
The Journey of Mitochondrial Protein Import and the Roadmap to Follow
 , , and
, , and
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
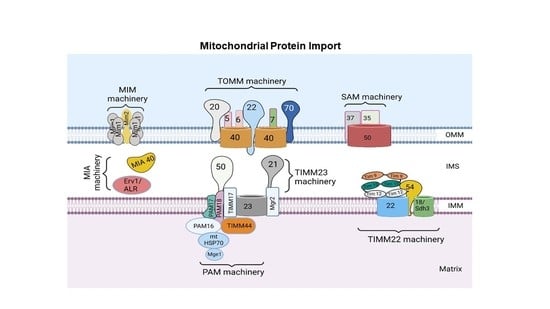
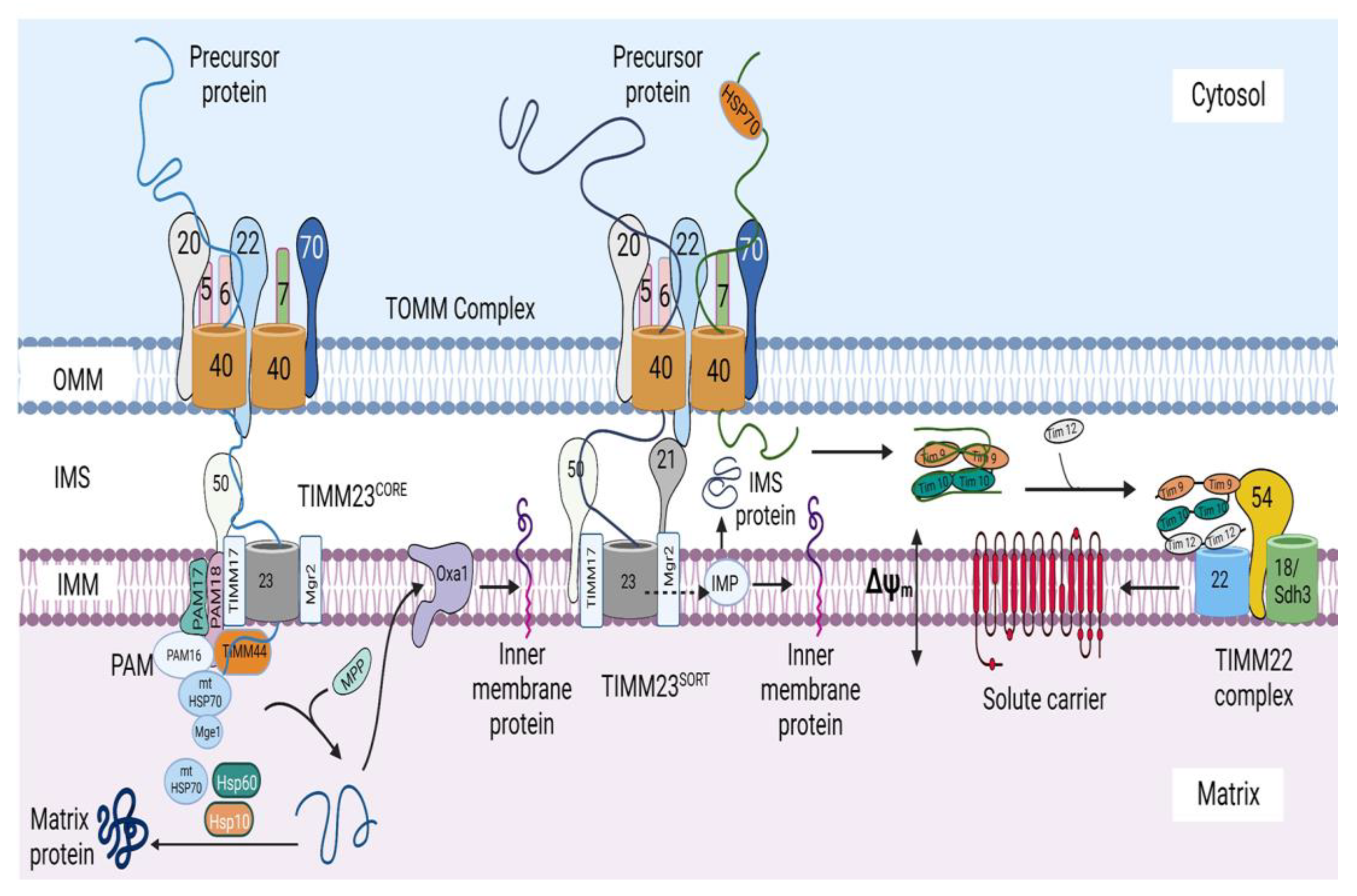
2. Mitochondrial Protein Import System
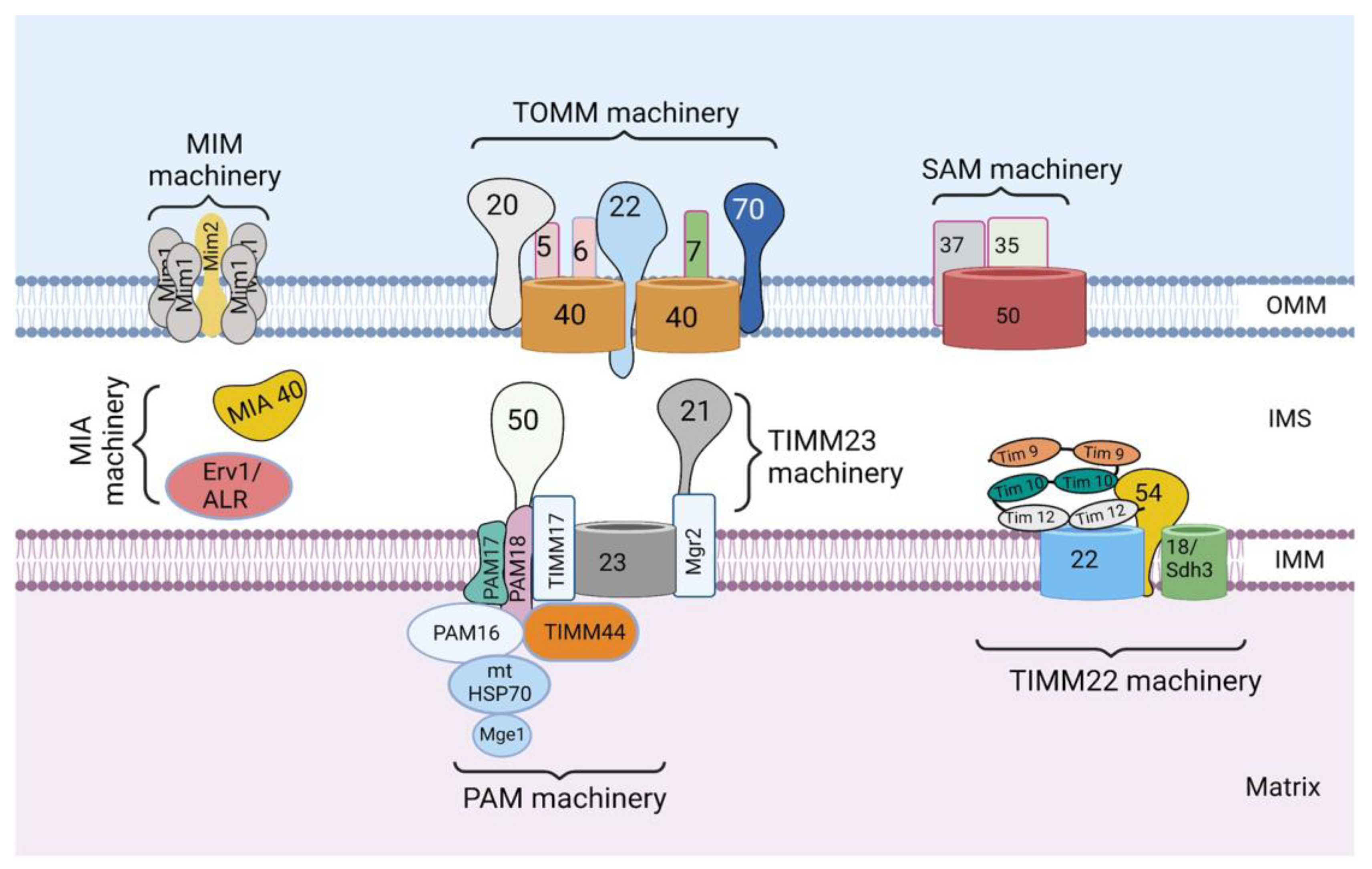
3. Protein Sorting at the Outer Mitochondrial Membrane
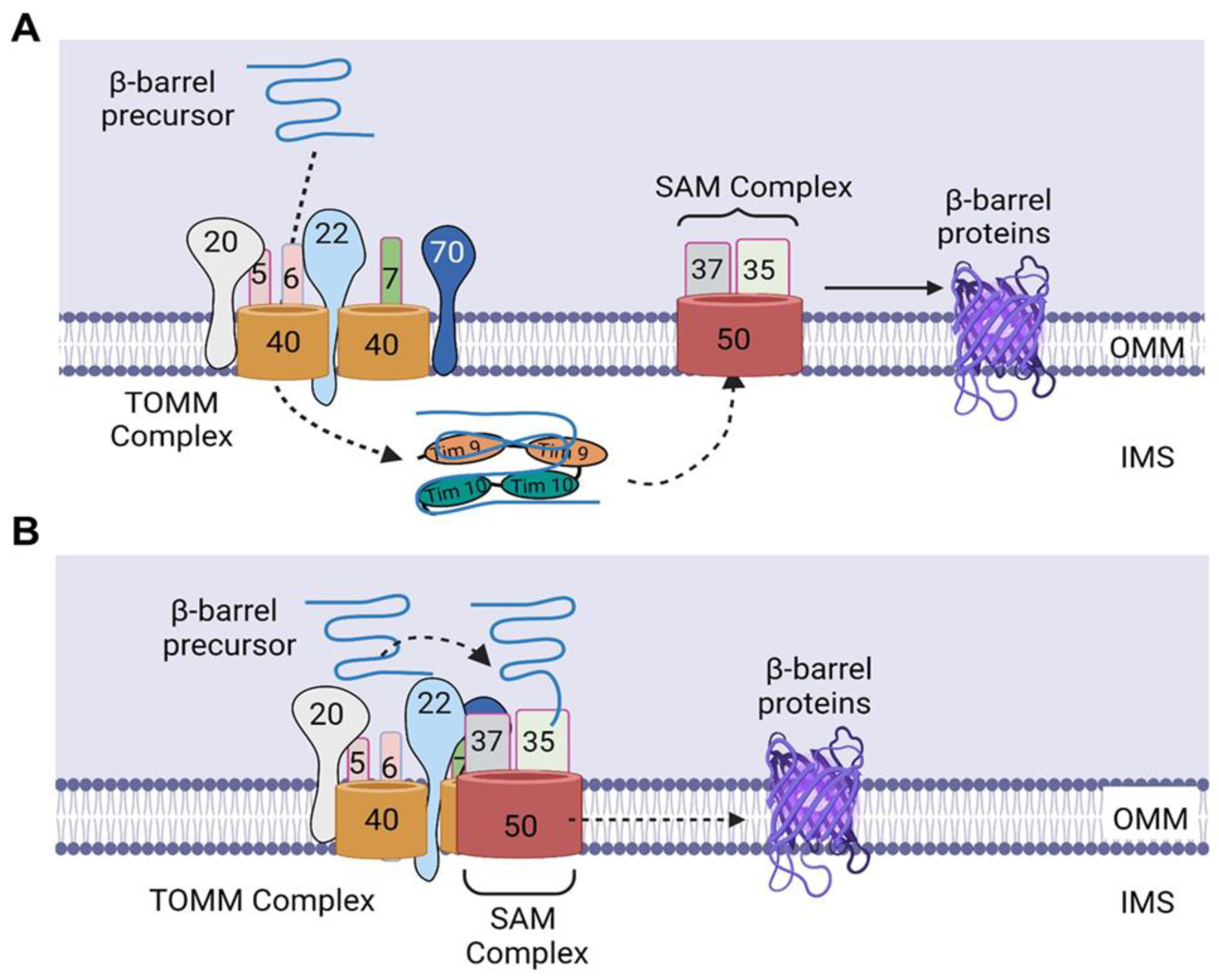
3.1. Sorting of β-Barrel Proteins into the Outer Mitochondrial Membrane
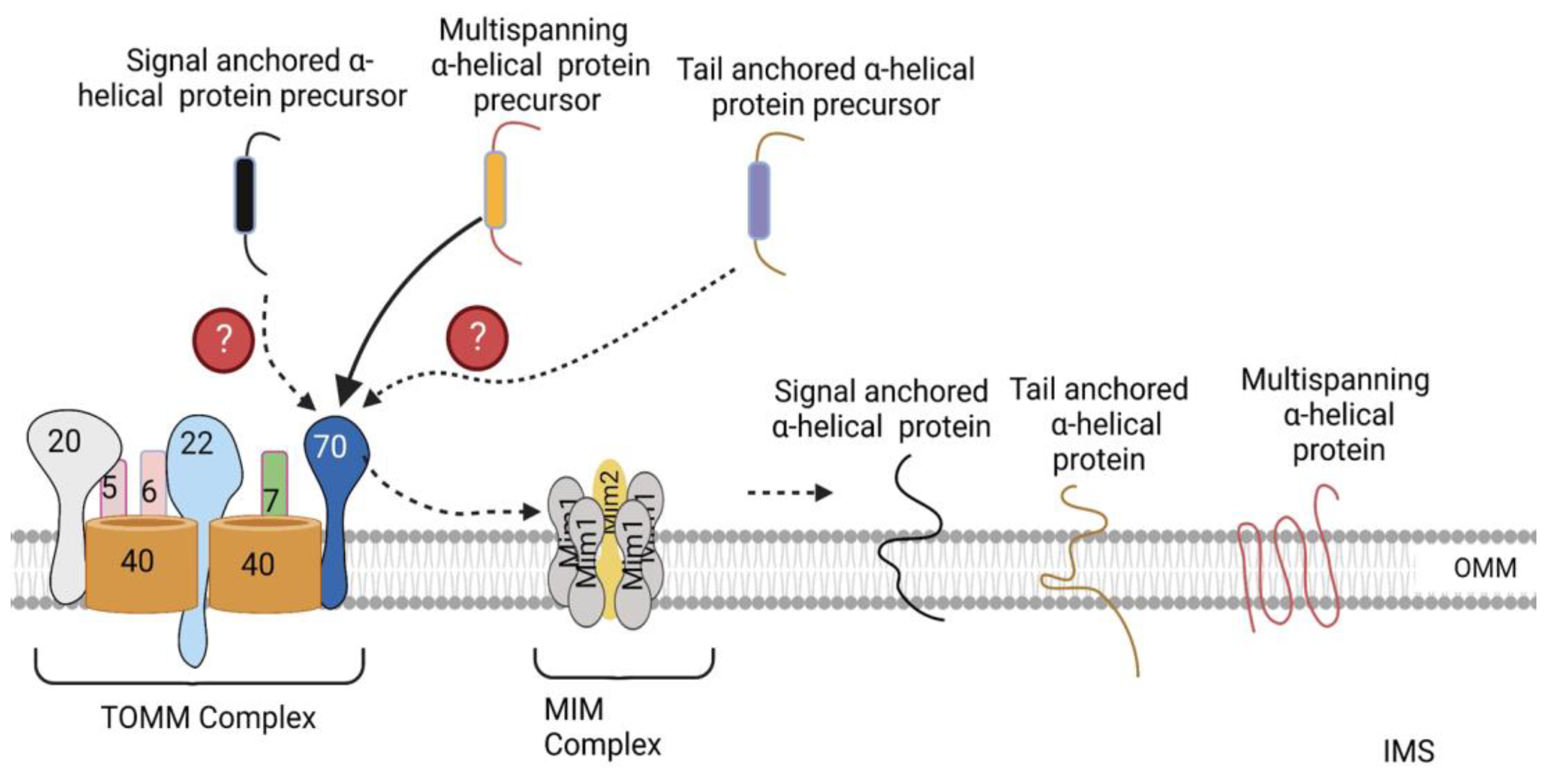
3.2. Sorting of α-Helical Proteins into the Outer Mitochondrial Membrane
4. Protein Sorting into the Intermembrane Space
5. Protein Sorting at the Inner Mitochondrial Membrane and Matrix
5.1. Import of Precursors Containing Presequences
5.1.1. Sorting of Precursors Containing Presequences into the Inner Mitochondrial Membrane
5.1.2. Sorting of Precursors Containing Presequences into the Matrix
5.2. Import of Carrier Precursors into the Inner Mitochondrial Membrane
6. Consequences of Defective Mitochondrial Protein Import
6.1. Pathobiological Implications of Altered Mitochondrial Unfolded Protein Response
6.1.1. Parkinson’s Disease
6.1.2. Alzheimer’s Disease
6.1.3. Cardiovascular Diseases
6.1.4. Cancer
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Wang, Z.; Ying, Z.; Bosy-Westphal, A.; Zhang, J.; Schautz, B.; Later, W.; Heymsfield, S.B.; Müller, M.J. Specific metabolic rates of major organs and tissues across adulthood: Evaluation by mechanistic model of resting energy expenditure. Am. J. Clin. Nutr. 2010, 92, 1369–1377. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pagliarini, D.J.; Calvo, S.E.; Chang, B.; Sheth, S.A.; Vafai, S.B.; Ong, S.-E.; Walford, G.A.; Sugiana, C.; Boneh, A.; Chen, W.K.; et al. A mitochondrial protein compendium elucidates complex I disease biology. Cell 2008, 134, 112–123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Archibald, J.M. Endosymbiosis and Eukaryotic Cell Evolution. Curr. Biol. 2015, 25, R911–R921. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wiedemann, N.; Pfanner, N. Mitochondrial Machineries for Protein Import and Assembly. Annu. Rev. Biochem. 2017, 86, 685–714. [Google Scholar] [CrossRef] [Green Version]
- Srinivasan, S.; Guha, M.; Kashina, A.; Avadhani, N.G. Mitochondrial dysfunction and mitochondrial dynamics-The cancer connection. Biochim. Biophys. Acta Bioenerg. 2017, 1858, 602–614. [Google Scholar] [CrossRef]
- Adebayo, M.; Singh, S.; Singh, A.P.; Dasgupta, S. Mitochondrial fusion and fission: The fine-tune balance for cellular homeostasis. FASEB J. 2021, 35, e21620. [Google Scholar] [CrossRef]
- O’Rourke, B. Mitochondrial ion channels. Annu. Rev. Physiol. 2007, 69, 19–49. [Google Scholar] [CrossRef] [Green Version]
- Lemasters, J.J. Modulation of mitochondrial membrane permeability in pathogenesis, autophagy and control of metabolism. J. Gastroenterol. Hepatol. 2007, 22, S31–S37. [Google Scholar] [CrossRef]
- Gilkerson, R.W. Mitochondrial DNA nucleoids determine mitochondrial genetics and dysfunction. Int. J. Biochem. Cell Biol. 2009, 41, 1899–1906. [Google Scholar] [CrossRef]
- Chacinska, A.; Koehler, C.M.; Milenkovic, D.; Lithgow, T.; Pfanner, N. Importing mitochondrial proteins: Machineries and mechanisms. Cell 2009, 138, 628–644. [Google Scholar] [CrossRef] [Green Version]
- Harbauer, A.B.; Zahedi, R.P.; Sickmann, A.; Pfanner, N.; Meisinger, C. The protein import machinery of mitochondria-a regulatory hub in metabolism, stress, and disease. Cell Metab. 2014, 19, 357–372. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dudek, J.; Rehling, P.; van der Laan, M. Mitochondrial protein import: Common principles and physiological networks. Biochim. Biophys. Acta 2013, 1833, 274–285. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, W.; Chen, X.; Zhang, L.; Yi, J.; Ma, Q.; Yin, J.; Zhuo, W.; Gu, J.; Yang, M. Atomic structure of human TOM core complex. Cell Discov. 2020, 6, 67. [Google Scholar] [CrossRef] [PubMed]
- Jores, T.; Klinger, A.; Groß, L.E.; Kawano, S.; Flinner, N.; Duchardt-Ferner, E.; Wöhnert, J.; Kalbacher, H.; Endo, T.; Schleiff, E.; et al. Characterization of the targeting signal in mitochondrial β-barrel proteins. Nat. Commun. 2016, 7, 12036. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qiu, J.; Wenz, L.-S.; Zerbes, R.M.; Oeljeklaus, S.; Bohnert, M.; Stroud, D.A.; Wirth, C.; Ellenrieder, L.; Thornton, N.; Kutik, S.; et al. Coupling of mitochondrial import and export translocases by receptor-mediated supercomplex formation. Cell 2013, 154, 596–608. [Google Scholar] [CrossRef] [Green Version]
- Krimmer, T.; Rapaport, D.; Ryan, M.; Meisinger, C.; Kassenbrock, C.K.; Blachly-Dyson, E.; Forte, M.; Douglas, M.G.; Neupert, W.; Nargang, F.E.; et al. Biogenesis of porin of the outer mitochondrial membrane involves an import pathway via receptors and the general import pore of the TOM complex. J. Cell Biol. 2001, 152, 289–300. [Google Scholar] [CrossRef]
