


Genetic Diversity and Population Structure of a Wide Pisum spp. Core Collection
 , ,
, ,  , and
, and 
Abstract
:1. Introduction
2. Results
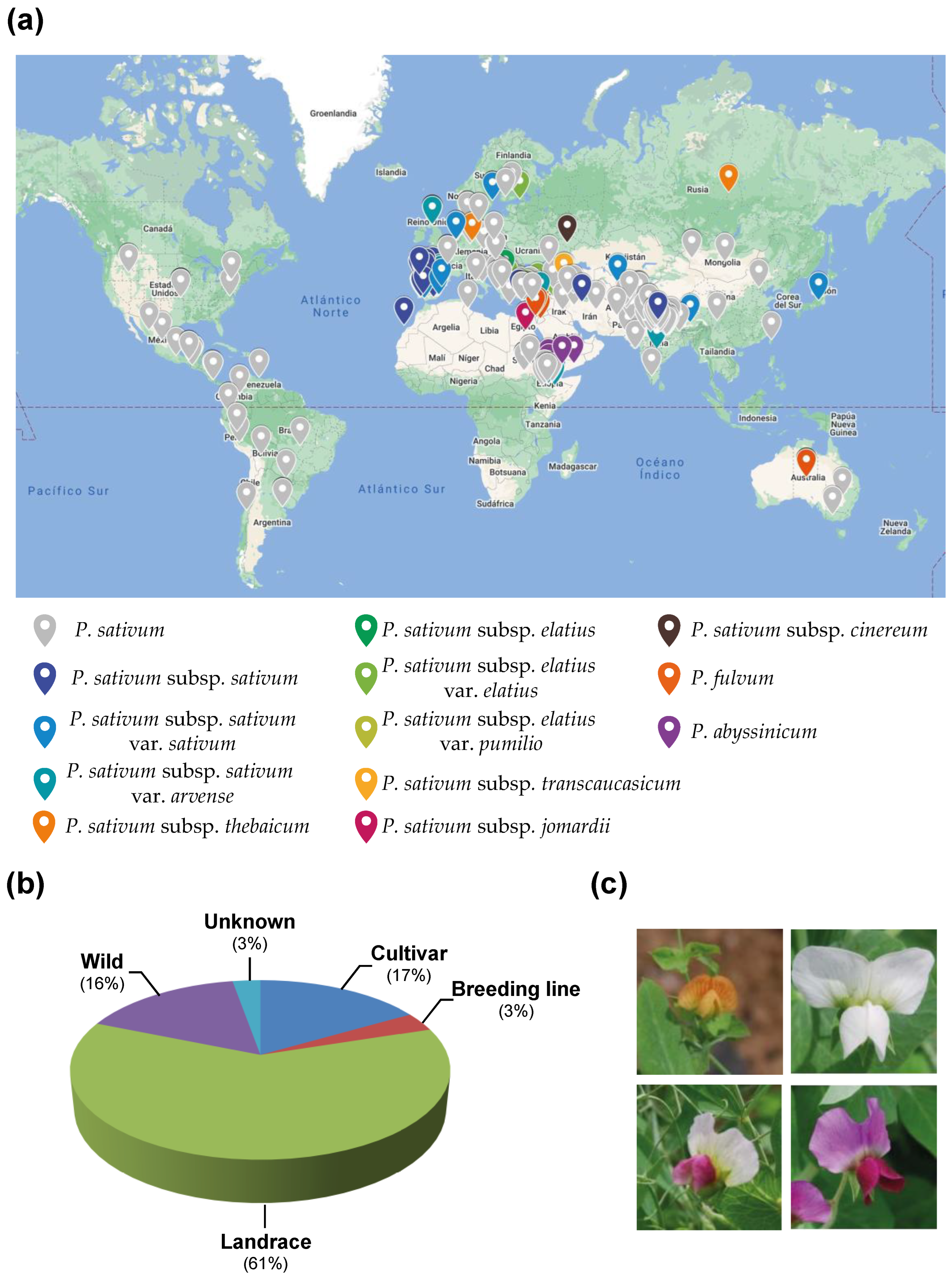
2.1. The Pea Core Collection
2.2. DArTSeq Marker Sequencing and Genetic Diversity Indices
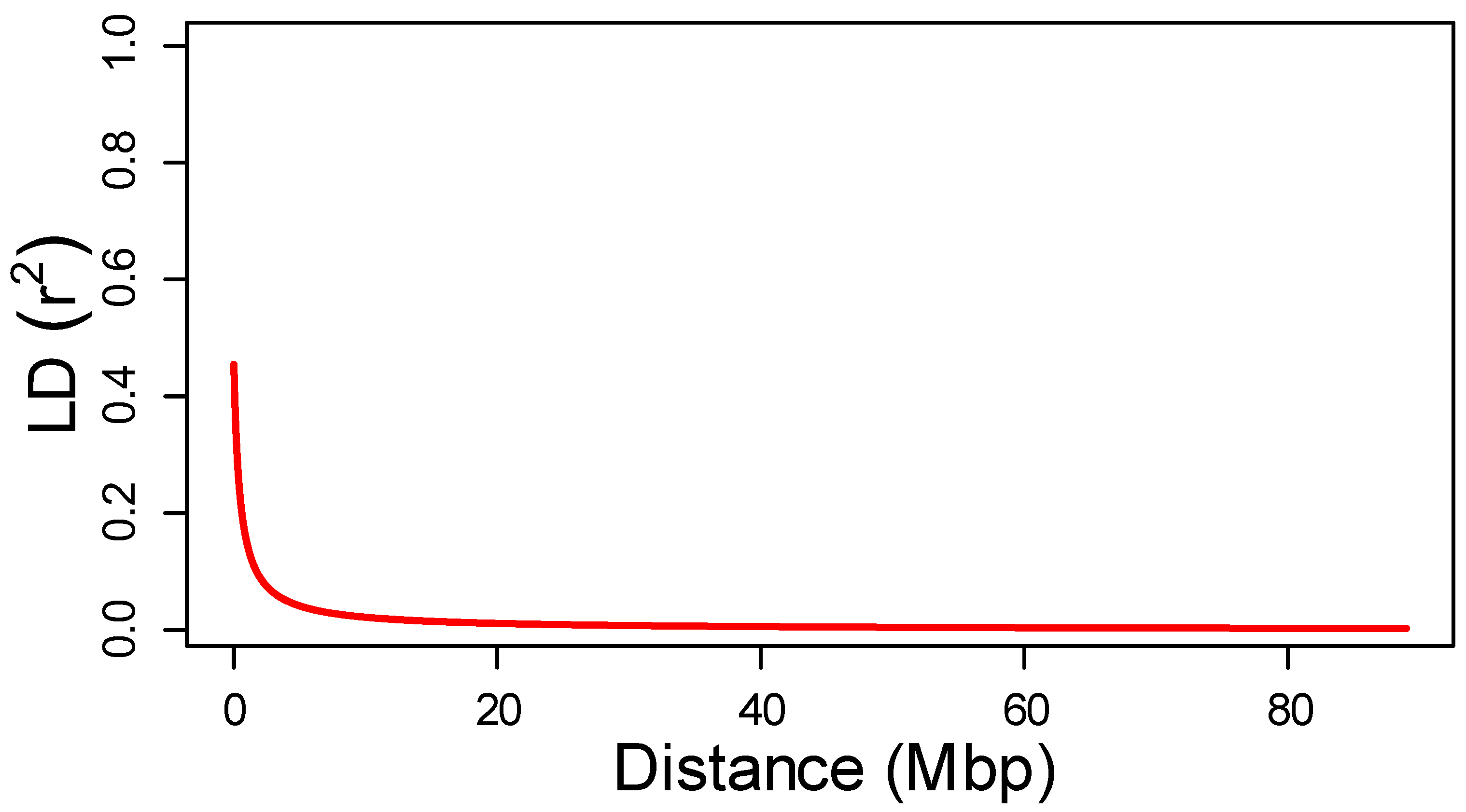
2.3. Marker Distribution and Linkage Disequilibrium
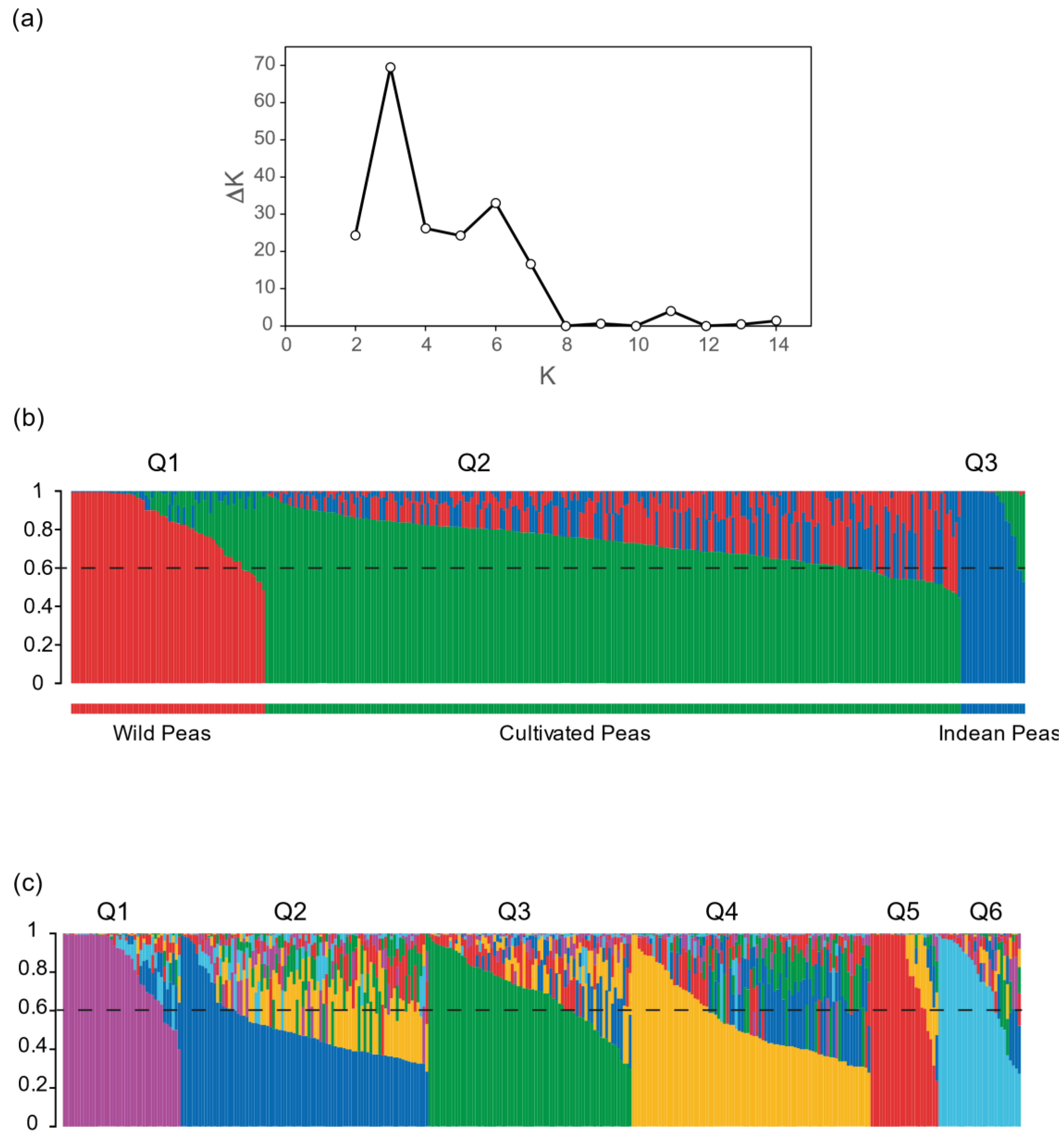
2.4. Genetic Structure of the Pea Core Collection
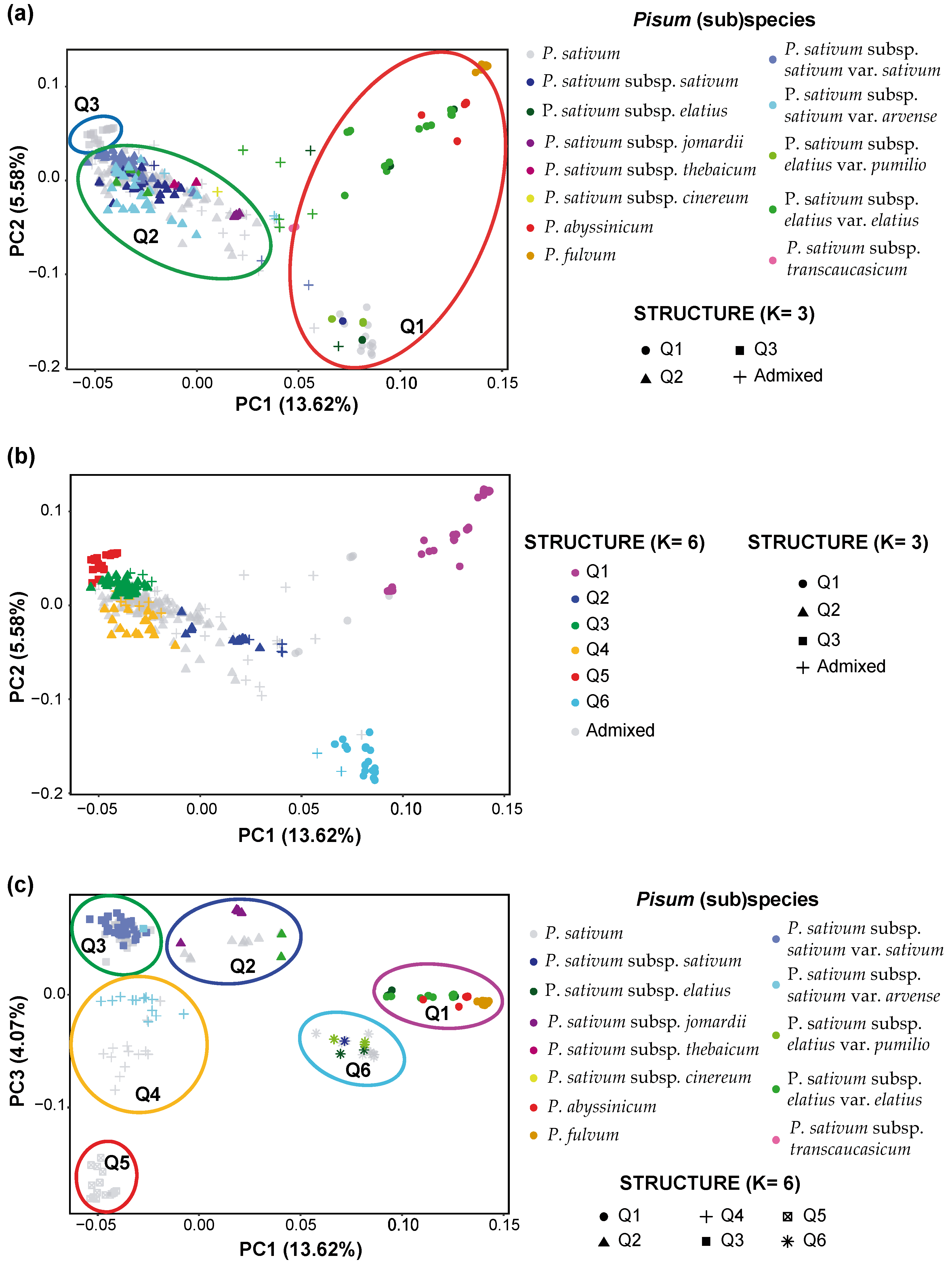
2.5. Principal Component Analysis
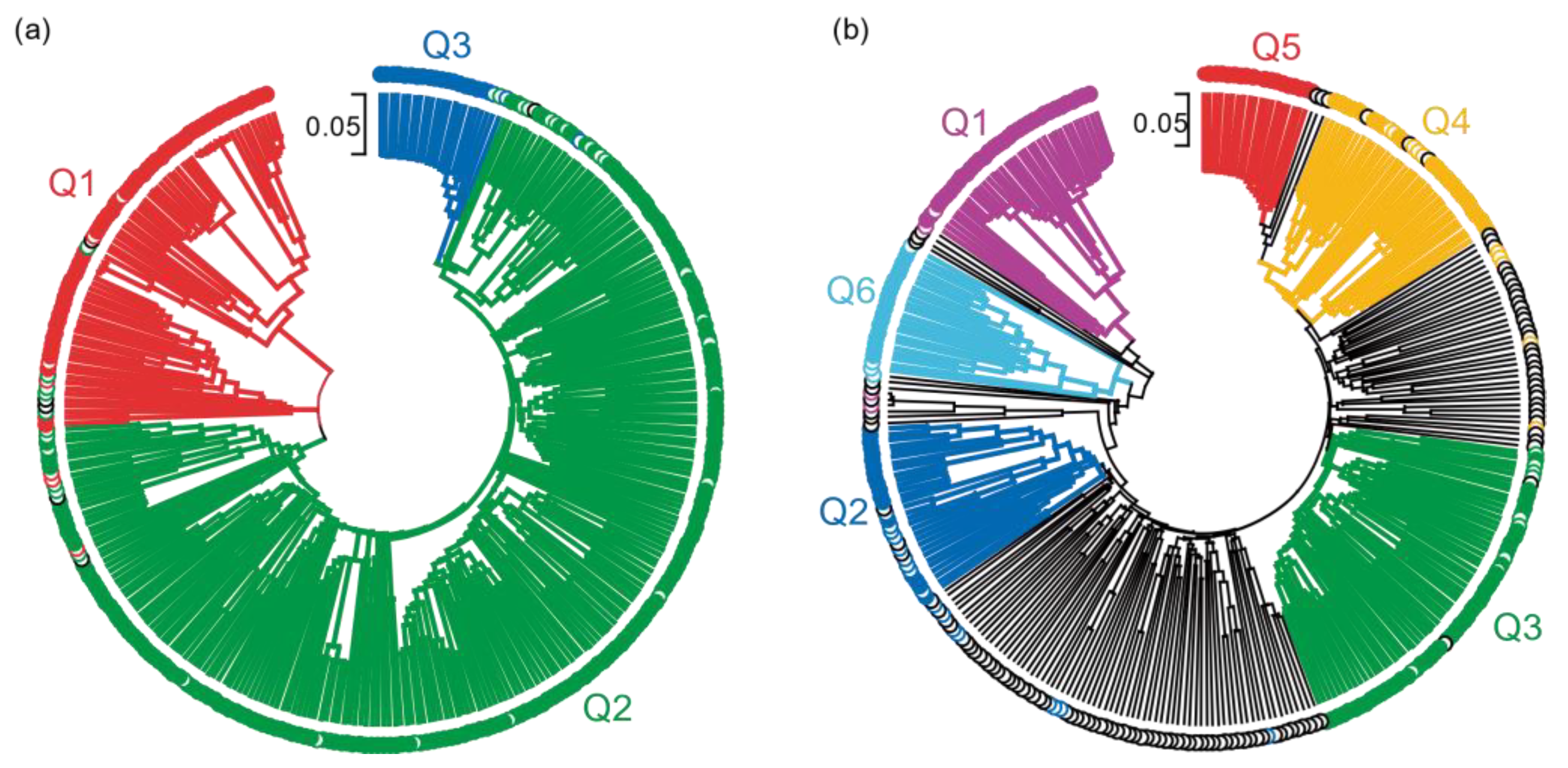
2.6. Phylogenetic Relationship of the Pisum Core Collection
3. Discussion
4. Materials and Methods
4.1. Plant Material
4.2. DNA Extraction, Library Construction, and Sequencing
4.3. Population Structure of the Pea Core Collection
4.4. Phylogenetic Relationship of the Pea Core Collection
4.5. Linkage Disequilibrium
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Rubiales, D.; Fondevilla, S.; Chen, W.D.; Gentzbittel, L.; Higgins, T.J.V.; Castillejo, M.A.; Singh, K.B.; Rispail, N. Achievements and challenges in legume breeding for pest and disease resistance. Crit. Rev. Plant Sci. 2015, 34, 195–236. [Google Scholar] [CrossRef] [Green Version]
- Pandey, A.K.; Rubiales, D.; Wang, Y.G.; Fang, P.P.; Sun, T.; Liu, N.; Xu, P. Omics resources and omics-enabled approaches for achieving high productivity and improved quality in pea (Pisum sativum L.). Theor. Appl. Genet. 2021, 134, 755–776. [Google Scholar] [CrossRef]
- Rubiales, D.; González-Bernal, M.J.; Warkentin, T.D.; Bueckert, R.; Patto, M.C.V.; McPhee, K.E.; McGee, R.; Smýkal, P. Advances in pea breeding. In Achieving Sustainable Cultivation of Vegetables, Hochmuth, G., Ed.; Burleigh Dodds Series in Agricultural Science; Burleigh Dodds Science Publishing: London, UK, 2019; p. 32. [Google Scholar]
- Poore, J.; Nemecek, T. Reducing food’s environmental impacts through producers and consumers. Science 2018, 360, 987–992. [Google Scholar] [CrossRef] [Green Version]
- Rubiales, D.; Fernandez-Aparicio, M.; Moral, A.; Barilli, E.; Sillero, J.C.; Fondevilla, S. Disease resistance in pea (Pisum sativum L.) types for autumn sowings in Mediterranean environments. Czech. J. Genet. Plant 2009, 45, 135–142. [Google Scholar] [CrossRef] [Green Version]
