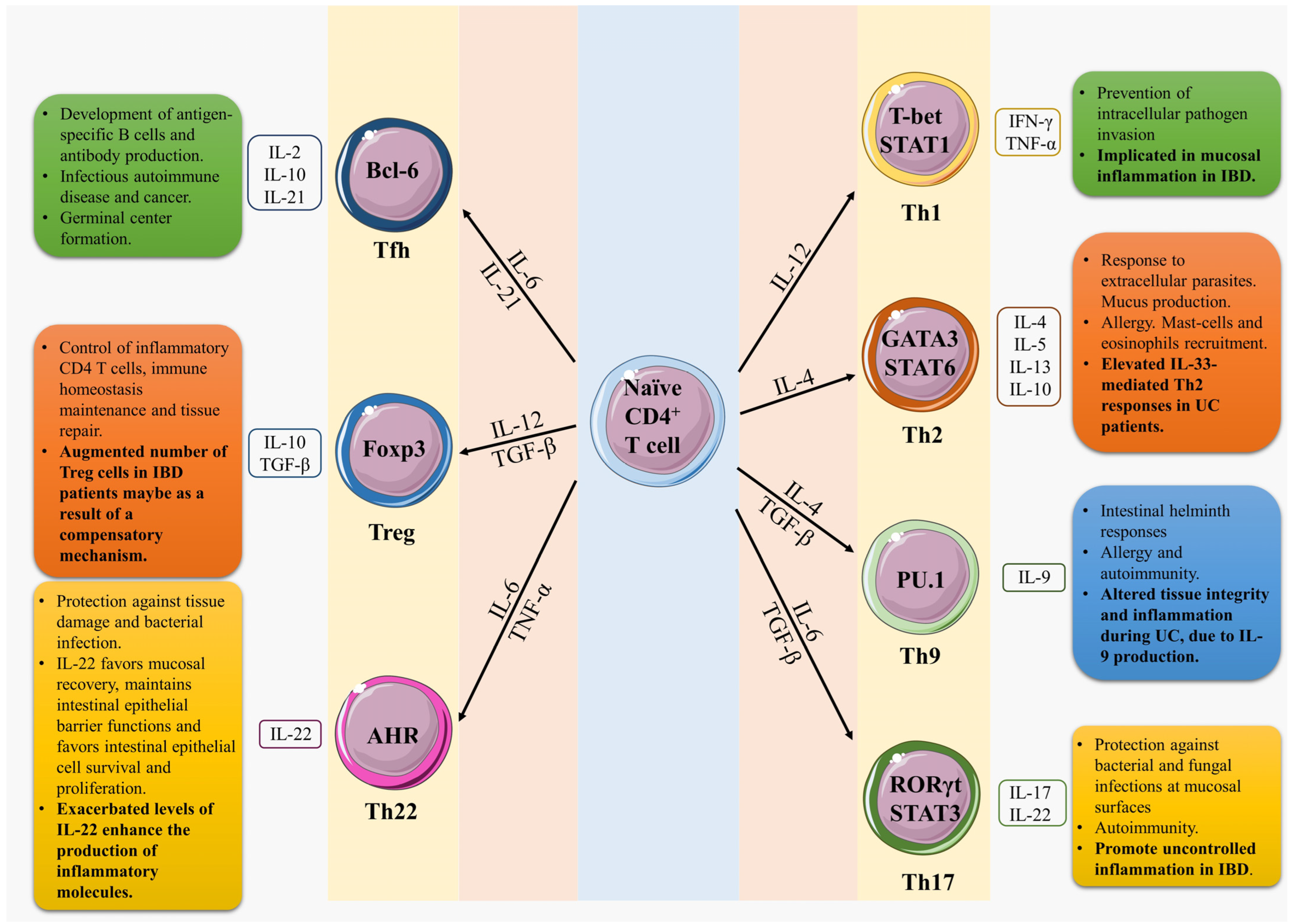
3. T Helper 1 (Th1) Cells
4. T Helper 2 (Th2) Cells
5. T Helper 9 (Th9) Cells
6. T Helper 17 (Th17) Cells
7. T Helper 22 (Th22) Cells
8. Regulatory T-Cells (Treg)
9. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| AHR | Aryl hydrocarbon receptor |
| AMP | Antimicrobial peptide |
| APC | Antigen presenting cell |
| BATF | Basic leucine zipper ATF-like transcription factor |
| CCL | Chemokine (C-C motif) ligand |
| CCR | C-C motif chemokine receptor |
| CD | Crohn’s disease |
| CD5L | CD5 molecule like |
| CTL | Cytotoxic T cells |
| CTLA-4 | Cytotoxic lymphocyte antigen 4 |
| CXCL | Chemokine (C-X-C motif) ligand |
| DAMPs | Damage-associated molecular patterns |
| DCs | Dendritic cells |
| DR | Death receptor |
| DSS | Dextran sodium sulfate |
| EpCAM | Epithelial cell adhesion molecule |
| Foxp3 | Forkhead box P3 |
| GATA-3 | GATA binding protein 3 |
| G-CSF | Granulocyte colony-stimulating factor |
| GM-CSF | Granulocyte-macrophage colony-stimulating factor |
| IBD | Inflammatory bowel disease |
| IEC | Intestinal epithelial cells |
| IFN | Interferon |
| Ig | Immunoglobulin |
| ILCs | Innate lymphoid cells |
| IL | Interleukin |
| IL-12Rβ2 | IL-12 receptor subunit β2 |
| IL-22BP | IL-22 binding protein |
| IRF | Interferon regulatory factor |
| ISC | Intestinal stem cell |
| JAK | Activates janus kinase |
| KLRG1 | Killer cell lectin like receptor G1 |
| KO | Knockout |
| MHC | Major histocompatibility complex |
| MMPs | Matrix metalloproteinases |
| NK | Natural killer |
| NKG2D | Natural killer group 2D receptor |
| NLR | Nod-like receptors |
| PAMPs | Pathogen-associated molecular patterns |
| PD-1 | Programmed cell death 1 |
| PRRs | Pattern recognition receptors |
| RAG | Recombination activating gene |
| RORγt | Retinoic acid receptor-related orphan receptor gamma t |
| scRNA-seq | Single cell RNA sequencing |
| STAT | Signal transducer and activator of transcription |
| T-bet | T-box-containing protein |
| TCR | T cell antigen receptor |
| Tfh | T follicular helper |
| TGF | Transforming growth factor |
| Th | T helper |
| TIGIT | T cell immunoreceptor with immunoglobulin and immunoreceptor tyrosine-based inhibition motif domains |
| TL | TNF-like factor |
| TL1A | TNF-like ligand 1 A |
| TLRs | Toll-like receptors |
| TNBS | 2,4,6-trinitrobenzene sulfonic acid |
| TNF | Tumor necrosis factor |
| Treg | T regulatory |
| Tr1 | T regulatory type 1 |
| UC | Ulcerative colitis |
References
- Herrero-Fernandez, B.; Gomez-Bris, R.; Somovilla-Crespo, B.; Gonzalez-Granado, J.M. Immunobiology of Atherosclerosis: A Complex Net of Interactions. Int. J. Mol. Sci. 2019, 20, 5293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yao, H.; Tang, G. Macrophages in intestinal fibrosis and regression. Cell. Immunol. 2022, 381, 104614. [Google Scholar] [CrossRef] [PubMed]
- Katsandegwaza, B.; Horsnell, W.; Smith, K. Inflammatory Bowel Disease: A Review of Pre-Clinical Murine Models of Human Disease. Int. J. Mol. Sci. 2022, 23, 9344. [Google Scholar] [CrossRef] [PubMed]
- Kang, L.; Fang, X.; Song, Y.H.; He, Z.X.; Wang, Z.J.; Wang, S.L.; Li, Z.S.; Bai, Y. Neutrophil-Epithelial Crosstalk During Intestinal Inflammation. Cell. Mol. Gastroenterol. Hepatol. 2022, 14, 1257–1267. [Google Scholar] [CrossRef]
- Kaluzna, A.; Olczyk, P.; Komosinska-Vassev, K. The Role of Innate and Adaptive Immune Cells in the Pathogenesis and Development of the Inflammatory Response in Ulcerative Colitis. J. Clin. Med. 2022, 11, 400. [Google Scholar] [CrossRef]
- He, X.; Tan, S.; Shao, Z.; Wang, X. Latitudinal and longitudinal regulation of tissue macrophages in inflammatory diseases. Genes Dis. 2022, 9, 1194–1207. [Google Scholar] [CrossRef]
- Ghilas, S.; O’Keefe, R.; Mielke, L.A.; Raghu, D.; Buchert, M.; Ernst, M. Crosstalk between epithelium, myeloid and innate lymphoid cells during gut homeostasis and disease. Front. Immunol. 2022, 13, 944982. [Google Scholar] [CrossRef]
- Wardell, C.M.; MacDonald, K.N.; Levsings, M.K.; Cook, L. Cross talk between human regulatory T cells and antigen-presenting cells: Lessons for clinical applications. Eur. J. Immunol. 2021, 51, 27–38. [Google Scholar] [CrossRef]
- Schulz-Kuhnt, A.; Neurath, M.F.; Wirtz, S.; Atreya, I. Innate Lymphoid Cells as Regulators of Epithelial Integrity: Therapeutic Implications for Inflammatory Bowel Diseases. Front. Med. (Lausanne) 2021, 8, 656745. [Google Scholar] [CrossRef]
- Saez, A.; Gomez-Bris, R.; Herrero-Fernandez, B.; Mingorance, C.; Rius, C.; Gonzalez-Granado, J.M. Innate Lymphoid Cells in Intestinal Homeostasis and Inflammatory Bowel Disease. Int. J. Mol. Sci. 2021, 22, 7618. [Google Scholar] [CrossRef]
- Peloquin, J.M.; Goel, G.; Villablanca, E.J.; Xavier, R.J. Mechanisms of Pediatric Inflammatory Bowel Disease. Annu. Rev. Immunol. 2016, 34, 31–64. [Google Scholar] [CrossRef]
- Liu, T.C.; Stappenbeck, T.S. Genetics and Pathogenesis of Inflammatory Bowel Disease. Annu. Rev. Pathol. 2016, 11, 127–148. [Google Scholar] [CrossRef] [Green Version]
- Torres, J.; Mehandru, S.; Colombel, J.F.; Peyrin-Biroulet, L. Crohn’s disease. Lancet 2017, 389, 1741–1755. [Google Scholar] [CrossRef]
- Ungaro, R.; Mehandru, S.; Allen, P.B.; Peyrin-Biroulet, L.; Colombel, J.F. Ulcerative colitis. Lancet 2017, 389, 1756–1770. [Google Scholar] [CrossRef]
- Kaplan, G.G.; Windsor, J.W. The four epidemiological stages in the global evolution of inflammatory bowel disease. Nat. Rev. Gastroenterol. Hepatol. 2021, 18, 56–66. [Google Scholar] [CrossRef]
- Olen, O.; Askling, J.; Sachs, M.C.; Neovius, M.; Smedby, K.E.; Ekbom, A.; Ludvigsson, J.F. Mortality in adult-onset and elderly-onset IBD: A nationwide register-based cohort study 1964–2014. Gut 2020, 69, 453–461. [Google Scholar] [CrossRef]
- Peyrin-Biroulet, L.; Panes, J.; Sandborn, W.J.; Vermeire, S.; Danese, S.; Feagan, B.G.; Colombel, J.F.; Hanauer, S.B.; Rycroft, B. Defining Disease Severity in Inflammatory Bowel Diseases: Current and Future Directions. Clin. Gastroenterol. Hepatol. 2016, 14, 348–354.e17. [Google Scholar] [CrossRef] [Green Version]
- Pabla, B.S.; Schwartz, D.A. Assessing Severity of Disease in Patients with Ulcerative Colitis. Gastroenterol. Clin. N. Am. 2020, 49, 671–688. [Google Scholar] [CrossRef]
- Cleynen, I.; Gonzalez, J.R.; Figueroa, C.; Franke, A.; McGovern, D.; Bortlik, M.; Crusius, B.J.; Vecchi, M.; Artieda, M.; Szczypiorska, M.; et al. Genetic factors conferring an increased susceptibility to develop Crohn’s disease also influence disease phenotype: Results from the IBDchip European Project. Gut 2013, 62, 1556–1565. [Google Scholar] [CrossRef] [Green Version]
- Satsangi, J.; Silverberg, M.S.; Vermeire, S.; Colombel, J.F. The Montreal classification of inflammatory bowel disease: Controversies, consensus, and implications. Gut 2006, 55, 749–753. [Google Scholar] [CrossRef]
- Louis, E.; Van Kemseke, C.; Reenaers, C. Necessity of phenotypic classification of inflammatory bowel disease. Best Pract. Res. Clin. Gastroenterol. 2011, 25 (Suppl. 1), S2–S7. [Google Scholar] [CrossRef] [PubMed]
- Levine, A.; Griffiths, A.; Markowitz, J.; Wilson, D.C.; Turner, D.; Russell, R.K.; Fell, J.; Ruemmele, F.M.; Walters, T.; Sherlock, M.; et al. Pediatric modification of the Montreal classification for inflammatory bowel disease: The Paris classification. Inflamm. Bowel Dis. 2011, 17, 1314–1321. [Google Scholar] [CrossRef] [PubMed]
- Gecse, K.B.; Vermeire, S. Differential diagnosis of inflammatory bowel disease: Imitations and complications. Lancet Gastroenterol. Hepatol. 2018, 3, 644–653. [Google Scholar] [CrossRef]
- de Souza, H.S.P.; Fiocchi, C.; Iliopoulos, D. The IBD interactome: An integrated view of aetiology, pathogenesis and therapy. Nat. Rev. Gastroenterol. Hepatol. 2017, 14, 739–749. [Google Scholar] [CrossRef] [PubMed]
- Choy, M.C.; Visvanathan, K.; De Cruz, P. An Overview of the Innate and Adaptive Immune System in Inflammatory Bowel Disease. Inflamm. Bowel Dis. 2017, 23, 2–13. [Google Scholar] [CrossRef]
- Tindemans, I.; Joosse, M.E.; Samsom, J.N. Dissecting the Heterogeneity in T-Cell Mediated Inflammation in IBD. Cells 2020, 9, 110. [Google Scholar] [CrossRef] [Green Version]
- Giuffrida, P.; Di Sabatino, A. Targeting T cells in inflammatory bowel disease. Pharmacol. Res. 2020, 159, 105040. [Google Scholar] [CrossRef]
- Zenewicz, L.A.; Antov, A.; Flavell, R.A. CD4 T-cell differentiation and inflammatory bowel disease. Trends Mol. Med. 2009, 15, 199–207. [Google Scholar] [CrossRef]
- Friedrich, M.; Pohin, M.; Powrie, F. Cytokine Networks in the Pathophysiology of Inflammatory Bowel Disease. Immunity 2019, 50, 992–1006. [Google Scholar] [CrossRef] [Green Version]
- Cao, H.; Diao, J.; Liu, H.; Liu, S.; Liu, J.; Yuan, J.; Lin, J. The Pathogenicity and Synergistic Action of Th1 and Th17 Cells in Inflammatory Bowel Diseases. Inflamm. Bowel Dis. 2022, izac199. [Google Scholar] [CrossRef]
- Zhang, N.; Bevan, M.J. CD8(+) T cells: Foot soldiers of the immune system. Immunity 2011, 35, 161–168. [Google Scholar] [CrossRef] [Green Version]
- Sakaguchi, S.; Miyara, M.; Costantino, C.M.; Hafler, D.A. FOXP3+ regulatory T cells in the human immune system. Nat. Rev. Immunol. 2010, 10, 490–500. [Google Scholar] [CrossRef]
- Elahi, S.; Horton, H. Association of HLA-alleles with the immune regulation of chronic viral infections. Int. J. Biochem. Cell Biol. 2012, 44, 1361–1365. [Google Scholar] [CrossRef] [Green Version]
- Morel, P.A. Differential T-cell receptor signals for T helper cell programming. Immunology 2018, 155, 63–71. [Google Scholar] [CrossRef] [Green Version]
- Lee, G.R.; Kim, S.T.; Spilianakis, C.G.; Fields, P.E.; Flavell, R.A. T helper cell differentiation: Regulation by cis elements and epigenetics. Immunity 2006, 24, 369–379. [Google Scholar] [CrossRef] [Green Version]
- Bustos-Moran, E.; Blas-Rus, N.; Martin-Cofreces, N.B.; Sanchez-Madrid, F. Orchestrating Lymphocyte Polarity in Cognate Immune Cell-Cell Interactions. Int. Rev. Cell Mol. Biol. 2016, 327, 195–261. [Google Scholar]