- Wiedemann, N.; Truscott, K.N.; Pfannschmidt, S.; Guiard, B.; Meisinger, C.; Pfanner, N. Biogenesis of the protein import channel Tom40 of the mitochondrial outer membrane: Intermembrane space components are involved in an early stage of the assembly pathway. J. Biol. Chem. 2004, 279, 18188–18194. [Google Scholar] [CrossRef] [Green Version]
- Kutik, S.; Stojanovski, D.; Becker, L.; Becker, T.; Meinecke, M.; Krüger, V.; Prinz, C.; Meisinger, C.; Guiard, B.; Wagner, R.; et al. Dissecting membrane insertion of mitochondrial beta-barrel proteins. Cell 2008, 132, 1011–1024. [Google Scholar] [CrossRef] [Green Version]
- Diederichs, K.A.; Ni, X.; Rollauer, S.E.; Botos, I.; Tan, X.; King, M.S.; Kunji, E.R.S.; Jiang, J.; Buchanan, S.K. Structural insight into mitochondrial β-barrel outer membrane protein biogenesis. Nat. Commun. 2020, 11, 3290. [Google Scholar] [CrossRef]
- Wenz, L.S.; Ellenrieder, L.; Qiu, J.; Bohnert, M.; Zufall, N.; van der Laan, M.; Pfanner, N.; Wiedemann, N.; Becker, T. Sam37 is crucial for formation of the mitochondrial TOM-SAM supercomplex, thereby promoting β-barrel biogenesis. J. Cell Biol. 2015, 210, 1047–1054. [Google Scholar] [CrossRef]
- Ahting, U.; Waizenegger, T.; Neupert, W.; Rapaport, D. Signal-anchored proteins follow a unique insertion pathway into the outer membrane of mitochondria. J. Biol. Chem. 2005, 280, 48–53. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meineke, B.; Engl, G.; Kemper, C.; Vasiljev-Neumeyer, A.; Paulitschke, H.; Rapaport, D. The outer membrane form of the mitochondrial protein Mcr1 follows a TOM-independent membrane insertion pathway. FEBS Lett. 2008, 582, 855–860. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Becker, T.; Wenz, L.-S.; Krüger, V.; Lehmann, W.; Müller, J.M.; Goroncy, L.; Zufall, N.; Lithgow, T.; Guiard, B.; Chacinska, A.; et al. The mitochondrial import protein Mim1 promotes biogenesis of multispanning outer membrane proteins. J. Cell Biol. 2011, 194, 387–395. [Google Scholar] [CrossRef] [Green Version]
- Papic, D.; Krumpe, K.; Dukanovic, J.; Dimmer, K.S.; Rapaport, D. Multispan mitochondrial outer membrane protein Ugo1 follows a unique Mim1-dependent import pathway. J. Cell Biol. 2011, 194, 397–405. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Doan, K.N.; Grevel, A.; Mårtensson, C.U.; Ellenrieder, L.; Thornton, N.; Wenz, L.-S.; Opaliński, Ł.; Guiard, B.; Pfanner, N.; Becker, T. The Mitochondrial Import Complex MIM Functions as Main Translocase for α-Helical Outer Membrane Proteins. Cell Rep. 2020, 31, 107567. [Google Scholar] [CrossRef]
- Kemper, C.; Habib, S.; Engl, G.; Heckmeyer, P.; Dimmer, K.S.; Rapaport, D. Integration of tail-anchored proteins into the mitochondrial outer membrane does not require any known import components. J. Cell Sci. 2008, 121, 1990–1998. [Google Scholar] [CrossRef] [Green Version]
- Vögtle, F.-N.; Keller, M.; Taskin, A.A.; Horvath, S.E.; Guan, X.L.; Prinz, C.; Opalińska, M.; Zorzin, C.; van der Laan, M.; Wenk, M.R.; et al. The fusogenic lipid phosphatidic acid promotes the biogenesis of mitochondrial outer membrane protein Ugo1. J. Cell Biol. 2015, 210, 951–960. [Google Scholar] [CrossRef] [Green Version]
- Sinzel, M.; Tan, T.; Wendling, P.; Kalbacher, H.; Özbalci, C.; Chelius, X.; Westermann, B.; Brügger, B.; Rapaport, D.; Dimmer, K.S. Mcp3 is a novel mitochondrial outer membrane protein that follows a unique IMP-dependent biogenesis pathway. EMBO Rep. 2016, 17, 965–981. [Google Scholar] [CrossRef] [Green Version]
- Stojanovski, D.; Müller, J.M.; Milenkovic, D.; Guiard, B.; Pfanner, N.; Chacinska, A. The MIA system for protein import into the mitochondrial intermembrane space. Biochim. Biophys. Acta 2008, 1783, 610–617. [Google Scholar] [CrossRef] [Green Version]
- Durigon, R.; Wang, Q.; Pavia, E.C.; Grant, C.M.; Lu, H. Cytosolic thioredoxin system facilitates the import of mitochondrial small Tim proteins. EMBO Rep. 2012, 13, 916–922. [Google Scholar] [CrossRef]
- Gornicka, A.; Bragoszewski, P.; Chroscicki, P.; Wenz, L.-S.; Schulz, C.; Rehling, P.; Chacinska, A. A discrete pathway for the transfer of intermembrane space proteins across the outer membrane of mitochondria. Mol. Biol. Cell 2014, 25, 3999–4009. [Google Scholar] [CrossRef] [PubMed]
- Kurz, M.; Martin, H.; Rassow, J.; Pfanner, N.; Ryan, M.T. Biogenesis of Tim proteins of the mitochondrial carrier import pathway: Differential targeting mechanisms and crossing over with the main import pathway. Mol. Biol. Cell 1999, 10, 2461–2474. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Milenkovic, D.; Ramming, T.; Müller, J.M.; Wenz, L.-S.; Gebert, N.; Schulze-Specking, A.; Stojanovski, D.; Rospert, S.; Chacinska, A. Identification of the signal directing Tim9 and Tim10 into the intermembrane space of mitochondria. Mol. Biol. Cell 2009, 20, 2530–2539. [Google Scholar] [CrossRef] [Green Version]
- Peleh, V.; Cordat, E.; Herrmann, J.M. Mia40 is a trans-site receptor that drives protein import into the mitochondrial intermembrane space by hydrophobic substrate binding. eLife 2016, 5, e16177. [Google Scholar] [CrossRef]