- Schaefer, H.; Hechenleitner, P.; Santos-Guerra, A.; de Sequeira, M.M.; Pennington, R.T.; Kenicer, G.; Carine, M.A. Systematics, biogeography, and character evolution of the legume tribe Fabeae with special focus on the middle-Atlantic island lineages. BMC Evol. Biol. 2012, 12, 250. [Google Scholar] [CrossRef] [Green Version]
- Maxted, N.; Ambrose, M. Peas (Pisum L.). In Plant Genetic Resources of Legumes in the Mediterranean; Maxted, N., Bennett, S.J., Eds.; Springer Nature: Dordrecht, The Netherlands, 2001; Volume 39, pp. 181–190. [Google Scholar]
- Smykal, P.; Coyne, C.J.; Ambrose, M.J.; Maxted, N.; Schaefer, H.; Blair, M.W.; Berger, J.; Greene, S.L.; Nelson, M.N.; Besharat, N.; et al. Legume crops phylogeny and genetic diversity for science and breeding. Crit. Rev. Plant Sci. 2015, 34, 43–104. [Google Scholar] [CrossRef] [Green Version]
- Yang, T.; Liu, R.; Luo, Y.; Hu, S.; Wang, D.; Wang, C.; Pandey, M.K.; Ge, S.; Xu, Q.; Li, N.; et al. Improved pea reference genome and pan-genome highlight genomic features and evolutionary characteristics. Nat. Genet. 2022, 54, 1553–1563. [Google Scholar] [CrossRef]
- Weeden, N.F. Domestication of pea (Pisum sativum L.): The case of the Abyssinian pea. Front. Plant Sci. 2018, 9, 515. [Google Scholar] [CrossRef] [PubMed]
- Kosterin, O.E.; Bogdanova, V.S. Reciprocal compatibility within the genus Pisum L. as studied in F1 hybrids: 1. Crosses involving P.sativum L. subsp sativum. Genet. Resour. Crop Evol. 2015, 62, 691–709. [Google Scholar] [CrossRef]
- Ladizinsky, G.; Abbo, S. The Search for Wild Relatives of Cool Season Legumes; Springer International Publishing: Cham, Switzerland, 2015; p. 110. [Google Scholar]
- Zong, X.X.; Redden, R.J.; Liu, Q.C.; Wang, S.M.; Guan, J.P.; Liu, J.; Xu, Y.H.; Liu, X.J.; Gu, J.; Yan, L.; et al. Analysis of a diverse global Pisum sp collection and comparison to a Chinese local collection with microsatellite markers. Theor. Appl. Genet. 2009, 118, 193–204. [Google Scholar] [CrossRef] [PubMed]
- Kosterin, O.E.; Bogdanova, V.S. Relationship of wild and cultivated forms of Pisum L. as inferred from an analysis of three markers, of the plastid, mitochondrial and nuclear genomes. Genet. Resour. Crop Evol. 2008, 55, 735–755. [Google Scholar] [CrossRef]