- Hirahara, K.; Nakayama, T. CD4+ T-cell subsets in inflammatory diseases: Beyond the Th1/Th2 paradigm. Int. Immunol. 2016, 28, 163–171. [Google Scholar] [CrossRef] [Green Version]
- DuPage, M.; Bluestone, J.A. Harnessing the plasticity of CD4(+) T cells to treat immune-mediated disease. Nat. Rev. Immunol. 2016, 16, 149–163. [Google Scholar] [CrossRef] [Green Version]
- Li, P.; Spolski, R.; Liao, W.; Leonard, W.J. Complex interactions of transcription factors in mediating cytokine biology in T cells. Immunol. Rev. 2014, 261, 141–156. [Google Scholar] [CrossRef] [Green Version]
- Mosmann, T.R.; Cherwinski, H.; Bond, M.W.; Giedlin, M.A.; Coffman, R.L. Two types of murine helper T cell clone. I. Definition according to profiles of lymphokine activities and secreted proteins. J. Immunol. 1986, 136, 2348–2357. [Google Scholar] [CrossRef]
- Allaire, J.M.; Crowley, S.M.; Law, H.T.; Chang, S.Y.; Ko, H.J.; Vallance, B.A. The Intestinal Epithelium: Central Coordinator of Mucosal Immunity. Trends Immunol. 2018, 39, 677–696. [Google Scholar] [CrossRef] [PubMed]
- Basso, L.; Boue, J.; Auge, C.; Deraison, C.; Blanpied, C.; Cenac, N.; Lluel, P.; Vergnolle, N.; Dietrich, G. Mobilization of CD4+ T lymphocytes in inflamed mucosa reduces pain in colitis mice: Toward a vaccinal strategy to alleviate inflammatory visceral pain. Pain 2018, 159, 331–341. [Google Scholar] [CrossRef] [PubMed]
- Hirata, I.; Berrebi, G.; Austin, L.L.; Keren, D.F.; Dobbins, W.O., 3rd. Immunohistological characterization of intraepithelial and lamina propria lymphocytes in control ileum and colon and in inflammatory bowel disease. Dig. Dis. Sci. 1986, 31, 593–603. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Selby, W.S.; Janossy, G.; Bofill, M.; Jewell, D.P. Intestinal lymphocyte subpopulations in inflammatory bowel disease: An analysis by immunohistological and cell isolation techniques. Gut 1984, 25, 32–40. [Google Scholar] [CrossRef] [Green Version]
- Schreiber, S.; MacDermott, R.P.; Raedler, A.; Pinnau, R.; Bertovich, M.J.; Nash, G.S. Increased activation of isolated intestinal lamina propria mononuclear cells in inflammatory bowel disease. Gastroenterology 1991, 101, 1020–1030. [Google Scholar] [CrossRef]
- Rabe, H.; Malmquist, M.; Barkman, C.; Ostman, S.; Gjertsson, I.; Saalman, R.; Wold, A.E. Distinct patterns of naive, activated and memory T and B cells in blood of patients with ulcerative colitis or Crohn’s disease. Clin. Exp. Immunol. 2019, 197, 111–129. [Google Scholar] [CrossRef] [Green Version]
- Muller, S.; Lory, J.; Corazza, N.; Griffiths, G.M.; Z’Graggen, K.; Mazzucchelli, L.; Kappeler, A.; Mueller, C. Activated CD4+ and CD8+ cytotoxic cells are present in increased numbers in the intestinal mucosa from patients with active inflammatory bowel disease. Am. J. Pathol. 1998, 152, 261–268. [Google Scholar]
- Corridoni, D.; Antanaviciute, A.; Gupta, T.; Fawkner-Corbett, D.; Aulicino, A.; Jagielowicz, M.; Parikh, K.; Repapi, E.; Taylor, S.; Ishikawa, D.; et al. Single-cell atlas of colonic CD8(+) T cells in ulcerative colitis. Nat. Med. 2020, 26, 1480–1490. [Google Scholar] [CrossRef]
- Page, M.J.; Poritz, L.S.; Tilberg, A.F.; Zhang, W.J.; Chorney, M.J.; Koltun, W.A. Cd1d-restricted cellular lysis by peripheral blood lymphocytes: Relevance to the inflammatory bowel diseases. J. Surg. Res. 2000, 92, 214–221. [Google Scholar] [CrossRef]
- Allez, M.; Tieng, V.; Nakazawa, A.; Treton, X.; Pacault, V.; Dulphy, N.; Caillat-Zucman, S.; Paul, P.; Gornet, J.M.; Douay, C.; et al. CD4+NKG2D+ T cells in Crohn’s disease mediate inflammatory and cytotoxic responses through MICA interactions. Gastroenterology 2007, 132, 2346–2358. [Google Scholar] [CrossRef]
- Pariente, B.; Mocan, I.; Camus, M.; Dutertre, C.A.; Ettersperger, J.; Cattan, P.; Gornet, J.M.; Dulphy, N.; Charron, D.; Lemann, M.; et al. Activation of the receptor NKG2D leads to production of Th17 cytokines in CD4+ T cells of patients with Crohn’s disease. Gastroenterology 2011, 141, 217–226.e2. [Google Scholar] [CrossRef]
- de Souza, H.S.; Fiocchi, C. Immunopathogenesis of IBD: Current state of the art. Nat. Rev. Gastroenterol. Hepatol. 2016, 13, 13–27. [Google Scholar] [CrossRef]
- Mahapatro, M.; Erkert, L.; Becker, C. Cytokine-Mediated Crosstalk between Immune Cells and Epithelial Cells in the Gut. Cells 2021, 10, 111. [Google Scholar] [CrossRef]
- Raphael, I.; Nalawade, S.; Eagar, T.N.; Forsthuber, T.G. T cell subsets and their signature cytokines in autoimmune and inflammatory diseases. Cytokine 2015, 74, 5–17. [Google Scholar] [CrossRef] [Green Version]
- Caza, T.; Landas, S. Functional and Phenotypic Plasticity of CD4(+) T Cell Subsets. Biomed. Res. Int. 2015, 2015, 521957. [Google Scholar] [CrossRef] [Green Version]
- Lazarevic, V.; Glimcher, L.H.; Lord, G.M. T-bet: A bridge between innate and adaptive immunity. Nat. Rev. Immunol. 2013, 13, 777–789. [Google Scholar] [CrossRef]
- Wang, P.; Zhang, Q.; Tan, L.; Xu, Y.; Xie, X.; Zhao, Y. The Regulatory Effects of mTOR Complexes in the Differentiation and Function of CD4(+) T Cell Subsets. J. Immunol. Res. 2020, 2020, 3406032. [Google Scholar] [CrossRef] [Green Version]
- Baumann, C.; Bonilla, W.V.; Frohlich, A.; Helmstetter, C.; Peine, M.; Hegazy, A.N.; Pinschewer, D.D.; Lohning, M. T-bet- and STAT4-dependent IL-33 receptor expression directly promotes antiviral Th1 cell responses. Proc. Natl. Acad. Sci. USA 2015, 112, 4056–4061. [Google Scholar] [CrossRef] [Green Version]
- Szabo, S.J.; Kim, S.T.; Costa, G.L.; Zhang, X.; Fathman, C.G.; Glimcher, L.H. A novel transcription factor, T-bet, directs Th1 lineage commitment. Cell 2000, 100, 655–669. [Google Scholar] [CrossRef] [Green Version]
- Yang, R.; Mele, F.; Worley, L.; Langlais, D.; Rosain, J.; Benhsaien, I.; Elarabi, H.; Croft, C.A.; Doisne, J.M.; Zhang, P.; et al. Human T-bet Governs Innate and Innate-like Adaptive IFN-gamma Immunity against Mycobacteria. Cell 2020, 183, 1826–1847.e31. [Google Scholar] [CrossRef]
- Wu, X.; Tian, J.; Wang, S. Insight Into Non-Pathogenic Th17 Cells in Autoimmune Diseases. Front. Immunol. 2018, 9, 1112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leppkes, M.; Neurath, M.F. Cytokines in inflammatory bowel diseases—Update 2020. Pharmacol. Res. 2020, 158, 104835. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Ueno, A.; Fort Gasia, M.; Luider, J.; Wang, T.; Hirota, C.; Jijon, H.B.; Deane, M.; Tom, M.; Chan, R.; et al. Profiles of Lamina Propria T Helper Cell Subsets Discriminate Between Ulcerative Colitis and Crohn’s Disease. Inflamm. Bowel Dis. 2016, 22, 1779–1792. [Google Scholar] [CrossRef] [PubMed]
- Ma, C.; Wu, W.; Lin, R.; Ge, Y.; Zhang, C.; Sun, S.; Cong, Y.; Li, X.; Liu, Z. Critical Role of CD6highCD4+ T Cells in Driving Th1/Th17 Cell Immune Responses and Mucosal Inflammation in IBD. J. Crohns Colitis 2019, 13, 510–524. [Google Scholar] [CrossRef]
- Monteleone, G.; Trapasso, F.; Parrello, T.; Biancone, L.; Stella, A.; Iuliano, R.; Luzza, F.; Fusco, A.; Pallone, F. Bioactive IL-18 expression is up-regulated in Crohn’s disease. J. Immunol. 1999, 163, 143–147. [Google Scholar] [CrossRef]
- Monteleone, G.; Biancone, L.; Marasco, R.; Morrone, G.; Marasco, O.; Luzza, F.; Pallone, F. Interleukin 12 is expressed and actively released by Crohn’s disease intestinal lamina propria mononuclear cells. Gastroenterology 1997, 112, 1169–1178. [Google Scholar] [CrossRef]
- Zorzi, F.; Monteleone, I.; Sarra, M.; Calabrese, E.; Marafini, I.; Cretella, M.; Sedda, S.; Biancone, L.; Pallone, F.; Monteleone, G. Distinct profiles of effector cytokines mark the different phases of Crohn’s disease. PLoS ONE 2013, 8, e54562. [Google Scholar] [CrossRef]
- Zimmermann, J.; Kuhl, A.A.; Weber, M.; Grun, J.R.; Loffler, J.; Haftmann, C.; Riedel, R.; Maschmeyer, P.; Lehmann, K.; Westendorf, K.; et al. T-bet expression by Th cells promotes type 1 inflammation but is dispensable for colitis. Mucosal Immunol. 2016, 9, 1487–1499. [Google Scholar] [CrossRef] [Green Version]
- Soderquest, K.; Hertweck, A.; Giambartolomei, C.; Henderson, S.; Mohamed, R.; Goldberg, R.; Perucha, E.; Franke, L.; Herrero, J.; Plagnol, V.; et al. Genetic variants alter T-bet binding and gene expression in mucosal inflammatory disease. PLoS Genet. 2017, 13, e1006587. [Google Scholar] [CrossRef] [Green Version]
- Alfen, J.S.; Larghi, P.; Facciotti, F.; Gagliani, N.; Bosotti, R.; Paroni, M.; Maglie, S.; Gruarin, P.; Vasco, C.M.; Ranzani, V.; et al. Intestinal IFN-gamma-producing type 1 regulatory T cells coexpress CCR5 and programmed cell death protein 1 and downregulate IL-10 in the inflamed guts of patients with inflammatory bowel disease. J. Allergy Clin. Immunol. 2018, 142, 1537–1547.e8. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Wang, Q.; Ding, L.; Wang, Y.X.; Zhao, Z.D.; Mao, N.; Wu, C.T.; Wang, H.; Zhu, H.; Ning, S.B. Intercellular adhesion molecule-1 enhances the therapeutic effects of MSCs in a dextran sulfate sodium-induced colitis models by promoting MSCs homing to murine colons and spleens. Stem Cell Res. Ther. 2019, 10, 267. [Google Scholar] [CrossRef]
- Ziegler, A.; Heidenreich, R.; Braumuller, H.; Wolburg, H.; Weidemann, S.; Mocikat, R.; Rocken, M. EpCAM, a human tumor-associated antigen promotes Th2 development and tumor immune evasion. Blood 2009, 113, 3494–3502. [Google Scholar] [CrossRef]
- Atanackovic, D.; Reinhard, H.; Meyer, S.; Spock, S.; Grob, T.; Luetkens, T.; Yousef, S.; Cao, Y.; Hildebrandt, Y.; Templin, J.; et al. The trifunctional antibody catumaxomab amplifies and shapes tumor-specific immunity when applied to gastric cancer patients in the adjuvant setting. Hum. Vaccin. Immunother. 2013, 9, 2533–2542. [Google Scholar] [CrossRef]
- Mashimo, M.; Fujii, T.; Ono, S.; Moriwaki, Y.; Misawa, H.; Kawashima, K. Minireview: Divergent roles of alpha7 nicotinic acetylcholine receptors expressed on antigen-presenting cells and CD4(+) T cells in the regulation of T cell differentiation. Int. Immunopharmacol. 2020, 82, 106306. [Google Scholar] [CrossRef]