- Mesecke, N.; Bihlmaier, K.; Grumbt, B.; Longen, S.; Terziyska, N.; Hell, K.; Herrmann, J.M. The zinc-binding protein Hot13 promotes oxidation of the mitochondrial import receptor Mia40. EMBO Rep. 2008, 9, 1107–1113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dabir, D.V.; Leverich, E.P.; Kim, S.-K.; Tsai, F.D.; Hirasawa, M.; Knaff, D.B.; Koehler, C.M. A role for cytochrome c and cytochrome c peroxidase in electron shuttling from Erv1. EMBO J. 2007, 26, 4801–4811. [Google Scholar] [CrossRef] [Green Version]
- Bihlmaier, K.; Mesecke, N.; Terziyska, N.; Bien, M.; Hell, K.; Herrmann, J.M. The disulfide relay system of mitochondria is connected to the respiratory chain. J. Cell Biol. 2007, 179, 389–395. [Google Scholar] [CrossRef] [Green Version]
- Ramesh, A.; Peleh, V.; Martinez-Caballero, S.; Wollweber, F.; Sommer, F.; van der Laan, M.; Schroda, M.; Alexander, R.T.; Campo, M.L.; Herrmann, J.M. A disulfide bond in the TIM23 complex is crucial for voltage gating and mitochondrial protein import. J. Cell Biol. 2016, 214, 417–431. [Google Scholar] [CrossRef] [Green Version]
- Zhao, F.; Zou, M.H. Role of the Mitochondrial Protein Import Machinery and Protein Processing in Heart Disease. Front. Cardiovasc. Med. 2021, 8, 749756. [Google Scholar] [CrossRef]
- Roise, D.; Horvath, S.J.; Tomich, J.M.; Richards, J.H.; Schatz, G. A chemically synthesized pre-sequence of an imported mitochondrial protein can form an amphiphilic helix and perturb natural and artificial phospholipid bilayers. EMBO J. 1986, 5, 1327–1334. [Google Scholar] [CrossRef]
- Abe, Y.; Shodai, T.; Muto, T.; Mihara, K.; Torii, H.; Nishikawa, S.-I.; Endo, T.; Kohda, D. Structural basis of presequence recognition by the mitochondrial protein import receptor Tom20. Cell 2000, 100, 551–560. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bolliger, L.; Junne, T.; Schatz, G.; Lithgow, T. Acidic receptor domains on both sides of the outer membrane mediate translocation of precursor proteins into yeast mitochondria. EMBO J. 1995, 14, 6318–6326. [Google Scholar] [CrossRef] [PubMed]
- Moczko, M.; Bömer, U.; Kübrich, M.; Zufall, N.; Hönlinger, A.; Pfanner, N. The intermembrane space domain of mitochondrial Tom22 functions as a trans binding site for preproteins with N-terminal targeting sequences. Mol. Cell. Biol. 1997, 17, 6574–6584. [Google Scholar] [CrossRef] [Green Version]
- Chacinska, A.; Lind, M.; Frazier, A.E.; Dudek, J.; Meisinger, C.; Geissler, A.; Sickmann, A.; Meyer, H.E.; Truscott, K.N.; Guiard, B.; et al. Mitochondrial presequence translocase: Switching between TOM tethering and motor recruitment involves Tim21 and Tim17. Cell 2005, 120, 817–829. [Google Scholar] [CrossRef] [Green Version]
- Albrecht, R.; Rehling, P.; Chacinska, A.; Brix, J.; Cadamuro, S.A.; Volkmer, R.; Guiard, B.; Pfanner, N.; Zeth, K. The Tim21 binding domain connects the preprotein translocases of both mitochondrial membranes. EMBO Rep. 2006, 7, 1233–1238. [Google Scholar] [CrossRef] [Green Version]
- Meinecke, M.; Wagner, R.; Kovermann, P.; Guiard, B.; Mick, D.U.; Hutu, D.P.; Voos, W.; Truscott, K.N.; Chacinska, A.; Pfanner, N.; et al. Tim50 maintains the permeability barrier of the mitochondrial inner membrane. Science 2006, 312, 1523–1526. [Google Scholar] [CrossRef]
- Naamati, A.; Regev-Rudzki, N.; Galperin, S.; Lill, R.; Pines, O. Dual targeting of Nfs1 and discovery of its novel processing enzyme, Icp55. J. Biol. Chem. 2009, 284, 30200–30208. [Google Scholar] [CrossRef] [Green Version]
- Ostermann, J.; Horwich, A.L.; Neupert, W.; Hartl, F.U. Protein folding in mitochondria requires complex formation with hsp60 and ATP hydrolysis. Nature 1989, 341, 125–130. [Google Scholar] [CrossRef] [Green Version]
- van der Laan, M.; Rissler, M.; Rehling, P. Mitochondrial preprotein translocases as dynamic molecular machines. FEMS Yeast Res. 2006, 6, 849–861. [Google Scholar] [CrossRef] [Green Version]
- Martin, J.; Mahlke, K.; Pfanner, N. Role of an energized inner membrane in mitochondrial protein import. Delta psi drives the movement of presequences. J. Biol. Chem. 1991, 266, 18051–18057. [Google Scholar] [CrossRef] [PubMed]
- Truscott, K.N.; Kovermann, P.; Geissler, A.; Merlin, A.; Meijer, M.; Driessen, A.J.; Rassow, J.; Pfanner, N.; Wagner, R. A presequence- and voltage-sensitive channel of the mitochondrial preprotein translocase formed by Tim23. Nat. Struct. Biol. 2001, 8, 1074–1082. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van der Laan, M.; Wiedemann, N.; Mick, D.U.; Guiard, B.; Rehling, P.; Pfanner, N. A role for Tim21 in membrane-potential-dependent preprotein sorting in mitochondria. Curr. Biol. 2006, 16, 2271–2276. [Google Scholar] [CrossRef] [PubMed]
- Bohnert, M.; Rehling, P.; Guiard, B.; Herrmann, J.M.; Pfanner, N.; van der Laan, M. Cooperation of stop-transfer and conservative sorting mechanisms in mitochondrial protein transport. Curr. Biol. 2010, 20, 1227–1232. [Google Scholar] [CrossRef] [Green Version]
- Stiller, S.B.; Höpker, J.; Oeljeklaus, S.; Schütze, C.; Schrempp, S.G.; Vent-Schmidt, J.; Horvath, S.E.; Frazier, A.; Gebert, N.; van der Laan, M.; et al. Mitochondrial OXA Translocase Plays a Major Role in Biogenesis of Inner-Membrane Proteins. Cell Metab. 2016, 23, 901–908. [Google Scholar] [CrossRef] [Green Version]