- Kosterin, O.E.; Bogdanova, V.S. Reciprocal compatibility within the genus Pisum L. as studied in F-1 hybrids: 4. Crosses within P. sativum L. subsp. elatius (Bieb.) Aschers. et Graebn. Genet. Resour. Crop Evol. 2021, 68, 2565–2590. [Google Scholar] [CrossRef]
- Smykal, P.; Kenicer, G.; Flavell, A.J.; Corander, J.; Kosterin, O.; Redden, R.J.; Ford, R.; Coyne, C.J.; Maxted, N.; Ambrose, M.J.; et al. Phylogeny, phylogeography and genetic diversity of the Pisum genus. Plant Genet. Resour. 2011, 9, 4–18. [Google Scholar] [CrossRef]
- Holdsworth, W.L.; Gazave, E.; Cheng, P.; Myers, J.R.; Gore, M.A.; Coyne, C.J.; McGee, R.J.; Mazourek, M. A community resource for exploring and utilizing genetic diversity in the USDA pea single plant plus collection. Hortic. Res. 2017, 4, 17017. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Varshney, R.K.; Roorkiwal, M.; Nguyen, H.T. Legume genomics: From genomic resources to molecular breeding. Plant Genome 2013, 6, plantgenome2013.12.0002in. [Google Scholar] [CrossRef] [Green Version]
- Jain, S.; Kumar, A.; Mamidi, S.; McPhee, K. Genetic diversity and population structure among pea (Pisum sativum L.) cultivars as revealed by simple sequence repeat and novel genic markers. Mol. Biotech. 2014, 56, 925–938. [Google Scholar] [CrossRef] [PubMed]
- Burstin, J.; Salloignon, P.; Chabert-Martinello, M.; Magnin-Robert, J.B.; Siol, M.; Jacquin, F.; Chauveau, A.; Pont, C.; Aubert, G.; Delaitre, C.; et al. Genetic diversity and trait genomic prediction in a pea diversity panel. BMC Genom. 2015, 16, 105. [Google Scholar] [CrossRef] [Green Version]
- Rispail, N.; Montilla-Bascon, G.; Sanchez-Martin, J.; Flores, F.; Howarth, C.; Langdon, T.; Rubiales, D.; Prats, E. Multi-environmental trials reveal genetic plasticity of oat agronomic traits associated with climate variable changes. Front. Plant Sci. 2018, 9, 1358. [Google Scholar] [CrossRef] [Green Version]
- Hellwig, T.; Abbo, S.; Sherman, A.; Ophir, R. Prospects for the natural distribution of crop wild-relatives with limited adaptability: The case of the wild pea Pisum fulvum. Plant Sci. 2021, 310, 110957. [Google Scholar] [CrossRef]
- Siol, M.; Jacquin, F.; Chabert-Martinello, M.; Smykal, P.; Le Paslier, M.C.; Aubert, G.; Burstin, J. Patterns of genetic structure and linkage disequilibrium in a large collection of pea germplasm. G3-Genes Genom. Genet. 2017, 7, 2461–2471. [Google Scholar] [CrossRef] [Green Version]