- Fuss, I.J.; Heller, F.; Boirivant, M.; Leon, F.; Yoshida, M.; Fichtner-Feigl, S.; Yang, Z.; Exley, M.; Kitani, A.; Blumberg, R.S.; et al. Nonclassical CD1d-restricted NK T cells that produce IL-13 characterize an atypical Th2 response in ulcerative colitis. J. Clin. Investig. 2004, 113, 1490–1497. [Google Scholar] [CrossRef] [Green Version]
- Heller, F.; Florian, P.; Bojarski, C.; Richter, J.; Christ, M.; Hillenbrand, B.; Mankertz, J.; Gitter, A.H.; Burgel, N.; Fromm, M.; et al. Interleukin-13 is the key effector Th2 cytokine in ulcerative colitis that affects epithelial tight junctions, apoptosis, and cell restitution. Gastroenterology 2005, 129, 550–564. [Google Scholar] [CrossRef]
- Camoglio, L.; Te Velde, A.A.; Tigges, A.J.; Das, P.K.; Van Deventer, S.J. Altered expression of interferon-gamma and interleukin-4 in inflammatory bowel disease. Inflamm. Bowel Dis. 1998, 4, 285–290. [Google Scholar] [CrossRef]
- Fuss, I.J.; Neurath, M.; Boirivant, M.; Klein, J.S.; de la Motte, C.; Strong, S.A.; Fiocchi, C.; Strober, W. Disparate CD4+ lamina propria (LP) lymphokine secretion profiles in inflammatory bowel disease. Crohn’s disease LP cells manifest increased secretion of IFN-gamma, whereas ulcerative colitis LP cells manifest increased secretion of IL-5. J. Immunol. 1996, 157, 1261–1270. [Google Scholar] [CrossRef]
- Biancheri, P.; Di Sabatino, A.; Ammoscato, F.; Facciotti, F.; Caprioli, F.; Curciarello, R.; Hoque, S.S.; Ghanbari, A.; Joe-Njoku, I.; Giuffrida, P.; et al. Absence of a role for interleukin-13 in inflammatory bowel disease. Eur. J. Immunol. 2014, 44, 370–385. [Google Scholar] [CrossRef] [Green Version]
- Rovedatti, L.; Kudo, T.; Biancheri, P.; Sarra, M.; Knowles, C.H.; Rampton, D.S.; Corazza, G.R.; Monteleone, G.; Di Sabatino, A.; Macdonald, T.T. Differential regulation of interleukin 17 and interferon gamma production in inflammatory bowel disease. Gut 2009, 58, 1629–1636. [Google Scholar] [CrossRef]
- Yan, J.; Pandey, S.P.; Barnes, B.J.; Turner, J.R.; Abraham, C. T Cell-Intrinsic IRF5 Regulates T Cell Signaling, Migration, and Differentiation and Promotes Intestinal Inflammation. Cell Rep. 2020, 31, 107820. [Google Scholar] [CrossRef] [PubMed]
- Rocha-Perugini, V.; Gonzalez-Granado, J.M. Nuclear envelope lamin-A as a coordinator of T cell activation. Nucleus 2014, 5, 396–401. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Granado, J.M.; Silvestre-Roig, C.; Rocha-Perugini, V.; Trigueros-Motos, L.; Cibrian, D.; Morlino, G.; Blanco-Berrocal, M.; Osorio, F.G.; Freije, J.M.P.; Lopez-Otin, C.; et al. Nuclear envelope lamin-A couples actin dynamics with immunological synapse architecture and T cell activation. Sci. Signal. 2014, 7, ra37. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Toribio-Fernandez, R.; Zorita, V.; Rocha-Perugini, V.; Iborra, S.; Martinez Del Hoyo, G.; Chevre, R.; Dorado, B.; Sancho, D.; Sanchez-Madrid, F.; Andres, V.; et al. Lamin A/C augments Th1 differentiation and response against vaccinia virus and Leishmania major. Cell Death Dis. 2018, 9, 9. [Google Scholar] [CrossRef] [Green Version]
- Saez, A.; Herrero-Fernandez, B.; Gomez-Bris, R.; Somovilla-Crespo, B.; Rius, C.; Gonzalez-Granado, J.M. Lamin A/C and the Immune System: One Intermediate Filament, Many Faces. Int. J. Mol. Sci. 2020, 21, 6109. [Google Scholar] [CrossRef]
- Toribio-Fernandez, R.; Herrero-Fernandez, B.; Zorita, V.; Lopez, J.A.; Vazquez, J.; Criado, G.; Pablos, J.L.; Collas, P.; Sanchez-Madrid, F.; Andres, V.; et al. Lamin A/C deficiency in CD4(+) T-cells enhances regulatory T-cells and prevents inflammatory bowel disease. J. Pathol. 2019, 249, 509–522. [Google Scholar] [CrossRef]
- Cepek, K.L.; Parker, C.M.; Madara, J.L.; Brenner, M.B. Integrin alpha E beta 7 mediates adhesion of T lymphocytes to epithelial cells. J. Immunol. 1993, 150 Pt 1, 3459–3470. [Google Scholar] [CrossRef]
- Lamb, C.A.; Mansfield, J.C.; Tew, G.W.; Gibbons, D.; Long, A.K.; Irving, P.; Diehl, L.; Eastham-Anderson, J.; Price, M.B.; O’Boyle, G.; et al. alphaEbeta7 Integrin Identifies Subsets of Pro-Inflammatory Colonic CD4+ T Lymphocytes in Ulcerative Colitis. J. Crohns Colitis 2017, 11, 610–620. [Google Scholar]
- Di Sabatino, A.; Rovedatti, L.; Kaur, R.; Spencer, J.P.; Brown, J.T.; Morisset, V.D.; Biancheri, P.; Leakey, N.A.; Wilde, J.I.; Scott, L.; et al. Targeting gut T cell Ca2+ release-activated Ca2+ channels inhibits T cell cytokine production and T-box transcription factor T-bet in inflammatory bowel disease. J. Immunol. 2009, 183, 3454–3462. [Google Scholar] [CrossRef] [Green Version]
- Zeng, W.P. ‘All things considered’: Transcriptional regulation of T helper type 2 cell differentiation from precursor to effector activation. Immunology 2013, 140, 31–38. [Google Scholar] [CrossRef]
- Zheng, W.; Flavell, R.A. The transcription factor GATA-3 is necessary and sufficient for Th2 cytokine gene expression in CD4 T cells. Cell 1997, 89, 587–596. [Google Scholar] [CrossRef] [Green Version]
- Zhang, D.H.; Cohn, L.; Ray, P.; Bottomly, K.; Ray, A. Transcription factor GATA-3 is differentially expressed in murine Th1 and Th2 cells and controls Th2-specific expression of the interleukin-5 gene. J. Biol. Chem. 1997, 272, 21597–21603. [Google Scholar] [CrossRef] [Green Version]
- Bryant, N.; Muehling, L.M. T-cell responses in asthma exacerbations. Ann. Allergy Asthma Immunol. 2022, 129, 709–718. [Google Scholar] [CrossRef]
- Shankar, A.; McAlees, J.W.; Lewkowich, I.P. Modulation of IL-4/IL-13 cytokine signaling in the context of allergic disease. J. Allergy Clin. Immunol. 2022, 150, 266–276. [Google Scholar] [CrossRef]
- Luo, W.; Hu, J.; Xu, W.; Dong, J. Distinct spatial and temporal roles for Th1, Th2, and Th17 cells in asthma. Front. Immunol. 2022, 13, 974066. [Google Scholar] [CrossRef]
- Habib, N.; Pasha, M.A.; Tang, D.D. Current Understanding of Asthma Pathogenesis and Biomarkers. Cells 2022, 11, 2764. [Google Scholar] [CrossRef]
- Salvati, L.; Liotta, F.; Annunziato, F.; Cosmi, L. Therapeutical Targets in Allergic Inflammation. Biomedicines 2022, 10, 2874. [Google Scholar] [CrossRef]
- Durham, S.R.; Shamji, M.H. Allergen immunotherapy: Past, present and future. Nat. Rev. Immunol. 2022, 1–12. [Google Scholar] [CrossRef]
- Boirivant, M.; Fuss, I.J.; Chu, A.; Strober, W. Oxazolone colitis: A murine model of T helper cell type 2 colitis treatable with antibodies to interleukin 4. J. Exp. Med. 1998, 188, 1929–1939. [Google Scholar] [CrossRef] [Green Version]
- Pushparaj, P.N.; Li, D.; Komai-Koma, M.; Guabiraba, R.; Alexander, J.; McSharry, C.; Xu, D. Interleukin-33 exacerbates acute colitis via interleukin-4 in mice. Immunology 2013, 140, 70–77. [Google Scholar] [CrossRef]
- Sedhom, M.A.K.; Pichery, M.; Murdoch, J.R.; Foligné, B.; Ortega, N.; Normand, S.; Mertz, K.; Sanmugalingam, D.; Brault, L.; Grandjean, T.; et al. Neutralisation of the interleukin-33/ST2 pathway ameliorates experimental colitis through enhancement of mucosal healing in mice. Gut 2013, 62, 1714–1723. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guan, Q.; Zhang, J. Recent Advances: The Imbalance of Cytokines in the Pathogenesis of Inflammatory Bowel Disease. Mediat. Inflamm. 2017, 2017, 4810258. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pastorelli, L.; De Salvo, C.; Vecchi, M.; Pizarro, T.T. The role of IL-33 in gut mucosal inflammation. Mediat. Inflamm. 2013, 2013, 608187. [Google Scholar] [CrossRef] [PubMed]
- Latiano, A.; Palmieri, O.; Pastorelli, L.; Vecchi, M.; Pizarro, T.T.; Bossa, F.; Merla, G.; Augello, B.; Latiano, T.; Corritore, G.; et al. Associations between genetic polymorphisms in IL-33, IL1R1 and risk for inflammatory bowel disease. PLoS ONE 2013, 8, e62144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, J.; Wang, Y.; Yang, F.; Sang, L.; Zhai, J.; Li, S.; Li, Y.; Wang, D.; Lu, C.; Sun, X. IL-33 alleviates DSS-induced chronic colitis in C57BL/6 mice colon lamina propria by suppressing Th17 cell response as well as Th1 cell response. Int. Immunopharmacol. 2015, 29, 846–853. [Google Scholar] [CrossRef]
- Zhu, J.; Xu, Y.; Zhu, C.; Zhao, J.; Meng, X.; Chen, S.; Wang, T.; Li, X.; Zhang, L.; Lu, C.; et al. IL-33 induces both regulatory B cells and regulatory T cells in dextran sulfate sodium-induced colitis. Int. Immunopharmacol. 2017, 46, 38–47. [Google Scholar] [CrossRef]
- Zhu, J.; Yang, F.; Sang, L.; Zhai, J.; Zhang, X.; Yue, D.; Li, S.; Li, Y.; Lu, C.; Sun, X. IL-33 Aggravates DSS-Induced Acute Colitis in Mouse Colon Lamina Propria by Enhancing Th2 Cell Responses. Mediat. Inflamm. 2015, 2015, 913041. [Google Scholar] [CrossRef] [Green Version]
- Monticelli, L.A.; Osborne, L.C.; Noti, M.; Tran, S.V.; Zaiss, D.M.; Artis, D. IL-33 promotes an innate immune pathway of intestinal tissue protection dependent on amphiregulin-EGFR interactions. Proc. Natl. Acad. Sci. USA 2015, 112, 10762–10767. [Google Scholar] [CrossRef] [Green Version]
- Duan, L.; Chen, J.; Zhang, H.; Yang, H.; Zhu, P.; Xiong, A.; Xia, Q.; Zheng, F.; Tan, Z.; Gong, F.; et al. Interleukin-33 ameliorates experimental colitis through promoting Th2/Foxp3(+) regulatory T-cell responses in mice. Mol. Med. 2012, 18, 753–761. [Google Scholar] [CrossRef]
- Reinisch, W.; Panes, J.; Khurana, S.; Toth, G.; Hua, F.; Comer, G.M.; Hinz, M.; Page, K.; O’Toole, M.; Moorehead, T.M.; et al. Anrukinzumab, an anti-interleukin 13 monoclonal antibody, in active UC: Efficacy and safety from a phase IIa randomised multicentre study. Gut 2015, 64, 894–900. [Google Scholar] [CrossRef]
- Danese, S.; Rudzinski, J.; Brandt, W.; Dupas, J.L.; Peyrin-Biroulet, L.; Bouhnik, Y.; Kleczkowski, D.; Uebel, P.; Lukas, M.; Knutsson, M.; et al. Tralokinumab for moderate-to-severe UC: A randomised, double-blind, placebo-controlled, phase IIa study. Gut 2015, 64, 243–249. [Google Scholar] [CrossRef]
- Zhu, J.; Xu, Y.; Li, Z.; Liu, S.; Fu, W.; Wei, Y. Interleukin-36beta exacerbates DSS-induce acute colitis via inhibiting Foxp3(+) regulatory T cell response and increasing Th2 cell response. Int. Immunopharmacol. 2022, 108, 108762. [Google Scholar] [CrossRef]