- Young, J.C.; Hoogenraad, N.J.; Hartl, F.U. Molecular chaperones Hsp90 and Hsp70 deliver preproteins to the mitochondrial import receptor Tom70. Cell 2003, 112, 41–50. [Google Scholar] [CrossRef] [Green Version]
- Wu, Y.; Sha, B. Crystal structure of yeast mitochondrial outer membrane translocon member Tom70p. Nat. Struct. Mol. Biol. 2006, 13, 589–593. [Google Scholar] [CrossRef] [PubMed]
- Curran, S.P.; Leuenberger, D.; Schmidt, E.; Koehler, C.M. The role of the Tim8p-Tim13p complex in a conserved import pathway for mitochondrial polytopic inner membrane proteins. J. Cell Biol. 2002, 158, 1017–1027. [Google Scholar] [CrossRef] [Green Version]
- Gebert, N.; Chacinska, A.; Wagner, K.; Guiard, B.; Koehler, C.M.; Rehling, P.; Pfanner, N.; Wiedemann, N. Assembly of the three small Tim proteins precedes docking to the mitochondrial carrier translocase. EMBO Rep. 2008, 9, 548–554. [Google Scholar] [CrossRef] [Green Version]
- Wagner, K.; Gebert, N.; Guiard, B.; Brandner, K.; Truscott, K.N.; Wiedemann, N.; Pfanner, N.; Rehling, P. The assembly pathway of the mitochondrial carrier translocase involves four preprotein translocases. Mol. Cell. Biol. 2008, 28, 4251–4260. [Google Scholar] [CrossRef] [Green Version]
- Rehling, P.; Model, K.; Brandner, K.; Kovermann, P.; Sickmann, A.; Meyer, H.E.; Kühlbrandt, W.; Wagner, R.; Truscott, K.N.; Pfanner, N. Protein insertion into the mitochondrial inner membrane by a twin-pore translocase. Science 2003, 299, 1747–1751. [Google Scholar] [CrossRef] [Green Version]
- Gebert, N.; Gebert, M.; Oeljeklaus, S.; von der Malsburg, K.; Stroud, D.A.; Kulawiak, B.; Wirth, C.; Zahedi, R.P.; Dolezal, P.; Wiese, S.; et al. Dual function of Sdh3 in the respiratory chain and TIM22 protein translocase of the mitochondrial inner membrane. Mol. Cell 2011, 44, 811–818. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bolender, N.; Sickmann, A.; Wagner, R.; Meisinger, C.; Pfanner, N. Multiple pathways for sorting mitochondrial precursor proteins. EMBO Rep. 2008, 9, 42–49. [Google Scholar] [CrossRef] [Green Version]
- Boos, F.; Labbadia, J.; Herrmann, J.M. How the Mitoprotein-Induced Stress Response Safeguards the Cytosol: A Unified View. Trends Cell Biol. 2020, 30, 241–254. [Google Scholar] [CrossRef] [PubMed]
- Nargund, A.M.; Pellegrino, M.W.; Fiorese, C.J.; Baker, B.M.; Haynes, C.M. Mitochondrial import efficiency of ATFS-1 regulates mitochondrial UPR activation. Science 2012, 337, 587–590. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Horibe, T.; Hoogenraad, N.J. The chop gene contains an element for the positive regulation of the mitochondrial unfolded protein response. PLoS ONE 2007, 2, e835. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lenkiewicz, A.M.; Krakowczyk, M.; Bragoszewski, P. Cytosolic Quality Control of Mitochondrial Protein Precursors-The Early Stages of the Organelle Biogenesis. Int. J. Mol. Sci. 2021, 23, 7. [Google Scholar] [CrossRef]
- Smyrnias, I.; Gray, S.P.; Okonko, D.O.; Sawyer, G.; Zoccarato, A.; Catibog, N.; López, B.; Gonzalez, A.; Ravassa, S.; Díez, J.; et al. Cardioprotective Effect of the Mitochondrial Unfolded Protein Response During Chronic Pressure Overload. J. Am. Coll. Cardiol. 2019, 73, 1795–1806. [Google Scholar] [CrossRef]
- Wang, X.; Chen, X.J. A cytosolic network suppressing mitochondria-mediated proteostatic stress and cell death. Nature 2015, 524, 481–484. [Google Scholar] [CrossRef] [Green Version]
- Coyne, L.P.; Chen, X.J. mPOS is a novel mitochondrial trigger of cell death-implications for neurodegeneration. FEBS Lett. 2018, 592, 759–775. [Google Scholar] [CrossRef] [Green Version]
- Weidberg, H.; Amon, A. MitoCPR-A surveillance pathway that protects mitochondria in response to protein import stress. Science 2018, 360, eaan4146. [Google Scholar] [CrossRef] [Green Version]
- Wrobel, L.; Topf, U.; Bragoszewski, P.; Wiese, S.; Sztolsztener, M.E.; Oeljeklaus, S.; Varabyova, A.; Lirski, M.; Chroscicki, P.; Mroczek, S.; et al. Mistargeted mitochondrial proteins activate a proteostatic response in the cytosol. Nature 2015, 524, 485–488. [Google Scholar] [CrossRef]
- Boos, F.; Krämer, L.; Groh, C.; Jung, F.; Haberkant, P.; Stein, F.; Wollweber, F.; Gackstatter, A.; Zöller, E.; Van Der Laan, M.; et al. Mitochondrial protein-induced stress triggers a global adaptive transcriptional programme. Nat. Cell Biol. 2019, 21, 442–451. [Google Scholar] [CrossRef] [PubMed]
- Schlagowski, A.M.; Knöringer, K.; Morlot, S.; Vicente, A.S.; Flohr, T.; Krämer, L.; Boos, F.; Khalid, N.; Ahmed, S.; Schramm, J.; et al. Increased levels of mitochondrial import factor Mia40 prevent the aggregation of polyQ proteins in the cytosol. EMBO J. 2021, 40, e107913. [Google Scholar] [CrossRef] [PubMed]
- Zhu, L.; Zhou, Q.; He, L.; Chen, L. Mitochondrial unfolded protein response: An emerging pathway in human diseases. Free Radic. Biol. Med. 2021, 163, 125–134. [Google Scholar] [CrossRef] [PubMed]