- Smykal, P.; Hradilova, I.; Trneny, O.; Brus, J.; Rathore, A.; Bariotakis, M.; Das, R.R.; Bhattacharyya, D.; Richards, C.; Coyne, C.J.; et al. Genomic diversity and macroecology of the crop wild relatives of domesticated pea. Sci. Rep. 2017, 7, 17384. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bani, M.; Rubiales, D.; Rispail, N. A detailed evaluation method to identify sources of quantitative resistance to Fusarium oxysporum f. sp. pisi race 2 within a Pisum spp. germplasm collection. Plant Pathol. 2012, 61, 532–542. [Google Scholar] [CrossRef] [Green Version]
- Barilli, E.; Sillero, J.C.; Fernandez-Aparicio, M.; Rubiales, D. Identification of resistance to Uromyces pisi (Pers.) Wint. in Pisum spp. germplasm. Field Crop Res. 2009, 114, 198–203. [Google Scholar] [CrossRef]
- Fondevilla, S.; Avila, C.M.; Cubero, J.I.; Rubiales, D. Response to Mycosphaerella pinodes in a germplasm collection of Pisum spp. Plant Breed. 2005, 124, 313–315. [Google Scholar] [CrossRef]
- Fondevilla, S.; Carver, T.L.W.; Moreno, M.T.; Rubiales, D. Identification and characterization of sources of resistance to Erysiphe pisi Syd. in Pisum spp. Plant Breed. 2007, 126, 113–119. [Google Scholar] [CrossRef]
- Rubiales, D.; Moreno, M.T.; Sillero, J.C. Search for resistance to crenate broomrape (Orobanche crenata Forsk.) in pea germplasm. Genet. Resour. Crop Evol. 2005, 52, 853–861. [Google Scholar] [CrossRef]
- Osuna-Caballero, S.; Rispail, N.; Barilli, E.; Rubiales, D. Identification and characterization of novel sources of resistance to rust caused by Uromyces pisi in Pisum spp. Plants 2022, 11, 2268. [Google Scholar] [CrossRef]
- Rispail, N.; Wohor, O.Z.; Osuna-caballero, S.; Barilli, E.; Rubiales, D. Dataset for genetic diversity and population structure of a wide Pisum spp. core collection. Zenodo 2022. [Google Scholar] [CrossRef]
- Bari, M.A.A.; Zheng, P.; Viera, I.; Worral, H.; Szwiec, S.; Ma, Y.; Main, D.; Coyne, C.J.; McGee, R.J.; Bandillo, N. Harnessing genetic diversity in the USDA pea germplasm collection through genomic prediction. Front. Genet. 2021, 12, 707754. [Google Scholar] [CrossRef]
- Li, H.H.; Rasheed, A.; Hickey, L.T.; He, Z.H. Fast-forwarding genetic gain. Trends Plant Sci. 2018, 23, 184–186. [Google Scholar] [CrossRef] [Green Version]