- Schmitt, E.; Bopp, T. Discovery and initial characterization of Th9 cells: The early years. Semin. Immunopathol. 2017, 39, 5–10. [Google Scholar] [CrossRef]
- Neurath, M.F.; Finotto, S. IL-9 signaling as key driver of chronic inflammation in mucosal immunity. Cytokine Growth Factor Rev. 2016, 29, 93–99. [Google Scholar] [CrossRef]
- Kaplan, M.H.; Schindler, U.; Smiley, S.T.; Grusby, M.J. Stat6 is required for mediating responses to IL-4 and for development of Th2 cells. Immunity 1996, 4, 313–319. [Google Scholar] [CrossRef] [Green Version]
- Lu, L.; Ma, J.; Wang, X.; Wang, J.; Zhang, F.; Yu, J.; He, G.; Xu, B.; Brand, D.D.; Horwitz, D.A.; et al. Synergistic effect of TGF-beta superfamily members on the induction of Foxp3+ Treg. Eur. J. Immunol. 2010, 40, 142–152. [Google Scholar] [CrossRef] [Green Version]
- Veldhoen, M.; Uyttenhove, C.; van Snick, J.; Helmby, H.; Westendorf, A.; Buer, J.; Martin, B.; Wilhelm, C.; Stockinger, B. Transforming growth factor-beta ‘reprograms’ the differentiation of T helper 2 cells and promotes an interleukin 9-producing subset. Nat. Immunol. 2008, 9, 1341–1346. [Google Scholar] [CrossRef]
- Dardalhon, V.; Awasthi, A.; Kwon, H.; Galileos, G.; Gao, W.; Sobel, R.A.; Mitsdoerffer, M.; Strom, T.B.; Elyaman, W.; Ho, I.C.; et al. IL-4 inhibits TGF-beta-induced Foxp3+ T cells and, together with TGF-beta, generates IL-9+ IL-10+ Foxp3(−) effector T cells. Nat. Immunol. 2008, 9, 1347–1355. [Google Scholar] [CrossRef] [Green Version]
- Cosmi, L.; Maggi, L.; Santarlasci, V.; Liotta, F.; Annunziato, F. T helper cells plasticity in inflammation. Cytom. Part A J. Int. Soc. Anal. Cytol. 2014, 85, 36–42. [Google Scholar] [CrossRef]
- Staudt, V.; Bothur, E.; Klein, M.; Lingnau, K.; Reuter, S.; Grebe, N.; Gerlitzki, B.; Hoffmann, M.; Ulges, A.; Taube, C.; et al. Interferon-regulatory factor 4 is essential for the developmental program of T helper 9 cells. Immunity 2010, 33, 192–202. [Google Scholar] [CrossRef] [Green Version]
- Chang, H.C.; Sehra, S.; Goswami, R.; Yao, W.; Yu, Q.; Stritesky, G.L.; Jabeen, R.; McKinley, C.; Ahyi, A.N.; Han, L.; et al. The transcription factor PU.1 is required for the development of IL-9-producing T cells and allergic inflammation. Nat. Immunol. 2010, 11, 527–534. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shohan, M.; Elahi, S.; Shirzad, H.; Rafieian-Kopaei, M.; Bagheri, N.; Soltani, E. Th9 Cells: Probable players in ulcerative colitis pathogenesis. Int. Rev. Immunol. 2018, 37, 192–205. [Google Scholar] [CrossRef] [PubMed]
- Xue, G.; Jin, G.; Fang, J.; Lu, Y. IL-4 together with IL-1beta induces antitumor Th9 cell differentiation in the absence of TGF-beta signaling. Nat. Commun. 2019, 10, 1376. [Google Scholar] [CrossRef] [PubMed]
- Wilhelm, C.; Turner, J.E.; Van Snick, J.; Stockinger, B. The many lives of IL-9: A question of survival? Nat. Immunol. 2012, 13, 637–641. [Google Scholar] [CrossRef]
- Schmitt, N.; Ueno, H. Regulation of human helper T cell subset differentiation by cytokines. Curr. Opin. Immunol. 2015, 34, 130–136. [Google Scholar] [CrossRef] [Green Version]
- Bauer, J.H.; Liu, K.D.; You, Y.; Lai, S.Y.; Goldsmith, M.A. Heteromerization of the gammac chain with the interleukin-9 receptor alpha subunit leads to STAT activation and prevention of apoptosis. J. Biol. Chem. 1998, 273, 9255–9260. [Google Scholar] [CrossRef] [Green Version]
- Demoulin, J.B.; Uyttenhove, C.; Van Roost, E.; DeLestre, B.; Donckers, D.; Van Snick, J.; Renauld, J.C. A single tyrosine of the interleukin-9 (IL-9) receptor is required for STAT activation, antiapoptotic activity, and growth regulation by IL-9. Mol. Cell. Biol. 1996, 16, 4710–4716. [Google Scholar] [CrossRef] [Green Version]
- Demoulin, J.B.; Van Roost, E.; Stevens, M.; Groner, B.; Renauld, J.C. Distinct roles for STAT1, STAT3, and STAT5 in differentiation gene induction and apoptosis inhibition by interleukin-9. J. Biol. Chem. 1999, 274, 25855–25861. [Google Scholar] [CrossRef] [Green Version]
- Gerlach, K.; Hwang, Y.; Nikolaev, A.; Atreya, R.; Dornhoff, H.; Steiner, S.; Lehr, H.A.; Wirtz, S.; Vieth, M.; Waisman, A.; et al. TH9 cells that express the transcription factor PU.1 drive T cell-mediated colitis via IL-9 receptor signaling in intestinal epithelial cells. Nat. Immunol. 2014, 15, 676–686. [Google Scholar] [CrossRef]
- Gerlach, K.; McKenzie, A.N.; Neurath, M.F.; Weigmann, B. IL-9 regulates intestinal barrier function in experimental T cell-mediated colitis. Tissue Barriers 2015, 3, e983777. [Google Scholar] [CrossRef] [Green Version]
- Turner, J.R. Intestinal mucosal barrier function in health and disease. Nat. Rev. Immunol. 2009, 9, 799–809. [Google Scholar] [CrossRef]
- Nalleweg, N.; Chiriac, M.T.; Podstawa, E.; Lehmann, C.; Rau, T.T.; Atreya, R.; Krauss, E.; Hundorfean, G.; Fichtner-Feigl, S.; Hartmann, A.; et al. IL-9 and its receptor are predominantly involved in the pathogenesis of UC. Gut 2015, 64, 743–755. [Google Scholar] [CrossRef] [Green Version]
- Atreya, R.; Neurath, M.F. IBD pathogenesis in 2014: Molecular pathways controlling barrier function in IBD. Nat. Rev. Gastroenterol. Hepatol. 2015, 12, 67–68. [Google Scholar] [CrossRef]
- Kim, H.S.; Chung, D.H. IL-9-producing invariant NKT cells protect against DSS-induced colitis in an IL-4-dependent manner. Mucosal Immunol. 2013, 6, 347–357. [Google Scholar] [CrossRef] [Green Version]
- Shohan, M.; Sabzevary-Ghahfarokhi, M.; Bagheri, N.; Shirzad, H.; Rahimian, G.; Soltani, A.; Ghatreh-Samani, M.; Deris, F.; Tahmasbi, K.; Shahverdi, E.; et al. Intensified Th9 Response is Associated with the Immunopathogenesis of Active Ulcerative Colitis. Immunol. Investig. 2018, 47, 700–711. [Google Scholar] [CrossRef]
- Singh, U.P.; Singh, N.P.; Murphy, E.A.; Price, R.L.; Fayad, R.; Nagarkatti, M.; Nagarkatti, P.S. Chemokine and cytokine levels in inflammatory bowel disease patients. Cytokine 2016, 77, 44–49. [Google Scholar] [CrossRef] [Green Version]
- Neurath, M.F.; Kaplan, M.H. Th9 cells in immunity and immunopathological diseases. Semin. Immunopathol. 2017, 39, 1–4. [Google Scholar] [CrossRef] [Green Version]
- Bilsborough, J.; Fiorino, M.F.; Henkle, B.W. Select animal models of colitis and their value in predicting clinical efficacy of biological therapies in ulcerative colitis. Expert Opin. Drug Discov. 2021, 16, 567–577. [Google Scholar] [CrossRef]
- Yuan, A.; Yang, H.; Qi, H.; Cui, J.; Hua, W.; Li, C.; Pang, Z.; Zheng, W.; Cui, G. IL-9 antibody injection suppresses the inflammation in colitis mice. Biochem. Biophys. Res. Commun. 2015, 468, 921–926. [Google Scholar] [CrossRef]
- Valatas, V.; Kolios, G.; Bamias, G. TL1A (TNFSF15) and DR3 (TNFRSF25): A Co-stimulatory System of Cytokines With Diverse Functions in Gut Mucosal Immunity. Front. Immunol. 2019, 10, 583. [Google Scholar] [CrossRef] [Green Version]
- Wang, D.; Li, H.; Duan, Y.Y.; Han, F.; Luo, Y.X.; Wu, M.Y.; Yang, M.Y.; Zhan, R.R.; Song, J.; Zhang, H.; et al. TL1A modulates the severity of colitis by promoting Th9 differentiation and IL-9 secretion. Life Sci. 2019, 231, 116536. [Google Scholar] [CrossRef] [PubMed]
- Perumal, N.B.; Kaplan, M.H. Regulating Il9 transcription in T helper cells. Trends Immunol. 2011, 32, 146–150. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Erle, D.J.; Briskin, M.J.; Butcher, E.C.; Garcia-Pardo, A.; Lazarovits, A.I.; Tidswell, M. Expression and function of the MAdCAM-1 receptor, integrin alpha 4 beta 7, on human leukocytes. J. Immunol. 1994, 153, 517–528. [Google Scholar] [CrossRef] [PubMed]
- Dotan, I.; Allez, M.; Danese, S.; Keir, M.; Tole, S.; McBride, J. The role of integrins in the pathogenesis of inflammatory bowel disease: Approved and investigational anti-integrin therapies. Med. Res. Rev. 2020, 40, 245–262. [Google Scholar] [CrossRef] [Green Version]
- Zundler, S.; Schillinger, D.; Fischer, A.; Atreya, R.; Lopez-Posadas, R.; Watson, A.; Neufert, C.; Atreya, I.; Neurath, M.F. Blockade of alphaEbeta7 integrin suppresses accumulation of CD8(+) and Th9 lymphocytes from patients with IBD in the inflamed gut in vivo. Gut 2017, 66, 1936–1948. [Google Scholar] [CrossRef] [Green Version]
- Acharya, S.; Timilshina, M.; Jiang, L.; Neupane, S.; Choi, D.Y.; Park, S.W.; Lee, S.Y.; Jeong, B.S.; Kim, J.A.; Nam, T.G.; et al. Amelioration of Experimental autoimmune encephalomyelitis and DSS induced colitis by NTG-A-009 through the inhibition of Th1 and Th17 cells differentiation. Sci. Rep. 2018, 8, 7799. [Google Scholar] [CrossRef] [Green Version]
- Korn, T.; Bettelli, E.; Oukka, M.; Kuchroo, V.K. IL-17 and Th17 Cells. Annu. Rev. Immunol. 2009, 27, 485–517. [Google Scholar] [CrossRef]
- Dong, C. Diversification of T-helper-cell lineages: Finding the family root of IL-17-producing cells. Nat. Rev. Immunol. 2006, 6, 329–333. [Google Scholar] [CrossRef]
- Weaver, C.T.; Harrington, L.E.; Mangan, P.R.; Gavrieli, M.; Murphy, K.M. Th17: An effector CD4 T cell lineage with regulatory T cell ties. Immunity 2006, 24, 677–688. [Google Scholar] [CrossRef] [Green Version]
- McGeachy, M.J.; Bak-Jensen, K.S.; Chen, Y.; Tato, C.M.; Blumenschein, W.; McClanahan, T.; Cua, D.J. TGF-beta and IL-6 drive the production of IL-17 and IL-10 by T cells and restrain T(H)-17 cell-mediated pathology. Nat. Immunol. 2007, 8, 1390–1397. [Google Scholar] [CrossRef]
- Omenetti, S.; Bussi, C.; Metidji, A.; Iseppon, A.; Lee, S.; Tolaini, M.; Li, Y.; Kelly, G.; Chakravarty, P.; Shoaie, S.; et al. The Intestine Harbors Functionally Distinct Homeostatic Tissue-Resident and Inflammatory Th17 Cells. Immunity 2019, 51, 77–89.e6. [Google Scholar] [CrossRef] [Green Version]
- Esplugues, E.; Huber, S.; Gagliani, N.; Hauser, A.E.; Town, T.; Wan, Y.Y.; O’Connor, W., Jr.; Rongvaux, A.; Van Rooijen, N.; Haberman, A.M.; et al. Control of TH17 cells occurs in the small intestine. Nature 2011, 475, 514–518. [Google Scholar] [CrossRef] [Green Version]
- Noster, R.; de Koning, H.D.; Maier, E.; Prelog, M.; Lainka, E.; Zielinski, C.E. Dysregulation of proinflammatory versus anti-inflammatory human T(H)17 cell functionalities in the autoinflammatory Schnitzler syndrome. J. Allergy Clin. Immunol. 2016, 138, 1161–1169.e6. [Google Scholar] [CrossRef]