- Suárez-Rivero, J.M.; Pastor-Maldonado, C.J.; Povea-Cabello, S.; Álvarez-Córdoba, M.; Villalón-García, I.; Talaverón-Rey, M.; Suárez-Carrillo, A.; Munuera-Cabeza, M.; Reche-López, D.; Cilleros-Holgado, P.; et al. Activation of the Mitochondrial Unfolded Protein Response: A New Therapeutic Target? Biomedicines 2022, 10, 1611. [Google Scholar] [CrossRef]
- Jolly, C.; Morimoto, R.I. Role of the heat shock response and molecular chaperones in oncogenesis and cell death. J. Natl. Cancer Inst. 2000, 92, 1564–1572. [Google Scholar] [CrossRef] [Green Version]
- Wawrzynow, B.; Zylicz, A.; Zylicz, M. Chaperoning the guardian of the genome. The two-faced role of molecular chaperones in p53 tumor suppressor action. Biochim. Biophys. Acta Rev. Cancer 2018, 1869, 161–174. [Google Scholar] [CrossRef]
- Arlt, A.; Bauer, I.; Schafmayer, C.; Tepel, J.; Müerköster, S.S.; Brosch, M.; Röder, C.; Kalthoff, H.; Hampe, J.; Moyer, M.P.; et al. Increased proteasome subunit protein expression and proteasome activity in colon cancer relate to an enhanced activation of nuclear factor E2-related factor 2 (Nrf2). Oncogene 2009, 28, 3983–3996. [Google Scholar] [CrossRef] [Green Version]
- Soave, C.L.; Guerin, T.; Liu, J.; Dou, Q.P. Targeting the ubiquitin-proteasome system for cancer treatment: Discovering novel inhibitors from nature and drug repurposing. Cancer Metastasis Rev. 2017, 36, 717–736. [Google Scholar] [CrossRef]
- Franco-Iborra, S.; Vila, M.; Perier, C. Mitochondrial Quality Control in Neurodegenerative Diseases: Focus on Parkinson’s Disease and Huntington’s Disease. Front. Neurosci. 2018, 12, 342. [Google Scholar] [CrossRef] [Green Version]
- Xie, W.; Wan, O.W.; Chung, K.K. New insights into the role of mitochondrial dysfunction and protein aggregation in Parkinson’s disease. Biochim. Biophys. Acta 2010, 1802, 935–941. [Google Scholar] [CrossRef] [Green Version]
- Perier, C.; Vila, M. Mitochondrial biology and Parkinson’s disease. Cold Spring Harb. Perspect. Med. 2012, 2, a009332. [Google Scholar] [CrossRef] [Green Version]
- Celardo, I.; Martins, L.M.; Gandhi, S. Unravelling mitochondrial pathways to Parkinson’s disease. Br. J. Pharmacol. 2014, 171, 1943–1957. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, M.; Yu, S.; Wang, J.; Qiao, J.; Liu, Y.; Wang, S.; Zhao, Y. Ginseng protein protects against mitochondrial dysfunction and neurodegeneration by inducing mitochondrial unfolded protein response in Drosophila melanogaster PINK1 model of Parkinson’s disease. J. Ethnopharmacol. 2020, 247, 112213. [Google Scholar] [CrossRef] [PubMed]
- Cooper, J.F.; Machiela, E.; Dues, D.J.; Spielbauer, K.K.; Senchuk, M.M.; Van Raamsdonk, J.M. Activation of the mitochondrial unfolded protein response promotes longevity and dopamine neuron survival in Parkinson’s disease models. Sci. Rep. 2017, 7, 16441. [Google Scholar] [CrossRef] [Green Version]
- Moisoi, N.; Klupsch, K.; Fedele, V.; East, P.; Sharma, S.; Renton, A.; Plun-Favreau, H.; Edwards, R.E.; Teismann, P.; Esposti, M.D.; et al. Mitochondrial dysfunction triggered by loss of HtrA2 results in the activation of a brain-specific transcriptional stress response. Cell Death Differ. 2009, 16, 449–464. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reeve, A.K.; Ludtmann, M.H.R.; Angelova, P.R.; Simcox, E.M.; Horrocks, M.H.; Klenerman, D.; Gandhi, S.C.; Turnbull, D.M.; Abramov, A.Y. Aggregated α-synuclein and complex I deficiency: Exploration of their relationship in differentiated neurons. Cell Death Dis. 2015, 6, e1820. [Google Scholar] [CrossRef] [Green Version]
- Spillantini, M.G.; Crowther, R.A.; Jakes, R.; Hasegawa, M.; Goedert, M. alpha-Synuclein in filamentous inclusions of Lewy bodies from Parkinson’s disease and dementia with lewy bodies. Proc. Natl. Acad. Sci. USA 1998, 95, 6469–6473. [Google Scholar] [CrossRef] [Green Version]
- Ludtmann, M.H.; Angelova, P.R.; Ninkina, N.N.; Gandhi, S.; Buchman, V.L.; Abramov, A.Y. Monomeric Alpha-Synuclein Exerts a Physiological Role on Brain ATP Synthase. J. Neurosci. 2016, 36, 10510–10521. [Google Scholar] [CrossRef] [Green Version]
- Devi, L.; Raghavendran, V.; Prabhu, B.M.; Avadhani, N.G.; Anandatheerthavarada, H.K. Mitochondrial import and accumulation of alpha-synuclein impair complex I in human dopaminergic neuronal cultures and Parkinson disease brain. J. Biol. Chem. 2008, 283, 9089–9100. [Google Scholar] [CrossRef] [Green Version]
- Di Maio, R.; Barrett, P.J.; Hoffman, E.K.; Barrett, C.W.; Zharikov, A.; Borah, A.; Hu, X.; McCoy, J.; Chu, C.T.; Burton, E.A.; et al. α-Synuclein binds to TOM20 and inhibits mitochondrial protein import in Parkinson’s disease. Sci. Transl. Med. 2016, 8, 342ra378. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, Y.F.; Haynes, C.M. Metabolism and the UPR(mt). Mol. Cell 2016, 61, 677–682. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martinez, B.A.; Petersen, D.A.; Gaeta, A.L.; Stanley, S.P.; Caldwell, G.A.; Caldwell, K.A. Dysregulation of the Mitochondrial Unfolded Protein Response Induces Non-Apoptotic Dopaminergic Neurodegeneration in. J. Neurosci. 2017, 37, 11085–11100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bali, J.; Gheinani, A.H.; Zurbriggen, S.; Rajendran, L. Role of genes linked to sporadic Alzheimer’s disease risk in the production of β-amyloid peptides. Proc. Natl. Acad. Sci. USA 2012, 109, 15307–15311. [Google Scholar] [CrossRef] [Green Version]