- Kreplak, J.; Madoui, M.A.; Capal, P.; Novak, P.; Labadie, K.; Aubert, G.; Bayer, P.E.; Gali, K.K.; Syme, R.A.; Main, D.; et al. A reference genome for pea provides insight into legume genome evolution. Nat. Genet. 2019, 51, 1411–1422. [Google Scholar] [CrossRef]
- Trneny, O.; Brus, J.; Hradilova, I.; Rathore, A.; Das, R.R.; Kopecky, P.; Coyne, C.J.; Reeves, P.; Richards, C.; Smykal, P. Molecular evidence for two domestication events in the pea crop. Genes 2018, 9, 535. [Google Scholar] [CrossRef] [Green Version]
- Alam, M.; Neal, J.; O’Connor, K.; Kilian, A.; Topp, B. Ultra-high-throughput DArTseq-based silicoDArT and SNP markers for genomic studies in macadamia. PLoS ONE 2018, 13, e0203465. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barilli, E.; Cobos, M.J.; Carrillo, E.; Kilian, A.; Carling, J.; Rubiales, D. A high-density integrated DArTseq SNP-based genetic map of Pisum fulvum and identification of QTLs controlling rust resistance. Front. Plant Sci. 2018, 9, 167. [Google Scholar] [CrossRef] [Green Version]
- Beji, S.; Fontaine, V.; Devaux, R.; Thomas, M.; Negro, S.S.; Bahrman, N.; Siol, M.; Aubert, G.; Burstin, J.; Hilbert, J.L.; et al. Genome-wide association study identifies favorable SNP alleles and candidate genes for frost tolerance in pea. BMC Genom. 2020, 21, 536. [Google Scholar] [CrossRef] [PubMed]
- Desgroux, A.; Baudais, V.N.; Aubert, V.; Le Roy, G.; de Larambergue, H.; Miteul, H.; Aubert, G.; Boutet, G.; Duc, G.; Baranger, A.; et al. Comparative genome-wide-association mapping identifies common loci controlling root system architecture and resistance to Aphanomyces euteiches in pea. Front. Plant Sci. 2018, 8, 2195. [Google Scholar] [CrossRef] [Green Version]
- Gali, K.K.; Sackville, A.; Tafesse, E.G.; Lachagari, V.B.R.; McPhee, K.; Hybl, M.; Mikic, A.; Smykal, P.; McGee, R.; Burstin, J.; et al. Genome-wide association mapping for agronomic and seed quality traits of field pea (Pisum sativum L.). Front. Plant Sci. 2019, 10, 1538. [Google Scholar] [CrossRef]