- Stockinger, B.; Omenetti, S. The dichotomous nature of T helper 17 cells. Nat. Rev. Immunol. 2017, 17, 535–544. [Google Scholar] [CrossRef]
- Ghoreschi, K.; Laurence, A.; Yang, X.P.; Tato, C.M.; McGeachy, M.J.; Konkel, J.E.; Ramos, H.L.; Wei, L.; Davidson, T.S.; Bouladoux, N.; et al. Generation of pathogenic T(H)17 cells in the absence of TGF-beta signalling. Nature 2010, 467, 967–971. [Google Scholar] [CrossRef] [Green Version]
- Lee, Y.; Awasthi, A.; Yosef, N.; Quintana, F.J.; Xiao, S.; Peters, A.; Wu, C.; Kleinewietfeld, M.; Kunder, S.; Hafler, D.A.; et al. Induction and molecular signature of pathogenic TH17 cells. Nat. Immunol. 2012, 13, 991–999. [Google Scholar] [CrossRef] [Green Version]
- Mangan, P.R.; Harrington, L.E.; O’Quinn, D.B.; Helms, W.S.; Bullard, D.C.; Elson, C.O.; Hatton, R.D.; Wahl, S.M.; Schoeb, T.R.; Weaver, C.T. Transforming growth factor-beta induces development of the T(H)17 lineage. Nature 2006, 441, 231–234. [Google Scholar] [CrossRef] [PubMed]
- Bettelli, E.; Carrier, Y.; Gao, W.; Korn, T.; Strom, T.B.; Oukka, M.; Weiner, H.L.; Kuchroo, V.K. Reciprocal developmental pathways for the generation of pathogenic effector TH17 and regulatory T cells. Nature 2006, 441, 235–238. [Google Scholar] [CrossRef] [PubMed]
- Veldhoen, M.; Hocking, R.J.; Atkins, C.J.; Locksley, R.M.; Stockinger, B. TGFbeta in the context of an inflammatory cytokine milieu supports de novo differentiation of IL-17-producing T cells. Immunity 2006, 24, 179–189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tesmer, L.A.; Lundy, S.K.; Sarkar, S.; Fox, D.A. Th17 cells in human disease. Immunol. Rev. 2008, 223, 87–113. [Google Scholar] [CrossRef]
- Yang, X.O.; Panopoulos, A.D.; Nurieva, R.; Chang, S.H.; Wang, D.; Watowich, S.S.; Dong, C. STAT3 regulates cytokine-mediated generation of inflammatory helper T cells. J. Biol. Chem. 2007, 282, 9358–9363. [Google Scholar] [CrossRef] [Green Version]
- Ghoreschi, K.; Laurence, A.; Yang, X.P.; Hirahara, K.; O’Shea, J.J. T helper 17 cell heterogeneity and pathogenicity in autoimmune disease. Trends Immunol. 2011, 32, 395–401. [Google Scholar] [CrossRef] [Green Version]
- Wang, C.; Yosef, N.; Gaublomme, J.; Wu, C.; Lee, Y.; Clish, C.B.; Kaminski, J.; Xiao, S.; Meyer Zu Horste, G.; Pawlak, M.; et al. CD5L/AIM Regulates Lipid Biosynthesis and Restrains Th17 Cell Pathogenicity. Cell 2015, 163, 1413–1427. [Google Scholar] [CrossRef]
- Gaublomme, J.T.; Yosef, N.; Lee, Y.; Gertner, R.S.; Yang, L.V.; Wu, C.; Pandolfi, P.P.; Mak, T.; Satija, R.; Shalek, A.K.; et al. Single-Cell Genomics Unveils Critical Regulators of Th17 Cell Pathogenicity. Cell 2015, 163, 1400–1412. [Google Scholar] [CrossRef] [Green Version]
- Singh, R.P.; Hasan, S.; Sharma, S.; Nagra, S.; Yamaguchi, D.T.; Wong, D.T.; Hahn, B.H.; Hossain, A. Th17 cells in inflammation and autoimmunity. Autoimmun. Rev. 2014, 13, 1174–1181. [Google Scholar] [CrossRef]
- Gaffen, S. IL-17 receptor composition. Nat. Rev. Immunol. 2016, 16, 4. [Google Scholar] [CrossRef]
- O’Connor, W., Jr.; Kamanaka, M.; Booth, C.J.; Town, T.; Nakae, S.; Iwakura, Y.; Kolls, J.K.; Flavell, R.A. A protective function for interleukin 17A in T cell-mediated intestinal inflammation. Nat. Immunol. 2009, 10, 603–609. [Google Scholar] [CrossRef]
- Franke, A.; Balschun, T.; Karlsen, T.H.; Hedderich, J.; May, S.; Lu, T.; Schuldt, D.; Nikolaus, S.; Rosenstiel, P.; Krawczak, M.; et al. Replication of signals from recent studies of Crohn’s disease identifies previously unknown disease loci for ulcerative colitis. Nat. Genet. 2008, 40, 713–715. [Google Scholar] [CrossRef]
- Song, X.; Dai, D.; He, X.; Zhu, S.; Yao, Y.; Gao, H.; Wang, J.; Qu, F.; Qiu, J.; Wang, H.; et al. Growth Factor FGF2 Cooperates with Interleukin-17 to Repair Intestinal Epithelial Damage. Immunity 2015, 43, 488–501. [Google Scholar] [CrossRef] [Green Version]
- Hegazy, A.N.; West, N.R.; Stubbington, M.J.T.; Wendt, E.; Suijker, K.I.M.; Datsi, A.; This, S.; Danne, C.; Campion, S.; Duncan, S.H.; et al. Circulating and Tissue-Resident CD4(+) T Cells With Reactivity to Intestinal Microbiota Are Abundant in Healthy Individuals and Function Is Altered During Inflammation. Gastroenterology 2017, 153, 1320–1337.e16. [Google Scholar] [CrossRef] [Green Version]
- Ueno, A.; Jeffery, L.; Kobayashi, T.; Hibi, T.; Ghosh, S.; Jijon, H. Th17 plasticity and its relevance to inflammatory bowel disease. J. Autoimmun. 2018, 87, 38–49. [Google Scholar] [CrossRef]
- Zhao, J.; Lu, Q.; Liu, Y.; Shi, Z.; Hu, L.; Zeng, Z.; Tu, Y.; Xiao, Z.; Xu, Q. Th17 Cells in Inflammatory Bowel Disease: Cytokines, Plasticity, and Therapies. J. Immunol. Res. 2021, 2021, 8816041. [Google Scholar] [CrossRef] [PubMed]
- Liang, S.C.; Tan, X.Y.; Luxenberg, D.P.; Karim, R.; Dunussi-Joannopoulos, K.; Collins, M.; Fouser, L.A. Interleukin (IL)-22 and IL-17 are coexpressed by Th17 cells and cooperatively enhance expression of antimicrobial peptides. J. Exp. Med. 2006, 203, 2271–2279. [Google Scholar] [CrossRef] [PubMed]
- Ye, P.; Rodriguez, F.H.; Kanaly, S.; Stocking, K.L.; Schurr, J.; Schwarzenberger, P.; Oliver, P.; Huang, W.; Zhang, P.; Zhang, J.; et al. Requirement of interleukin 17 receptor signaling for lung CXC chemokine and granulocyte colony-stimulating factor expression, neutrophil recruitment, and host defense. J. Exp. Med. 2001, 194, 519–527. [Google Scholar] [CrossRef] [PubMed]
- Jiang, P.; Zheng, C.; Xiang, Y.; Malik, S.; Su, D.; Xu, G.; Zhang, M. The involvement of TH17 cells in the pathogenesis of IBD. Cytokine Growth Factor Rev. 2022. [Google Scholar] [CrossRef] [PubMed]
- Schmitt, H.; Neurath, M.F.; Atreya, R. Role of the IL23/IL17 Pathway in Crohn’s Disease. Front. Immunol. 2021, 12, 622934. [Google Scholar] [CrossRef]
- Yan, J.B.; Luo, M.M.; Chen, Z.Y.; He, B.H. The Function and Role of the Th17/Treg Cell Balance in Inflammatory Bowel Disease. J. Immunol. Res. 2020, 2020, 8813558. [Google Scholar] [CrossRef]
- Raza, A.; Shata, M.T. Letter: Pathogenicity of Th17 cells may differ in ulcerative colitis compared with Crohn’s disease. Aliment. Pharmacol. Ther. 2012, 36, 204. [Google Scholar] [CrossRef]
- Sakuraba, A.; Sato, T.; Kamada, N.; Kitazume, M.; Sugita, A.; Hibi, T. Th1/Th17 immune response is induced by mesenteric lymph node dendritic cells in Crohn’s disease. Gastroenterology 2009, 137, 1736–1745. [Google Scholar] [CrossRef]
- Fujino, S.; Andoh, A.; Bamba, S.; Ogawa, A.; Hata, K.; Araki, Y.; Bamba, T.; Fujiyama, Y. Increased expression of interleukin 17 in inflammatory bowel disease. Gut 2003, 52, 65–70. [Google Scholar] [CrossRef]
- Monteleone, I.; Sarra, M.; Pallone, F.; Monteleone, G. Th17-related cytokines in inflammatory bowel diseases: Friends or foes? Curr. Mol. Med. 2012, 12, 592–597. [Google Scholar] [CrossRef]
- Zhang, Z.; Zheng, M.; Bindas, J.; Schwarzenberger, P.; Kolls, J.K. Critical role of IL-17 receptor signaling in acute TNBS-induced colitis. Inflamm. Bowel Dis. 2006, 12, 382–388. [Google Scholar] [CrossRef]
- Yang, X.O.; Chang, S.H.; Park, H.; Nurieva, R.; Shah, B.; Acero, L.; Wang, Y.H.; Schluns, K.S.; Broaddus, R.R.; Zhu, Z.; et al. Regulation of inflammatory responses by IL-17F. J. Exp. Med. 2008, 205, 1063–1075. [Google Scholar] [CrossRef]
- Strober, W.; Fuss, I.J.; Blumberg, R.S. The immunology of mucosal models of inflammation. Annu. Rev. Immunol. 2002, 20, 495–549. [Google Scholar] [CrossRef]
- Sandborn, W.J.; Feagan, B.G.; Fedorak, R.N.; Scherl, E.; Fleisher, M.R.; Katz, S.; Johanns, J.; Blank, M.; Rutgeerts, P.; Ustekinumab Crohn’s Disease Study Group. A randomized trial of Ustekinumab, a human interleukin-12/23 monoclonal antibody, in patients with moderate-to-severe Crohn’s disease. Gastroenterology 2008, 135, 1130–1141. [Google Scholar] [CrossRef] [Green Version]
- Fina, D.; Sarra, M.; Fantini, M.C.; Rizzo, A.; Caruso, R.; Caprioli, F.; Stolfi, C.; Cardolini, I.; Dottori, M.; Boirivant, M.; et al. Regulation of gut inflammation and th17 cell response by interleukin-21. Gastroenterology 2008, 134, 1038–1048. [Google Scholar] [CrossRef]
- Strengell, M.; Sareneva, T.; Foster, D.; Julkunen, I.; Matikainen, S. IL-21 up-regulates the expression of genes associated with innate immunity and Th1 response. J. Immunol. 2002, 169, 3600–3605. [Google Scholar] [CrossRef] [Green Version]
- Gahring, L.C.; Carlson, N.G.; Kulmar, R.A.; Rogers, S.W. Neuronal expression of tumor necrosis factor alpha in the murine brain. Neuroimmunomodulation 1996, 3, 289–303. [Google Scholar] [CrossRef]
- Perrier, C.; de Hertogh, G.; Cremer, J.; Vermeire, S.; Rutgeerts, P.; Van Assche, G.; Szymkowski, D.E.; Ceuppens, J.L. Neutralization of membrane TNF, but not soluble TNF, is crucial for the treatment of experimental colitis. Inflamm. Bowel Dis. 2013, 19, 246–253. [Google Scholar] [CrossRef]
- Annunziato, F.; Cosmi, L.; Santarlasci, V.; Maggi, L.; Liotta, F.; Mazzinghi, B.; Parente, E.; Fili, L.; Ferri, S.; Frosali, F.; et al. Phenotypic and functional features of human Th17 cells. J. Exp. Med. 2007, 204, 1849–1861. [Google Scholar] [CrossRef] [Green Version]
- Lee, Y.K.; Turner, H.; Maynard, C.L.; Oliver, J.R.; Chen, D.; Elson, C.O.; Weaver, C.T. Late developmental plasticity in the T helper 17 lineage. Immunity 2009, 30, 92–107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hueber, W.; Sands, B.E.; Lewitzky, S.; Vandemeulebroecke, M.; Reinisch, W.; Higgins, P.D.; Wehkamp, J.; Feagan, B.G.; Yao, M.D.; Karczewski, M.; et al. Secukinumab, a human anti-IL-17A monoclonal antibody, for moderate to severe Crohn’s disease: Unexpected results of a randomised, double-blind placebo-controlled trial. Gut 2012, 61, 1693–1700. [Google Scholar] [CrossRef] [Green Version]
- Targan, S.R.; Feagan, B.; Vermeire, S.; Panaccione, R.; Melmed, G.Y.; Landers, C.; Li, D.; Russell, C.; Newmark, R.; Zhang, N.; et al. A Randomized, Double-Blind, Placebo-Controlled Phase 2 Study of Brodalumab in Patients With Moderate-to-Severe Crohn’s Disease. Am. J. Gastroenterol. 2016, 111, 1599–1607. [Google Scholar] [CrossRef] [PubMed]