- Swerdlow, R.H. Pathogenesis of Alzheimer’s disease. Clin. Interv. Aging 2007, 2, 347–359. [Google Scholar] [PubMed]
- Shen, Y.; Ding, M.; Xie, Z.; Liu, X.; Yang, H.; Jin, S.; Xu, S.; Zhu, Z.; Wang, Y.; Wang, D.; et al. Activation of Mitochondrial Unfolded Protein Response in SHSY5Y Expressing APP Cells and APP/PS1 Mice. Front. Cell. Neurosci. 2019, 13, 568. [Google Scholar] [CrossRef]
- Swerdlow, R.H. Mitochondria and Mitochondrial Cascades in Alzheimer’s Disease. J. Alzheimers Dis. 2018, 62, 1403–1416. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Wang, W.; Li, L.; Perry, G.; Lee, H.G.; Zhu, X. Oxidative stress and mitochondrial dysfunction in Alzheimer’s disease. Biochim. Biophys. Acta 2014, 1842, 1240–1247. [Google Scholar] [CrossRef] [Green Version]
- Kim, D.K.; Mook-Jung, I. The role of cell type-specific mitochondrial dysfunction in the pathogenesis of Alzheimer’s disease. BMB Rep. 2019, 52, 679–688. [Google Scholar] [CrossRef]
- Devi, L.; Prabhu, B.M.; Galati, D.F.; Avadhani, N.G.; Anandatheerthavarada, H.K. Accumulation of amyloid precursor protein in the mitochondrial import channels of human Alzheimer’s disease brain is associated with mitochondrial dysfunction. J. Neurosci. 2006, 26, 9057–9068. [Google Scholar] [CrossRef] [Green Version]
- Lustbader, J.W.; Cirilli, M.; Lin, C.; Xu, H.W.; Takuma, K.; Wang, N.; Caspersen, C.; Chen, X.; Pollak, S.; Chaney, M.; et al. ABAD directly links Abeta to mitochondrial toxicity in Alzheimer’s disease. Science 2004, 304, 448–452. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, Y.; Bai, F. The Association of Tau With Mitochondrial Dysfunction in Alzheimer’s Disease. Front. Neurosci. 2018, 12, 163. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Szabo, L.; Eckert, A.; Grimm, A. Insights into Disease-Associated Tau Impact on Mitochondria. Int. J. Mol. Sci. 2020, 21, 6344. [Google Scholar] [CrossRef] [PubMed]
- Chong, W.C.; Shastri, M.D.; Eri, R. Endoplasmic Reticulum Stress and Oxidative Stress: A Vicious Nexus Implicated in Bowel Disease Pathophysiology. Int. J. Mol. Sci. 2017, 18, 771. [Google Scholar] [CrossRef] [PubMed]
- Hou, M.; Bao, W.; Gao, Y.; Chen, J.; Song, G. Honokiol improves cognitive impairment in APP/PS1 mice through activating mitophagy and mitochondrial unfolded protein response. Chem. Biol. Interact. 2022, 351, 109741. [Google Scholar] [CrossRef] [PubMed]
- Sorrentino, V.; Romani, M.; Mouchiroud, L.; Beck, J.S.; Zhang, H.; D’Amico, D.; Moullan, N.; Potenza, F.; Schmid, A.W.; Rietsch, S.; et al. Enhancing mitochondrial proteostasis reduces amyloid-β proteotoxicity. Nature 2017, 552, 187–193. [Google Scholar] [CrossRef]
- Pérez, M.J.; Ivanyuk, D.; Panagiotakopoulou, V.; Di Napoli, G.; Kalb, S.; Brunetti, D.; Al-Shaana, R.; Kaeser, S.A.; Fraschka, S.A.-K.; Jucker, M.; et al. Loss of function of the mitochondrial peptidase PITRM1 induces proteotoxic stress and Alzheimer’s disease-like pathology in human cerebral organoids. Mol. Psychiatry 2021, 26, 5733–5750. [Google Scholar] [CrossRef]
- Beck, J.S.; Mufson, E.J.; Counts, S.E. Evidence for Mitochondrial UPR Gene Activation in Familial and Sporadic Alzheimer’s Disease. Curr. Alzheimer. Res. 2016, 13, 610–614. [Google Scholar] [CrossRef] [PubMed]
- Svaguša, T.; Martinić, M.; Martinić, M.; Kovačević, L.; Šepac, A.; Miličić, D.; Bulum, J.; Starčević, B.; Sirotković-Skerlev, M.; Seiwerth, F.; et al. Mitochondrial unfolded protein response, mitophagy and other mitochondrial quality control mechanisms in heart disease and aged heart. Croat. Med. J. 2020, 61, 126–138. [Google Scholar] [CrossRef] [PubMed]
- Dietl, A.; Maack, C. Targeting Mitochondrial Calcium Handling and Reactive Oxygen Species in Heart Failure. Curr. Heart Fail Rep. 2017, 14, 338–349. [Google Scholar] [CrossRef]
- Wang, Y.T.; Im, Y.; McCall, M.N.; Huang, K.-T.; Haynes, C.M.; Nehrke, K.; Brookes, P.S. Cardioprotection by the mitochondrial unfolded protein response requires ATF5. Am. J. Physiol. Heart Circ. Physiol. 2019, 317, H472–H478. [Google Scholar] [CrossRef] [PubMed]
- Venkatesh, S.; Li, M.; Saito, T.; Tong, M.; Rashed, E.; Mareedu, S.; Zhai, P.; Bárcena, C.; López-Otín, C.; Yehia, G.; et al. Mitochondrial LonP1 protects cardiomyocytes from ischemia/reperfusion injury in vivo. J. Mol. Cell. Cardiol. 2019, 128, 38–50. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoshino, A.; Okawa, Y.; Ariyoshi, M.; Kaimoto, S.; Uchihashi, M.; Fukai, K.; Iwai-Kanai, E.; Matoba, S. Oxidative post-translational modifications develop LONP1 dysfunction in pressure overload heart failure. Circ. Heart Fail 2014, 7, 500–509. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nandi, S.S.; Katsurada, K.; Mahata, S.K.; Patel, K.P. Neurogenic Hypertension Mediated Mitochondrial Abnormality Leads to Cardiomyopathy: Contribution of UPR. Front. Physiol. 2021, 12, 718982. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Lei, J.; Wang, K.; Ma, L.; Liu, D.; Du, Y.; Wu, Y.; Zhang, S.; Wang, W.; Ma, X.; et al. Mitochondrial Omi/HtrA2 Promotes Caspase Activation Through Cleavage of HAX-1 in Aging Heart. Rejuvenation Res. 2017, 20, 183–192. [Google Scholar] [CrossRef]