- Robbana, C.; Kehel, Z.; Ben Naceur, M.; Sansaloni, C.; Bassi, F.; Amri, A. Genome-wide genetic diversity and population structure of Tunisian durum wheat landraces based on DArTSeq technology. Int. J. Mol. Sci. 2019, 20, 1352. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smykal, P.; Trneny, O.; Brus, J.; Hanacek, P.; Rathore, A.; Das Roma, R.; Pechanec, V.; Duchoslav, M.; Bhattacharyya, D.; Bariotakis, M.; et al. Genetic structure of wild pea (Pisum sativum subsp elatius) populations in the northern part of the Fertile Crescent reflects moderate cross-pollination and strong effect of geographic but not environmental distance. PLoS ONE 2018, 13, e0194056. [Google Scholar] [CrossRef] [Green Version]
- Gali, K.K.; Liu, Y.; Sindhu, A.; Diapari, M.; Shunmugam, A.S.K.; Arganosa, G.; Daba, K.; Caron, C.; Lachagari, R.V.B.; Tar’an, B.; et al. Construction of high-density linkage maps for mapping quantitative trait loci for multiple traits in field pea (Pisum sativum L.). BMC Plant Biol. 2018, 18, 172. [Google Scholar] [CrossRef]
- Flint-Garcia, S.A.; Thornsberry, J.M.; Buckler, E.S. Structure of linkage disequilibrium in plants. Annu. Rev. Plant Biol. 2003, 54, 357–374. [Google Scholar] [CrossRef] [Green Version]
- Kosterin, O.E.; Bogdanova, V.S.; Galieva, E.R. Reciprocal compatibility within the genus Pisum L. as studied in F-1 hybrids: 2. Crosses involving P. fulvum Sibth. et Smith. Genet. Resour. Crop Evol. 2019, 66, 383–399. [Google Scholar] [CrossRef]
- Frantz, A.C.; Cellina, S.; Krier, A.; Schley, L.; Burke, T. Using spatial Bayesian methods to determine the genetic structure of a continuously distributed population: Clusters or isolation by distance? J. Appl. Ecol. 2009, 46, 493–505. [Google Scholar] [CrossRef]
- Iorizzo, M.; Senalik, D.A.; Ellison, S.L.; Grzebelus, D.; Cavagnaro, P.F.; Allender, C.; Brunet, J.; Spooner, D.M.; Van Deynze, A.; Simon, P.W. Genetic structure and domesticationof carrot (Daucus carota subsp sativus) (Apaciae). Am. J. Bot. 2013, 100, 930–938. [Google Scholar] [CrossRef]
- Vigouroux, Y.; Glaubitz, J.C.; Matsuoka, Y.; Goodman, M.M.; Jesus, S.G.; Doebley, J. Population structure and genetic diversity of new world maize races assessed by DNA microsatellites. Am. J. Bot. 2008, 95, 1240–1253. [Google Scholar] [CrossRef] [PubMed]
- Waples, R.S.; Gaggiotti, O. What is a population? An empirical evaluation of some genetic methods for identifying the number of gene pools and their degree of connectivity. Mol. Ecol. 2006, 15, 1419–1439. [Google Scholar] [CrossRef]