- Ogawa, A.; Andoh, A.; Araki, Y.; Bamba, T.; Fujiyama, Y. Neutralization of interleukin-17 aggravates dextran sulfate sodium-induced colitis in mice. Clin. Immunol. 2004, 110, 55–62. [Google Scholar] [CrossRef] [PubMed]
- Maxwell, J.R.; Zhang, Y.; Brown, W.A.; Smith, C.L.; Byrne, F.R.; Fiorino, M.; Stevens, E.; Bigler, J.; Davis, J.A.; Rottman, J.B.; et al. Differential Roles for Interleukin-23 and Interleukin-17 in Intestinal Immunoregulation. Immunity 2015, 43, 739–750. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.S.; Tato, C.M.; Joyce-Shaikh, B.; Gulen, M.F.; Cayatte, C.; Chen, Y.; Blumenschein, W.M.; Judo, M.; Ayanoglu, G.; McClanahan, T.K.; et al. Interleukin-23-Independent IL-17 Production Regulates Intestinal Epithelial Permeability. Immunity 2015, 43, 727–738. [Google Scholar] [CrossRef] [Green Version]
- Arnold, I.C.; Mathisen, S.; Schulthess, J.; Danne, C.; Hegazy, A.N.; Powrie, F. CD11c(+) monocyte/macrophages promote chronic Helicobacter hepaticus-induced intestinal inflammation through the production of IL-23. Mucosal Immunol. 2016, 9, 352–363. [Google Scholar] [CrossRef] [Green Version]
- Becker, C.; Dornhoff, H.; Neufert, C.; Fantini, M.C.; Wirtz, S.; Huebner, S.; Nikolaev, A.; Lehr, H.A.; Murphy, A.J.; Valenzuela, D.M.; et al. Cutting edge: IL-23 cross-regulates IL-12 production in T cell-dependent experimental colitis. J. Immunol. 2006, 177, 2760–2764. [Google Scholar] [CrossRef] [Green Version]
- Yen, D.; Cheung, J.; Scheerens, H.; Poulet, F.; McClanahan, T.; McKenzie, B.; Kleinschek, M.A.; Owyang, A.; Mattson, J.; Blumenschein, W.; et al. IL-23 is essential for T cell-mediated colitis and promotes inflammation via IL-17 and IL-6. J. Clin. Investig. 2006, 116, 1310–1316. [Google Scholar] [CrossRef] [Green Version]
- Karaboga, I.; Demirtas, S.; Karaca, T. Investigation of the relationship between the Th17/IL-23 pathway and innate-adaptive immune system in TNBS-induced colitis in rats. Iran. J. Basic Med. Sci. 2017, 20, 870–879. [Google Scholar]
- Noviello, D.; Mager, R.; Roda, G.; Borroni, R.G.; Fiorino, G.; Vetrano, S. The IL23-IL17 Immune Axis in the Treatment of Ulcerative Colitis: Successes, Defeats, and Ongoing Challenges. Front. Immunol. 2021, 12, 611256. [Google Scholar] [CrossRef]
- Zheng, Y.; Valdez, P.A.; Danilenko, D.M.; Hu, Y.; Sa, S.M.; Gong, Q.; Abbas, A.R.; Modrusan, Z.; Ghilardi, N.; de Sauvage, F.J.; et al. Interleukin-22 mediates early host defense against attaching and effacing bacterial pathogens. Nat. Med. 2008, 14, 282–289. [Google Scholar] [CrossRef]
- Pickert, G.; Neufert, C.; Leppkes, M.; Zheng, Y.; Wittkopf, N.; Warntjen, M.; Lehr, H.A.; Hirth, S.; Weigmann, B.; Wirtz, S.; et al. STAT3 links IL-22 signaling in intestinal epithelial cells to mucosal wound healing. J. Exp. Med. 2009, 206, 1465–1472. [Google Scholar] [CrossRef] [Green Version]
- Basu, R.; O’Quinn, D.B.; Silberger, D.J.; Schoeb, T.R.; Fouser, L.; Ouyang, W.; Hatton, R.D.; Weaver, C.T. Th22 cells are an important source of IL-22 for host protection against enteropathogenic bacteria. Immunity 2012, 37, 1061–1075. [Google Scholar] [CrossRef]
- Dudakov, J.A.; Mertelsmann, A.M.; O’Connor, M.H.; Jenq, R.R.; Velardi, E.; Young, L.F.; Smith, O.M.; Boyd, R.L.; van den Brink, M.R.M.; Hanash, A.M. Loss of thymic innate lymphoid cells leads to impaired thymopoiesis in experimental graft-versus-host disease. Blood 2017, 130, 933–942. [Google Scholar] [CrossRef] [Green Version]
- Shohan, M.; Dehghani, R.; Khodadadi, A.; Dehnavi, S.; Ahmadi, R.; Joudaki, N.; Houshmandfar, S.; Shamshiri, M.; Shojapourian, S.; Bagheri, N. Interleukin-22 and intestinal homeostasis: Protective or destructive? IUBMB Life 2020, 72, 1585–1602. [Google Scholar] [CrossRef]
- Plank, M.W.; Kaiko, G.E.; Maltby, S.; Weaver, J.; Tay, H.L.; Shen, W.; Wilson, M.S.; Durum, S.K.; Foster, P.S. Th22 Cells Form a Distinct Th Lineage from Th17 Cells In Vitro with Unique Transcriptional Properties and Tbet-Dependent Th1 Plasticity. J. Immunol. 2017, 198, 2182–2190. [Google Scholar] [CrossRef] [Green Version]
- Duhen, T.; Geiger, R.; Jarrossay, D.; Lanzavecchia, A.; Sallusto, F. Production of interleukin 22 but not interleukin 17 by a subset of human skin-homing memory T cells. Nat. Immunol. 2009, 10, 857–863. [Google Scholar] [CrossRef]
- Li, J.; Chen, S.; Xiao, X.; Zhao, Y.; Ding, W.; Li, X.C. IL-9 and Th9 cells in health and diseases-From tolerance to immunopathology. Cytokine Growth Factor Rev. 2017, 37, 47–55. [Google Scholar] [CrossRef]
- Mock, B.A.; Krall, M.; Kozak, C.A.; Nesbitt, M.N.; McBride, O.W.; Renauld, J.C.; Van Snick, J. IL9 maps to mouse chromosome 13 and human chromosome 5. Immunogenetics 1990, 31, 265–270. [Google Scholar] [CrossRef]
- Stassen, M.; Schmitt, E.; Bopp, T. From interleukin-9 to T helper 9 cells. Ann. N. Y. Acad. Sci. 2012, 1247, 56–68. [Google Scholar] [CrossRef] [PubMed]
- Steenwinckel, V.; Louahed, J.; Orabona, C.; Huaux, F.; Warnier, G.; McKenzie, A.; Lison, D.; Levitt, R.; Renauld, J.C. IL-13 mediates in vivo IL-9 activities on lung epithelial cells but not on hematopoietic cells. J. Immunol. 2007, 178, 3244–3251. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arshad, T.; Mansur, F.; Palek, R.; Manzoor, S.; Liska, V. A Double Edged Sword Role of Interleukin-22 in Wound Healing and Tissue Regeneration. Front. Immunol. 2020, 11, 2148. [Google Scholar] [CrossRef] [PubMed]
- Lindemans, C.A.; Calafiore, M.; Mertelsmann, A.M.; O’Connor, M.H.; Dudakov, J.A.; Jenq, R.R.; Velardi, E.; Young, L.F.; Smith, O.M.; Lawrence, G.; et al. Interleukin-22 promotes intestinal-stem-cell-mediated epithelial regeneration. Nature 2015, 528, 560–564. [Google Scholar] [CrossRef]
- Sugimoto, K.; Ogawa, A.; Mizoguchi, E.; Shimomura, Y.; Andoh, A.; Bhan, A.K.; Blumberg, R.S.; Xavier, R.J.; Mizoguchi, A. IL-22 ameliorates intestinal inflammation in a mouse model of ulcerative colitis. J. Clin. Investig. 2008, 118, 534–544. [Google Scholar] [CrossRef] [Green Version]
- Gibson, D.L.; Montero, M.; Ropeleski, M.J.; Bergstrom, K.S.; Ma, C.; Ghosh, S.; Merkens, H.; Huang, J.; Mansson, L.E.; Sham, H.P.; et al. Interleukin-11 reduces TLR4-induced colitis in TLR2-deficient mice and restores intestinal STAT3 signaling. Gastroenterology 2010, 139, 1277–1288. [Google Scholar] [CrossRef]
- Andoh, A.; Zhang, Z.; Inatomi, O.; Fujino, S.; Deguchi, Y.; Araki, Y.; Tsujikawa, T.; Kitoh, K.; Kim-Mitsuyama, S.; Takayanagi, A.; et al. Interleukin-22, a member of the IL-10 subfamily, induces inflammatory responses in colonic subepithelial myofibroblasts. Gastroenterology 2005, 129, 969–984. [Google Scholar] [CrossRef]
- Pavlidis, P.; Tsakmaki, A.; Pantazi, E.; Li, K.; Cozzetto, D.; Digby-Bell, J.; Yang, F.; Lo, J.W.; Alberts, E.; Sa, A.C.C.; et al. Interleukin-22 regulates neutrophil recruitment in ulcerative colitis and is associated with resistance to ustekinumab therapy. Nat. Commun. 2022, 13, 5820. [Google Scholar] [CrossRef]
- Huber, S.; Gagliani, N.; Zenewicz, L.A.; Huber, F.J.; Bosurgi, L.; Hu, B.; Hedl, M.; Zhang, W.; O’Connor, W., Jr.; Murphy, A.J.; et al. IL-22BP is regulated by the inflammasome and modulates tumorigenesis in the intestine. Nature 2012, 491, 259–263. [Google Scholar] [CrossRef] [Green Version]
- Pelczar, P.; Witkowski, M.; Perez, L.G.; Kempski, J.; Hammel, A.G.; Brockmann, L.; Kleinschmidt, D.; Wende, S.; Haueis, C.; Bedke, T.; et al. A pathogenic role for T cell-derived IL-22BP in inflammatory bowel disease. Science 2016, 354, 358–362. [Google Scholar] [CrossRef]
- Jinnohara, T.; Kanaya, T.; Hase, K.; Sakakibara, S.; Kato, T.; Tachibana, N.; Sasaki, T.; Hashimoto, Y.; Sato, T.; Watarai, H.; et al. IL-22BP dictates characteristics of Peyer’s patch follicle-associated epithelium for antigen uptake. J. Exp. Med. 2017, 214, 1607–1618. [Google Scholar] [CrossRef]
- Martin, J.C.; Beriou, G.; Heslan, M.; Chauvin, C.; Utriainen, L.; Aumeunier, A.; Scott, C.L.; Mowat, A.; Cerovic, V.; Houston, S.A.; et al. Interleukin-22 binding protein (IL-22BP) is constitutively expressed by a subset of conventional dendritic cells and is strongly induced by retinoic acid. Mucosal Immunol. 2014, 7, 101–113. [Google Scholar] [CrossRef] [Green Version]
- Savage, A.K.; Liang, H.E.; Locksley, R.M. The Development of Steady-State Activation Hubs between Adult LTi ILC3s and Primed Macrophages in Small Intestine. J. Immunol. 2017, 199, 1912–1922. [Google Scholar] [CrossRef] [Green Version]
- Zenewicz, L.A. IL-22 Binding Protein (IL-22BP) in the Regulation of IL-22 Biology. Front. Immunol. 2021, 12, 766586. [Google Scholar] [CrossRef]
- Kotenko, S.V.; Izotova, L.S.; Mirochnitchenko, O.V.; Esterova, E.; Dickensheets, H.; Donnelly, R.P.; Pestka, S. Identification, cloning, and characterization of a novel soluble receptor that binds IL-22 and neutralizes its activity. J. Immunol. 2001, 166, 7096–7103. [Google Scholar] [CrossRef] [Green Version]
- Schmechel, S.; Konrad, A.; Diegelmann, J.; Glas, J.; Wetzke, M.; Paschos, E.; Lohse, P.; Goke, B.; Brand, S. Linking genetic susceptibility to Crohn’s disease with Th17 cell function: IL-22 serum levels are increased in Crohn’s disease and correlate with disease activity and IL23R genotype status. Inflamm. Bowel Dis. 2008, 14, 204–212. [Google Scholar] [CrossRef]
- Wolk, K.; Witte, E.; Hoffmann, U.; Doecke, W.D.; Endesfelder, S.; Asadullah, K.; Sterry, W.; Volk, H.D.; Wittig, B.M.; Sabat, R. IL-22 induces lipopolysaccharide-binding protein in hepatocytes: A potential systemic role of IL-22 in Crohn’s disease. J. Immunol. 2007, 178, 5973–5981. [Google Scholar] [CrossRef] [Green Version]
- Martin, J.C.; Beriou, G.; Heslan, M.; Bossard, C.; Jarry, A.; Abidi, A.; Hulin, P.; Menoret, S.; Thinard, R.; Anegon, I.; et al. IL-22BP is produced by eosinophils in human gut and blocks IL-22 protective actions during colitis. Mucosal Immunol. 2016, 9, 539–549. [Google Scholar] [CrossRef] [Green Version]
- Gui, X.; Li, J.; Ueno, A.; Iacucci, M.; Qian, J.; Ghosh, S. Histopathological Features of Inflammatory Bowel Disease are Associated With Different CD4+ T Cell Subsets in Colonic Mucosal Lamina Propria. J. Crohns Colitis 2018, 12, 1448–1458. [Google Scholar] [CrossRef]