- Wang, K.; Yuan, Y.; Liu, X.; Lau, W.B.; Zuo, L.; Wang, X.; Ma, L.; Jiao, K.; Shang, J.; Wang, W.; et al. Cardiac Specific Overexpression of Mitochondrial Omi/HtrA2 Induces Myocardial Apoptosis and Cardiac Dysfunction. Sci. Rep. 2016, 6, 37927. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Malley, J.; Kumar, R.; Inigo, J.; Yadava, N.; Chandra, D. Mitochondrial Stress Response and Cancer. Trends Cancer 2020, 6, 688–701. [Google Scholar] [CrossRef]
- Chen, F.-M.; Huang, L.-J.; Ou-Yang, F.; Kan, J.-Y.; Kao, L.-C.; Hou, M.-F. Activation of mitochondrial unfolded protein response is associated with Her2-overexpression breast cancer. Breast Cancer Res. Treat. 2020, 183, 61–70. [Google Scholar] [CrossRef]
- Kenny, T.C.; Craig, A.J.; Villanueva, A.; Germain, D. Mitohormesis Primes Tumor Invasion and Metastasis. Cell Rep. 2019, 27, 2292–2303.e2296. [Google Scholar] [CrossRef] [Green Version]
- Feldheim, J.; Kessler, A.F.; Schmitt, D.; Wilczek, L.; Linsenmann, T.; Dahlmann, M.; Monoranu, C.M.; Ernestus, R.-I.; Hagemann, C.; Löhr, M. Expression of activating transcription factor 5 (ATF5) is increased in astrocytomas of different WHO grades and correlates with survival of glioblastoma patients. Onco Targets Ther. 2018, 11, 8673–8684. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Monaco, S.E.; Angelastro, J.M.; Szabolcs, M.; Greene, L.A. The transcription factor ATF5 is widely expressed in carcinomas, and interference with its function selectively kills neoplastic, but not nontransformed, breast cell lines. Int. J. Cancer 2007, 120, 1883–1890. [Google Scholar] [CrossRef] [PubMed]
- Hu, M.; Wang, B.; Qian, D.; Li, L.; Zhang, L.; Song, X.; Liu, D.X. Interference with ATF5 function enhances the sensitivity of human pancreatic cancer cells to paclitaxel-induced apoptosis. Anticancer Res. 2012, 32, 4385–4394. [Google Scholar] [PubMed]
- Kong, X.; Meng, W.; Zhou, Z.; Li, Y.; Zhou, B.; Wang, R.; Zhan, L. Overexpression of activating transcription factor 5 in human rectal cancer. Exp. Ther. Med. 2011, 2, 827–831. [Google Scholar] [CrossRef] [Green Version]
- Chen, A.; Qian, D.; Wang, B.; Hu, M.; Lu, J.; Qi, Y.; Liu, D.X. ATF5 is overexpressed in epithelial ovarian carcinomas and interference with its function increases apoptosis through the downregulation of Bcl-2 in SKOV-3 cells. Int. J. Gynecol. Pathol. 2012, 31, 532–537. [Google Scholar] [CrossRef]
- Sheng, Z.; Li, L.; Zhu, L.J.; Smith, T.W.; Demers, A.; Ross, A.H.; Moser, R.P.; Green, M.R. A genome-wide RNA interference screen reveals an essential CREB3L2-ATF5-MCL1 survival pathway in malignant glioma with therapeutic implications. Nat. Med. 2010, 16, 671–677. [Google Scholar] [CrossRef] [Green Version]
- Ishihara, S.; Yasuda, M.; Ishizu, A.; Ishikawa, M.; Shirato, H.; Haga, H. Activating transcription factor 5 enhances radioresistance and malignancy in cancer cells. Oncotarget 2015, 6, 4602–4614. [Google Scholar] [CrossRef] [Green Version]
- Liu, D.X.; Qian, D.; Wang, B.; Yang, J.M.; Lu, Z. p300-Dependent ATF5 acetylation is essential for Egr-1 gene activation and cell proliferation and survival. Mol. Cell. Biol. 2011, 31, 3906–3916. [Google Scholar] [CrossRef] [Green Version]
- Dluzen, D.; Li, G.; Tacelosky, D.; Moreau, M.; Liu, D.X. BCL-2 is a downstream target of ATF5 that mediates the prosurvival function of ATF5 in a cell type-dependent manner. J. Biol. Chem. 2011, 286, 7705–7713. [Google Scholar] [CrossRef] [Green Version]
- Luo, J.; Zeng, B.; Tao, C.; Lu, M.; Ren, G. ClpP regulates breast cancer cell proliferation, invasion and apoptosis by modulating the Src/PI3K/Akt signaling pathway. PeerJ 2020, 8, e8754. [Google Scholar] [CrossRef]
- Seo, J.H.; Rivadeneira, D.B.; Caino, M.C.; Chae, Y.C.; Speicher, D.W.; Tang, H.Y.; Vaira, V.; Bosari, S.; Palleschi, A.; Rampini, P.; et al. The Mitochondrial Unfoldase-Peptidase Complex ClpXP Controls Bioenergetics Stress and Metastasis. PLoS Biol. 2016, 14, e1002507. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cole, A.; Wang, Z.; Coyaud, E.; Voisin, V.; Gronda, M.; Jitkova, Y.; Mattson, R.; Hurren, R.; Babovic, S.; Maclean, N.; et al. Inhibition of the Mitochondrial Protease ClpP as a Therapeutic Strategy for Human Acute Myeloid Leukemia. Cancer Cell 2015, 27, 864–876. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quirós, P.M.; Español, Y.; Acín-Pérez, R.; Rodríguez, F.; Bárcena, C.; Watanabe, K.; Calvo, E.; Loureiro, M.; Fernández-García, M.S.; Fueyo, A.; et al. ATP-dependent Lon protease controls tumor bioenergetics by reprogramming mitochondrial activity. Cell Rep. 2014, 8, 542–556. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, C.; Wang, H.; Li, H.; Chen, X.; Wu, X.; Lu, B.; Zhang, W.; Zhou, Y.; Xiao, G.G.; Gao, G. Inhibition of LONP1 Suppresses Pancreatic Cancer Progression Via c-Jun N-Terminal Kinase Pathway-Meditated Epithelial-Mesenchymal Transition. Pancreas 2019, 48, 629–635. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, J.C.; Seo, J.H.; Agarwal, E.; Wang, Y.; Kossenkov, A.V.; Tang, H.Y.; Speicher, D.W.; Altieri, D.C. Akt phosphorylation of mitochondrial Lonp1 protease enables oxidative metabolism and advanced tumor traits. Oncogene 2019, 38, 6926–6939. [Google Scholar] [CrossRef]