- Hellwig, T.; Abbo, S.; Ophir, R. Drivers of genetic differentiation and recent evolutionary history of an Eurasian wild pea. J. Biogeogr. 2022, 49, 794–808. [Google Scholar] [CrossRef]
- Coyne, C.J.; McGee, R.J.; Redden, R.J.; Ambrose, M.J.; Furman, B.J.; Miles, C.A. Genetic Adjustment to Changing Climates: Pea. In Crop Adaptation to Climate Change; Yadav, S.S., Redden, R.J., Hatfield, J.L., Lotze-Campen, H., Hall, A.E., Eds.; John Wiley and Sons Inc.: Chichester, UK, 2011; pp. 238–250. [Google Scholar]
- Jing, R.C.; Vershinin, A.; Grzebyta, J.; Shaw, P.; Smykal, P.; Marshall, D.; Ambrose, M.J.; Ellis, T.H.N.; Flavell, A.J. The genetic diversity and evolution of field pea (Pisum) studied by high throughput retrotransposon based insertion polymorphism (RBIP) marker analysis. BMC Evol. Biol. 2010, 10, 44. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kosterin, O.E.; Zaytseva, O.O.; Bogdanova, V.S.; Ambrose, M.J. New data on three molecular markers from different cellular genomes in Mediterranean accessions reveal new insights into phylogeography of Pisum sativum L. subsp elatius (Bieb.) Schmalh. Genet. Resour. Crop Evol. 2010, 57, 733–739. [Google Scholar] [CrossRef]
- Hellwig, T.; Abbo, S.; Ophir, R. Phylogeny and disparate selection signatures suggest two genetically independent domestication events in pea (Pisum L.). Plant J. 2022, 110, 419–439. [Google Scholar] [CrossRef]
- Schrank, F. Flora Monacensis; Bände: Munich, Germany, 1818; Volume t. 4. [Google Scholar]
- Montilla-Bascon, G.; Rispail, N.; Sanchez-Martin, J.; Rubiales, D.; Mur, L.A.J.; Langdon, T.; Howarth, C.J.; Prats, E. Genome-wide association study for crown rust (Puccinia coronata f. sp avenae) and powdery mildew (Blumeria graminis f. sp avenae) resistance in an oat (Avena sativa) collection of commercial varieties and landraces. Front. Plant Sci. 2015, 6, 103. [Google Scholar] [CrossRef] [Green Version]
- Ouellette, L.A.; Reid, R.W.; Blanchard, S.G.; Brouwer, C.R. LinkageMapView-rendering high-resolution linkage and QTL maps. Bioinformatics 2018, 34, 306–307. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gosselin, T. radiator: RADSeq Data Exploration, Manipulation and Visualization Using R, Version 1.1.9. 2020. Available online: https://thierrygosselin.github.io/radiator/ (accessed on 16 January 2023).