- Fang, L.; Pang, Z.; Shu, W.; Wu, W.; Sun, M.; Cong, Y.; Liu, Z. Anti-TNF Therapy Induces CD4+ T-Cell Production of IL-22 and Promotes Epithelial Repairs in Patients With Crohn’s Disease. Inflamm. Bowel Dis. 2018, 24, 1733–1744. [Google Scholar] [CrossRef]
- Josefowicz, S.Z.; Lu, L.F.; Rudensky, A.Y. Regulatory T cells: Mechanisms of differentiation and function. Annu. Rev. Immunol. 2012, 30, 531–564. [Google Scholar] [CrossRef] [PubMed]
- Itoh, M.; Takahashi, T.; Sakaguchi, N.; Kuniyasu, Y.; Shimizu, J.; Otsuka, F.; Sakaguchi, S. Thymus and autoimmunity: Production of CD25+CD4+ naturally anergic and suppressive T cells as a key function of the thymus in maintaining immunologic self-tolerance. J. Immunol. 1999, 162, 5317–5326. [Google Scholar] [CrossRef] [PubMed]
- Fontenot, J.D.; Gavin, M.A.; Rudensky, A.Y. Foxp3 programs the development and function of CD4+CD25+ regulatory T cells. Nat. Immunol. 2003, 4, 330–336. [Google Scholar] [CrossRef] [PubMed]
- Hori, S.; Nomura, T.; Sakaguchi, S. Control of regulatory T cell development by the transcription factor Foxp3. Science 2003, 299, 1057–1061. [Google Scholar] [CrossRef]
- Sakaguchi, S.; Sakaguchi, N.; Asano, M.; Itoh, M.; Toda, M. Immunologic self-tolerance maintained by activated T cells expressing IL-2 receptor alpha-chains (CD25). Breakdown of a single mechanism of self-tolerance causes various autoimmune diseases. J. Immunol. 1995, 155, 1151–1164. [Google Scholar] [CrossRef]
- Josefowicz, S.Z.; Rudensky, A. Control of regulatory T cell lineage commitment and maintenance. Immunity 2009, 30, 616–625. [Google Scholar] [CrossRef] [Green Version]
- Abbas, A.K.; Benoist, C.; Bluestone, J.A.; Campbell, D.J.; Ghosh, S.; Hori, S.; Jiang, S.; Kuchroo, V.K.; Mathis, D.; Roncarolo, M.G.; et al. Regulatory T cells: Recommendations to simplify the nomenclature. Nat. Immunol. 2013, 14, 307–308. [Google Scholar] [CrossRef] [Green Version]
- Sakaguchi, S.; Mikami, N.; Wing, J.B.; Tanaka, A.; Ichiyama, K.; Ohkura, N. Regulatory T Cells and Human Disease. Annu. Rev. Immunol. 2020, 38, 541–566. [Google Scholar] [CrossRef] [Green Version]
- Hsieh, C.S.; Lee, H.M.; Lio, C.W. Selection of regulatory T cells in the thymus. Nat. Rev. Immunol. 2012, 12, 157–167. [Google Scholar] [CrossRef]
- Lancaster, J.N.; Li, Y.; Ehrlich, L.I.R. Chemokine-Mediated Choreography of Thymocyte Development and Selection. Trends Immunol. 2018, 39, 86–98. [Google Scholar] [CrossRef]
- Lancaster, J.N.; Thyagarajan, H.M.; Srinivasan, J.; Li, Y.; Hu, Z.; Ehrlich, L.I.R. Live-cell imaging reveals the relative contributions of antigen-presenting cell subsets to thymic central tolerance. Nat. Commun. 2019, 10, 2220. [Google Scholar] [CrossRef] [Green Version]
- Caramalho, I.; Nunes-Cabaco, H.; Foxall, R.B.; Sousa, A.E. Regulatory T-Cell Development in the Human Thymus. Front. Immunol. 2015, 6, 395. [Google Scholar] [CrossRef] [Green Version]
- Sakaguchi, S.; Yamaguchi, T.; Nomura, T.; Ono, M. Regulatory T cells and immune tolerance. Cell 2008, 133, 775–787. [Google Scholar] [CrossRef] [Green Version]
- Takaba, H.; Takayanagi, H. The Mechanisms of T Cell Selection in the Thymus. Trends Immunol. 2017, 38, 805–816. [Google Scholar] [CrossRef]
- Veenbergen, S.; Samsom, J.N. Maintenance of small intestinal and colonic tolerance by IL-10-producing regulatory T cell subsets. Curr. Opin. Immunol. 2012, 24, 269–276. [Google Scholar] [CrossRef]
- Worbs, T.; Bode, U.; Yan, S.; Hoffmann, M.W.; Hintzen, G.; Bernhardt, G.; Forster, R.; Pabst, O. Oral tolerance originates in the intestinal immune system and relies on antigen carriage by dendritic cells. J. Exp. Med. 2006, 203, 519–527. [Google Scholar] [CrossRef] [Green Version]
- Hauet-Broere, F.; Unger, W.W.; Garssen, J.; Hoijer, M.A.; Kraal, G.; Samsom, J.N. Functional CD25− and CD25+ mucosal regulatory T cells are induced in gut-draining lymphoid tissue within 48 h after oral antigen application. Eur. J. Immunol. 2003, 33, 2801–2810. [Google Scholar] [CrossRef]
- Davidson, T.S.; DiPaolo, R.J.; Andersson, J.; Shevach, E.M. Cutting Edge: IL-2 is essential for TGF-beta-mediated induction of Foxp3+ T regulatory cells. J. Immunol. 2007, 178, 4022–4026. [Google Scholar] [CrossRef] [Green Version]
- Kretschmer, K.; Apostolou, I.; Hawiger, D.; Khazaie, K.; Nussenzweig, M.C.; von Boehmer, H. Inducing and expanding regulatory T cell populations by foreign antigen. Nat. Immunol. 2005, 6, 1219–1227. [Google Scholar] [CrossRef]
- Chen, W.; Jin, W.; Hardegen, N.; Lei, K.J.; Li, L.; Marinos, N.; McGrady, G.; Wahl, S.M. Conversion of peripheral CD4+CD25− naive T cells to CD4+CD25+ regulatory T cells by TGF-beta induction of transcription factor Foxp3. J. Exp. Med. 2003, 198, 1875–1886. [Google Scholar] [CrossRef]
- Coombes, J.L.; Siddiqui, K.R.; Arancibia-Carcamo, C.V.; Hall, J.; Sun, C.M.; Belkaid, Y.; Powrie, F. A functionally specialized population of mucosal CD103+ DCs induces Foxp3+ regulatory T cells via a TGF-beta and retinoic acid-dependent mechanism. J. Exp. Med. 2007, 204, 1757–1764. [Google Scholar] [CrossRef] [PubMed]
- Mucida, D.; Park, Y.; Kim, G.; Turovskaya, O.; Scott, I.; Kronenberg, M.; Cheroutre, H. Reciprocal TH17 and regulatory T cell differentiation mediated by retinoic acid. Science 2007, 317, 256–260. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.; Ioan-Facsinay, A.; van der Voort, E.I.; Huizinga, T.W.; Toes, R.E. Transient expression of FOXP3 in human activated nonregulatory CD4+ T cells. Eur. J. Immunol. 2007, 37, 129–138. [Google Scholar] [CrossRef] [PubMed]
- Jacobse, J.; Li, J.; Rings, E.; Samsom, J.N.; Goettel, J.A. Intestinal Regulatory T Cells as Specialized Tissue-Restricted Immune Cells in Intestinal Immune Homeostasis and Disease. Front. Immunol. 2021, 12, 716499. [Google Scholar] [CrossRef]
- Nelson, R.W.; Beisang, D.; Tubo, N.J.; Dileepan, T.; Wiesner, D.L.; Nielsen, K.; Wuthrich, M.; Klein, B.S.; Kotov, D.I.; Spanier, J.A.; et al. T cell receptor cross-reactivity between similar foreign and self peptides influences naive cell population size and autoimmunity. Immunity 2015, 42, 95–107. [Google Scholar] [CrossRef] [Green Version]
- Tanoue, T.; Atarashi, K.; Honda, K. Development and maintenance of intestinal regulatory T cells. Nat. Rev. Immunol. 2016, 16, 295–309. [Google Scholar] [CrossRef]
- Atarashi, K.; Tanoue, T.; Shima, T.; Imaoka, A.; Kuwahara, T.; Momose, Y.; Cheng, G.; Yamasaki, S.; Saito, T.; Ohba, Y.; et al. Induction of colonic regulatory T cells by indigenous Clostridium species. Science 2011, 331, 337–341. [Google Scholar] [CrossRef] [Green Version]
- Wing, J.B.; Tanaka, A.; Sakaguchi, S. Human FOXP3(+) Regulatory T Cell Heterogeneity and Function in Autoimmunity and Cancer. Immunity 2019, 50, 302–316. [Google Scholar] [CrossRef] [Green Version]
- Miyara, M.; Yoshioka, Y.; Kitoh, A.; Shima, T.; Wing, K.; Niwa, A.; Parizot, C.; Taflin, C.; Heike, T.; Valeyre, D.; et al. Functional delineation and differentiation dynamics of human CD4+ T cells expressing the FoxP3 transcription factor. Immunity 2009, 30, 899–911. [Google Scholar] [CrossRef] [Green Version]
- Fisson, S.; Darrasse-Jeze, G.; Litvinova, E.; Septier, F.; Klatzmann, D.; Liblau, R.; Salomon, B.L. Continuous activation of autoreactive CD4+ CD25+ regulatory T cells in the steady state. J. Exp. Med. 2003, 198, 737–746. [Google Scholar] [CrossRef]
- Asseman, C.; Mauze, S.; Leach, M.W.; Coffman, R.L.; Powrie, F. An essential role for interleukin 10 in the function of regulatory T cells that inhibit intestinal inflammation. J. Exp. Med. 1999, 190, 995–1004. [Google Scholar] [CrossRef]
- Wirtz, S.; Billmeier, U.; McHedlidze, T.; Blumberg, R.S.; Neurath, M.F. Interleukin-35 mediates mucosal immune responses that protect against T-cell-dependent colitis. Gastroenterology 2011, 141, 1875–1886. [Google Scholar] [CrossRef] [Green Version]
- Nakamura, K.; Kitani, A.; Fuss, I.; Pedersen, A.; Harada, N.; Nawata, H.; Strober, W. TGF-beta 1 plays an important role in the mechanism of CD4+CD25+ regulatory T cell activity in both humans and mice. J. Immunol. 2004, 172, 834–842. [Google Scholar] [CrossRef] [Green Version]
- Powrie, F.; Carlino, J.; Leach, M.W.; Mauze, S.; Coffman, R.L. A critical role for transforming growth factor-beta but not interleukin 4 in the suppression of T helper type 1-mediated colitis by CD45RB(low) CD4+ T cells. J. Exp. Med. 1996, 183, 2669–2674. [Google Scholar] [CrossRef]
- Read, S.; Greenwald, R.; Izcue, A.; Robinson, N.; Mandelbrot, D.; Francisco, L.; Sharpe, A.H.; Powrie, F. Blockade of CTLA-4 on CD4+CD25+ regulatory T cells abrogates their function in vivo. J. Immunol. 2006, 177, 4376–4383. [Google Scholar] [CrossRef] [Green Version]
- Read, S.; Malmstrom, V.; Powrie, F. Cytotoxic T lymphocyte-associated antigen 4 plays an essential role in the function of CD25(+)CD4(+) regulatory cells that control intestinal inflammation. J. Exp. Med. 2000, 192, 295–302. [Google Scholar] [CrossRef] [Green Version]
- Burzyn, D.; Kuswanto, W.; Kolodin, D.; Shadrach, J.L.; Cerletti, M.; Jang, Y.; Sefik, E.; Tan, T.G.; Wagers, A.J.; Benoist, C.; et al. A special population of regulatory T cells potentiates muscle repair. Cell 2013, 155, 1282–1295. [Google Scholar] [CrossRef] [Green Version]
- Maul, J.; Loddenkemper, C.; Mundt, P.; Berg, E.; Giese, T.; Stallmach, A.; Zeitz, M.; Duchmann, R. Peripheral and intestinal regulatory CD4+ CD25(high) T cells in inflammatory bowel disease. Gastroenterology 2005, 128, 1868–1878. [Google Scholar] [CrossRef]
- Wang, Y.; Song, W.; Yu, S.; Liu, Y.; Chen, Y.G. Intestinal cellular heterogeneity and disease development revealed by single-cell technology. Cell Regen. 2022, 11, 26. [Google Scholar] [CrossRef]
- Huang, B.; Chen, Z.; Geng, L.; Wang, J.; Liang, H.; Cao, Y.; Chen, H.; Huang, W.; Su, M.; Wang, H.; et al. Mucosal Profiling of Pediatric-Onset Colitis and IBD Reveals Common Pathogenics and Therapeutic Pathways. Cell 2019, 179, 1160–1176.e24. [Google Scholar] [CrossRef]