- Fan, W.; Fan, S.-S.; Feng, J.; Xiao, D.; Fan, S.; Luo, J. Elevated expression of HSP10 protein inhibits apoptosis and associates with poor prognosis of astrocytoma. PLoS ONE 2017, 12, e0185563. [Google Scholar] [CrossRef] [Green Version]
- Feng, J.; Luo, J.; Wang, H.; Lu, J.; Zhan, Y.; Zang, H.; Wen, Q.; Wang, W.; Chen, L.; Xu, L.; et al. High expression of heat shock protein 10 (Hsp10) is associated with poor prognosis in oral squamous cell carcinoma. Int. J. Clin. Exp. Pathol. 2017, 10, 7784–7791. [Google Scholar]
- Feng, J.; Zhan, Y.; Zhang, Y.; Zheng, H.; Wang, W.; Fan, S. Increased expression of heat shock protein (HSP) 10 and HSP70 correlates with poor prognosis of nasopharyngeal carcinoma. Cancer Manag. Res. 2019, 11, 8219–8227. [Google Scholar] [CrossRef] [Green Version]
- Hjerpe, E.; Egyhazi, S.; Carlson, J.; Stolt, M.F.; Schedvins, K.; Johansson, H.; Shoshan, M.; Avall-Lundqvist, E. HSP60 predicts survival in advanced serous ovarian cancer. Int. J. Gynecol. Cancer 2013, 23, 448–455. [Google Scholar] [CrossRef]
- Ghosh, J.C.; Dohi, T.; Kang, B.H.; Altieri, D.C. Hsp60 regulation of tumor cell apoptosis. J. Biol. Chem. 2008, 283, 5188–5194. [Google Scholar] [CrossRef] [Green Version]
- Cappello, F.; de Macario, E.C.; Marasà, L.; Zummo, G.; Macario, A.J. Hsp60 expression, new locations, functions and perspectives for cancer diagnosis and therapy. Cancer Biol. Ther. 2008, 7, 801–809. [Google Scholar] [CrossRef]
- Li, X.S.; Xu, Q.; Fu, X.Y.; Luo, W.S. Heat shock protein 60 overexpression is associated with the progression and prognosis in gastric cancer. PLoS ONE 2014, 9, e107507. [Google Scholar] [CrossRef]
- Inigo, J.R.; Chandra, D. The mitochondrial unfolded protein response (UPRmt): Shielding against toxicity to mitochondria in cancer. J. Hematol. Oncol. 2022, 15, 98. [Google Scholar] [CrossRef] [PubMed]
- Zhou, C.; Sun, H.; Zheng, C.; Gao, J.; Fu, Q.; Hu, N.; Shao, X.; Zhou, Y.; Xiong, J.; Nie, K.; et al. Oncogenic HSP60 regulates mitochondrial oxidative phosphorylation to support Erk1/2 activation during pancreatic cancer cell growth. Cell Death Dis. 2018, 9, 161. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, S.; O’Malley, J.; Chaudhary, A.K.; Inigo, J.R.; Yadav, N.; Kumar, R.; Chandra, D. Hsp60 and IL-8 axis promotes apoptosis resistance in cancer. Br. J. Cancer 2019, 121, 934–943. [Google Scholar] [CrossRef]
- Chun, J.N.; Choi, B.; Lee, K.W.; Lee, D.J.; Kang, D.H.; Lee, J.Y.; Song, I.S.; Kim, H.I.; Lee, S.H.; Kim, H.S.; et al. Cytosolic Hsp60 is involved in the NF-kappaB-dependent survival of cancer cells via IKK regulation. PLoS ONE 2010, 5, e9422. [Google Scholar] [CrossRef]
- Cappello, F.; Di Stefano, A.; D’Anna, S.E.; Donner, C.; Zummo, G. Immunopositivity of heat shock protein 60 as a biomarker of bronchial carcinogenesis. Lancet Oncol. 2005, 6, 816. [Google Scholar] [CrossRef] [Green Version]
- Ruan, W.; Wang, Y.; Ma, Y.; Xing, X.; Lin, J.; Cui, J.; Lai, M. HSP60, a protein downregulated by IGFBP7 in colorectal carcinoma. J. Exp. Clin. Cancer Res. 2010, 29, 41. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Zhou, X.; Chang, H.; Huang, X.; Guo, X.; Du, X.; Tian, S.; Wang, L.; Lyv, Y.; Yuan, P.; et al. Hsp60 exerts a tumor suppressor function by inducing cell differentiation and inhibiting invasion in hepatocellular carcinoma. Oncotarget 2016, 7, 68976–68989. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, H.; Chen, Y.; Liu, X.; Wang, S.; Lv, Y.; Wu, D.; Wang, Q.; Luo, M.; Deng, H. Downregulation of HSP60 disrupts mitochondrial proteostasis to promote tumorigenesis and progression in clear cell renal cell carcinoma. Oncotarget 2016, 7, 38822–38834. [Google Scholar] [CrossRef] [Green Version]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Haastrup, M.O.; Vikramdeo, K.S.; Singh, S.; Singh, A.P.; Dasgupta, S. The Journey of Mitochondrial Protein Import and the Roadmap to Follow. Int. J. Mol. Sci. 2023, 24, 2479. https://doi.org/10.3390/ijms24032479
Haastrup MO, Vikramdeo KS, Singh S, Singh AP, Dasgupta S. The Journey of Mitochondrial Protein Import and the Roadmap to Follow. International Journal of Molecular Sciences. 2023; 24(3):2479. https://doi.org/10.3390/ijms24032479
Chicago/Turabian StyleHaastrup, Mary Oluwadamilola, Kunwar Somesh Vikramdeo, Seema Singh, Ajay Pratap Singh, and Santanu Dasgupta. 2023. "The Journey of Mitochondrial Protein Import and the Roadmap to Follow" International Journal of Molecular Sciences 24, no. 3: 2479. https://doi.org/10.3390/ijms24032479