- Jaeger, N.; Gamini, R.; Cella, M.; Schettini, J.L.; Bugatti, M.; Zhao, S.; Rosadini, C.V.; Esaulova, E.; Di Luccia, B.; Kinnett, B.; et al. Single-cell analyses of Crohn’s disease tissues reveal intestinal intraepithelial T cells heterogeneity and altered subset distributions. Nat. Commun. 2021, 12, 1921. [Google Scholar] [CrossRef] [PubMed]
- Kinchen, J.; Chen, H.H.; Parikh, K.; Antanaviciute, A.; Jagielowicz, M.; Fawkner-Corbett, D.; Ashley, N.; Cubitt, L.; Mellado-Gomez, E.; Attar, M.; et al. Structural Remodeling of the Human Colonic Mesenchyme in Inflammatory Bowel Disease. Cell 2018, 175, 372–386.e17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martin, J.C.; Chang, C.; Boschetti, G.; Ungaro, R.; Giri, M.; Grout, J.A.; Gettler, K.; Chuang, L.S.; Nayar, S.; Greenstein, A.J.; et al. Single-Cell Analysis of Crohn’s Disease Lesions Identifies a Pathogenic Cellular Module Associated with Resistance to Anti-TNF Therapy. Cell 2019, 178, 1493–1508.e20. [Google Scholar] [CrossRef] [PubMed]
- Parikh, K.; Antanaviciute, A.; Fawkner-Corbett, D.; Jagielowicz, M.; Aulicino, A.; Lagerholm, C.; Davis, S.; Kinchen, J.; Chen, H.H.; Alham, N.K.; et al. Colonic epithelial cell diversity in health and inflammatory bowel disease. Nature 2019, 567, 49–55. [Google Scholar] [CrossRef] [PubMed]
- Smillie, C.S.; Biton, M.; Ordovas-Montanes, J.; Sullivan, K.M.; Burgin, G.; Graham, D.B.; Herbst, R.H.; Rogel, N.; Slyper, M.; Waldman, J.; et al. Intra- and Inter-cellular Rewiring of the Human Colon during Ulcerative Colitis. Cell 2019, 178, 714–730.e22. [Google Scholar] [CrossRef]
- West, N.R.; Hegazy, A.N.; Owens, B.M.J.; Bullers, S.J.; Linggi, B.; Buonocore, S.; Coccia, M.; Gortz, D.; This, S.; Stockenhuber, K.; et al. Oncostatin M drives intestinal inflammation and predicts response to tumor necrosis factor-neutralizing therapy in patients with inflammatory bowel disease. Nat. Med. 2017, 23, 579–589. [Google Scholar] [CrossRef] [Green Version]
- Mitsialis, V.; Wall, S.; Liu, P.; Ordovas-Montanes, J.; Parmet, T.; Vukovic, M.; Spencer, D.; Field, M.; McCourt, C.; Toothaker, J.; et al. Single-Cell Analyses of Colon and Blood Reveal Distinct Immune Cell Signatures of Ulcerative Colitis and Crohn’s Disease. Gastroenterology 2020, 159, 591–608.e10. [Google Scholar] [CrossRef]
- Luoma, A.M.; Suo, S.; Williams, H.L.; Sharova, T.; Sullivan, K.; Manos, M.; Bowling, P.; Hodi, F.S.; Rahma, O.; Sullivan, R.J.; et al. Molecular Pathways of Colon Inflammation Induced by Cancer Immunotherapy. Cell 2020, 182, 655–671.e22. [Google Scholar] [CrossRef]
- Devlin, J.C.; Axelrad, J.; Hine, A.M.; Chang, S.; Sarkar, S.; Lin, J.D.; Ruggles, K.V.; Hudesman, D.; Cadwell, K.; Loke, P. Single-Cell Transcriptional Survey of Ileal-Anal Pouch Immune Cells From Ulcerative Colitis Patients. Gastroenterology 2021, 160, 1679–1693. [Google Scholar] [CrossRef]
- Hovhannisyan, Z.; Treatman, J.; Littman, D.R.; Mayer, L. Characterization of interleukin-17-producing regulatory T cells in inflamed intestinal mucosa from patients with inflammatory bowel diseases. Gastroenterology 2011, 140, 957–965. [Google Scholar] [CrossRef] [Green Version]
- Rubtsov, Y.P.; Niec, R.E.; Josefowicz, S.; Li, L.; Darce, J.; Mathis, D.; Benoist, C.; Rudensky, A.Y. Stability of the regulatory T cell lineage in vivo. Science 2010, 329, 1667–1671. [Google Scholar] [CrossRef] [Green Version]
- Wing, J.B.; Sakaguchi, S. Multiple treg suppressive modules and their adaptability. Front. Immunol. 2012, 3, 178. [Google Scholar] [CrossRef] [Green Version]
- Levine, A.G.; Mendoza, A.; Hemmers, S.; Moltedo, B.; Niec, R.E.; Schizas, M.; Hoyos, B.E.; Putintseva, E.V.; Chaudhry, A.; Dikiy, S.; et al. Stability and function of regulatory T cells expressing the transcription factor T-bet. Nature 2017, 546, 421–425. [Google Scholar] [CrossRef] [Green Version]
- Koch, M.A.; Tucker-Heard, G.; Perdue, N.R.; Killebrew, J.R.; Urdahl, K.B.; Campbell, D.J. The transcription factor T-bet controls regulatory T cell homeostasis and function during type 1 inflammation. Nat. Immunol. 2009, 10, 595–602. [Google Scholar] [CrossRef]
- Zheng, Y.; Chaudhry, A.; Kas, A.; deRoos, P.; Kim, J.M.; Chu, T.T.; Corcoran, L.; Treuting, P.; Klein, U.; Rudensky, A.Y. Regulatory T-cell suppressor program co-opts transcription factor IRF4 to control T(H)2 responses. Nature 2009, 458, 351–356. [Google Scholar] [CrossRef] [Green Version]
- Cretney, E.; Xin, A.; Shi, W.; Minnich, M.; Masson, F.; Miasari, M.; Belz, G.T.; Smyth, G.K.; Busslinger, M.; Nutt, S.L.; et al. The transcription factors Blimp-1 and IRF4 jointly control the differentiation and function of effector regulatory T cells. Nat. Immunol. 2011, 12, 304–311. [Google Scholar] [CrossRef]
- Wohlfert, E.A.; Grainger, J.R.; Bouladoux, N.; Konkel, J.E.; Oldenhove, G.; Ribeiro, C.H.; Hall, J.A.; Yagi, R.; Naik, S.; Bhairavabhotla, R.; et al. GATA3 controls Foxp3(+) regulatory T cell fate during inflammation in mice. J. Clin. Investig. 2011, 121, 4503–4515. [Google Scholar] [CrossRef] [Green Version]
- Chaudhry, A.; Rudra, D.; Treuting, P.; Samstein, R.M.; Liang, Y.; Kas, A.; Rudensky, A.Y. CD4+ regulatory T cells control TH17 responses in a Stat3-dependent manner. Science 2009, 326, 986–991. [Google Scholar] [CrossRef] [Green Version]
- Ohnmacht, C.; Park, J.H.; Cording, S.; Wing, J.B.; Atarashi, K.; Obata, Y.; Gaboriau-Routhiau, V.; Marques, R.; Dulauroy, S.; Fedoseeva, M.; et al. MUCOSAL IMMUNOLOGY. The microbiota regulates type 2 immunity through RORgammat(+) T cells. Science 2015, 349, 989–993. [Google Scholar] [CrossRef]
- Sefik, E.; Geva-Zatorsky, N.; Oh, S.; Konnikova, L.; Zemmour, D.; McGuire, A.M.; Burzyn, D.; Ortiz-Lopez, A.; Lobera, M.; Yang, J.; et al. MUCOSAL IMMUNOLOGY. Individual intestinal symbionts induce a distinct population of RORgamma(+) regulatory T cells. Science 2015, 349, 993–997. [Google Scholar] [CrossRef] [Green Version]
- Linterman, M.A.; Pierson, W.; Lee, S.K.; Kallies, A.; Kawamoto, S.; Rayner, T.F.; Srivastava, M.; Divekar, D.P.; Beaton, L.; Hogan, J.J.; et al. Foxp3+ follicular regulatory T cells control the germinal center response. Nat. Med. 2011, 17, 975–982. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chung, Y.; Tanaka, S.; Chu, F.; Nurieva, R.I.; Martinez, G.J.; Rawal, S.; Wang, Y.H.; Lim, H.; Reynolds, J.M.; Zhou, X.H.; et al. Follicular regulatory T cells expressing Foxp3 and Bcl-6 suppress germinal center reactions. Nat. Med. 2011, 17, 983–988. [Google Scholar] [CrossRef] [PubMed]
- Wollenberg, I.; Agua-Doce, A.; Hernandez, A.; Almeida, C.; Oliveira, V.G.; Faro, J.; Graca, L. Regulation of the germinal center reaction by Foxp3+ follicular regulatory T cells. J. Immunol. 2011, 187, 4553–4560. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schiering, C.; Krausgruber, T.; Chomka, A.; Frohlich, A.; Adelmann, K.; Wohlfert, E.A.; Pott, J.; Griseri, T.; Bollrath, J.; Hegazy, A.N.; et al. The alarmin IL-33 promotes regulatory T-cell function in the intestine. Nature 2014, 513, 564–568. [Google Scholar] [CrossRef]
- Kim, K.S.; Hong, S.W.; Han, D.; Yi, J.; Jung, J.; Yang, B.G.; Lee, J.Y.; Lee, M.; Surh, C.D. Dietary antigens limit mucosal immunity by inducing regulatory T cells in the small intestine. Science 2016, 351, 858–863. [Google Scholar] [CrossRef]
- Negi, S.; Saini, S.; Tandel, N.; Sahu, K.; Mishra, R.P.N.; Tyagi, R.K. Translating Treg Therapy for Inflammatory Bowel Disease in Humanized Mice. Cells 2021, 10, 1847. [Google Scholar] [CrossRef]
- Cassinotti, A.; Passamonti, F.; Segato, S. Cell Therapy in Inflammatory Bowel Disease. Pharmacol. Res. 2021, 163, 105247. [Google Scholar] [CrossRef]
- Bacchetta, R.; Bigler, M.; Touraine, J.L.; Parkman, R.; Tovo, P.A.; Abrams, J.; de Waal Malefyt, R.; de Vries, J.E.; Roncarolo, M.G. High levels of interleukin 10 production in vivo are associated with tolerance in SCID patients transplanted with HLA mismatched hematopoietic stem cells. J. Exp. Med. 1994, 179, 493–502. [Google Scholar] [CrossRef]
- He, J.; Tsai, L.M.; Leong, Y.A.; Hu, X.; Ma, C.S.; Chevalier, N.; Sun, X.; Vandenberg, K.; Rockman, S.; Ding, Y.; et al. Circulating precursor CCR7(lo)PD-1(hi) CXCR5(+) CD4(+) T cells indicate Tfh cell activity and promote antibody responses upon antigen reexposure. Immunity 2013, 39, 770–781. [Google Scholar] [CrossRef] [Green Version]
- Ueno, H.; Banchereau, J.; Vinuesa, C.G. Pathophysiology of T follicular helper cells in humans and mice. Nat. Immunol. 2015, 16, 142–152. [Google Scholar] [CrossRef] [Green Version]
- Moore, K.W.; de Waal Malefyt, R.; Coffman, R.L.; O’Garra, A. Interleukin-10 and the interleukin-10 receptor. Annu. Rev. Immunol. 2001, 19, 683–765. [Google Scholar] [CrossRef]
- Jia, X.; Zhai, T.; Wang, B.; Yao, Q.; Li, Q.; Mu, K.; Zhang, J.A. Decreased number and impaired function of type 1 regulatory T cells in autoimmune diseases. J. Cell. Physiol. 2019, 234, 12442–12450. [Google Scholar] [CrossRef]
- Sun, L.; Kong, R.; Li, H.; Wang, D. The Role of T Follicular Helper Cells and Interleukin-21 in the Pathogenesis of Inflammatory Bowel Disease. Gastroenterol. Res. Pract. 2021, 2021, 9621738. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gomez-Bris, R.; Saez, A.; Herrero-Fernandez, B.; Rius, C.; Sanchez-Martinez, H.; Gonzalez-Granado, J.M. CD4 T-Cell Subsets and the Pathophysiology of Inflammatory Bowel Disease. Int. J. Mol. Sci. 2023, 24, 2696. https://doi.org/10.3390/ijms24032696
Gomez-Bris R, Saez A, Herrero-Fernandez B, Rius C, Sanchez-Martinez H, Gonzalez-Granado JM. CD4 T-Cell Subsets and the Pathophysiology of Inflammatory Bowel Disease. International Journal of Molecular Sciences. 2023; 24(3):2696. https://doi.org/10.3390/ijms24032696
Chicago/Turabian StyleGomez-Bris, Raquel, Angela Saez, Beatriz Herrero-Fernandez, Cristina Rius, Hector Sanchez-Martinez, and Jose M. Gonzalez-Granado. 2023. "CD4 T-Cell Subsets and the Pathophysiology of Inflammatory Bowel Disease" International Journal of Molecular Sciences 24, no. 3: 2696. https://doi.org/10.3390/ijms24032696