- Keenan, K.; McGinnity, P.; Cross, T.F.; Crozier, W.W.; Prodohl, P.A. diveRsity: An R package for the estimation and exploration of population genetics parameters and their associated errors. Methods Ecol. Evol. 2013, 4, 782–788. [Google Scholar] [CrossRef] [Green Version]
- Chang, C.C.; Chow, C.C.; Tellier, L.; Vattikuti, S.; Purcell, S.M.; Lee, J.J. Second-generation PLINK: Rising to the challenge of larger and richer datasets. Gigascience 2015, 4, s13742-015. [Google Scholar] [CrossRef] [PubMed]
- Pritchard, J.K.; Stephens, M.; Donnelly, P. Inference of population structure using multilocus genotype data. Genetics 2000, 155, 945–959. [Google Scholar] [CrossRef]
- Falush, D.; Stephens, M.; Pritchard, J.K. Inference of population structure using multilocus genotype data: Linked loci and correlated allele frequencies. Genetics 2003, 164, 1567–1587. [Google Scholar] [CrossRef]
- Earl, D.A.; Vonholdt, B.M. STRUCTURE HARVESTER: A website and program for visualizing structure output and implementing the Evanno method. Conserv. Genet. Resour. 2012, 4, 359–361. [Google Scholar] [CrossRef]
- Evanno, G.; Regnaut, S.; Goudet, J. Detecting the number of clusters of individuals using the software structure: A simulation study. Mol. Ecol. 2005, 14, 2611–2620. [Google Scholar] [CrossRef] [Green Version]
- Ramasamy, R.K.; Ramasamy, S.; Bindroo, B.B.; Naik, V.G. Structure Plot: A program for drawing elegant structure bar plots in user friendly interface. Springerplus 2014, 3, 431. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- The R Core Team. R: A Language and Environment for Statistical Computing; The R Core Team: Vienna, Austria, 2020. [Google Scholar]
- Tang, Y.; Horikoshi, M.; Li, W.X. ggfortify: Unified interface to visualize statistical results of popular R packages. R J. 2016, 8, 474–485. [Google Scholar] [CrossRef] [Green Version]
- Wickham, H. ggplot2: Elegant Graphics for Data Analysis; Springer: New York, NY, USA, 2009. [Google Scholar]
- R Studio Team. R Studio: Integrated Development Environment for R; R Studio Team: Boston, MA, USA, 2015. [Google Scholar]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular evolutionary genetics analysis across computing platforms. Mol Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef]
- Nei, M.; Kumar, S. Molecular Evolution and Phylogenetics; Oxford University Press: New York, NY, USA, 2000; p. 352. [Google Scholar]
- Hasegawa, M.; Kishino, H.; Yano, T.A. Dating of the human ape splitting by a molecular clock of mitochondrial DNA. J. Mol. Evol. 1985, 22, 160–174. [Google Scholar] [CrossRef] [PubMed]
- Saitou, N.; Nei, M. The neighbor-joining method: A new method for reconstructing phylogenetic trees. Mol. Biol. Evol. 1987, 4, 406–425. [Google Scholar] [CrossRef] [PubMed]
- Bradbury, P.J.; Zhang, Z.; Kroon, D.E.; Casstevens, T.M.; Ramdoss, Y.; Buckler, E.S. Tassel: Software for association mapping of complex traits in diverse samples. Bioinformatics 2007, 23, 2633–2635. [Google Scholar] [CrossRef] [PubMed]
- Marroni, F.; Pinosio, S.; Zaina, G.; Fogolari, F.; Felice, N.; Cattonaro, F.; Morgante, M. Nucleotide diversity and linkage disequilibrium in Populus nigra cinnamyl alcohol dehydrogenase (CAD4) gene. Tree Genet. Genomes 2011, 7, 1011–1023. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Species | Number of Accessions |
|---|---|
| P. sativum | 167 |
| P. sativum subsp. sativum | 23 |
| P. sativum subsp. sativum var. sativum | 37 |
| P. sativum subsp. sativum var. arvense | 32 |
| P. sativum subsp. elatius | 7 |
| P. sativum subsp. elatius var. elatius | 24 |
| P. sativum subsp. elatius var. pumilio | 3 |
| P. sativum subsp. jomardii | 7 |
| P. sativum subsp. transcaucasicum | 2 |
| P. sativum subsp. thebaicum | 2 |
| P. sativum subsp. cinereum | 1 |
| P. abyssinicum | 7 |
| P. fulvum | 13 |
| Total | 325 |
| SilicoDArT | SNP | |||||
|---|---|---|---|---|---|---|
| Mean | Min | Max | Mean | Min | Max | |
| PIC | 0.295 | 0.006 | 0.499 | 0.267 | 0.013 | 0.594 |
| MAF | 0.41 | 0.052 | 0.997 | 0.532 | 0.046 | 0.975 |
| Ar | 1.783 | 1 | 2.91 | 1.515 | 0.4 | 2 |
| Ho | 0.06 | 0 | 0.7 | 0.035 | 0 | 0.78 |
| He | 0.217 | 0 | 0.5 | 0.148 | 0 | 0.5 |
| FIS | 0.692 | −0.102 | 1 | 0.707 | −0.592 | 1 |
| FIS Low | 0.615 | −0.675 | 1 | 0.588 | −0.905 | 1 |
| FIS High | 0.762 | −0.058 | 1.1412 | 0.815 | −0.375 | 1.305 |
| Total Lenght (Mbp) | Marker Number | Chromosome Coverage | Mean Distance Between Markers | Marker Density | |||||
|---|---|---|---|---|---|---|---|---|---|
| SilicoDarT | SNP | SilicoDarT | SNP | SilicoDarT | SNP | SilicoDarT | SNP | ||
| Chr1 | 372.17 | 2388 | 1284 | 0.04–372.1 | 0.24–372.0 | 0.156 | 0.290 | 6.4 | 3.5 |
| Chr2 | 427.6 | 2157 | 1068 | 0.27–427.4 | 0.03–427.4 | 0.198 | 0.401 | 5.0 | 2.5 |
| Chr3 | 437.56 | 2331 | 1194 | 0.09–437.5 | 0.09–437.5 | 0.188 | 0.367 | 5.3 | 2.7 |
| Chr4 | 446.35 | 2716 | 1341 | 0.03–446.3 | 0.03–446.3 | 0.164 | 0.333 | 6.1 | 3.0 |
| Chr5 | 579.27 | 3696 | 1993 | 0.09–579.1 | 0.14–579.1 | 0.157 | 0.291 | 6.4 | 3.4 |
| Chr6 | 480.42 | 2970 | 1672 | 0.23–480.4 | 0.21–480.4 | 0.162 | 0.287 | 6.2 | 3.5 |
| Chr7 | 491.38 | 3253 | 1573 | 0.05–491.3 | 0.05–491.1 | 0.151 | 0.312 | 6.6 | 3.2 |
| Whole genome | 19,514 | 11,511 | 0.166 | 0.319 | 6.0 | 3.1 | |||
| MeanLD | LD90 | Dist LD50 (Mbp) | ||||
|---|---|---|---|---|---|---|
| SilicoDarT | SNP | SilicoDarT | SNP | SilicoDarT | SNP | |
| Chr1 | 0.104 | 0.12 | 0.244 | 0.28 | 0.60 | 1.58 |
| Chr2 | 0.091 | 0.104 | 0.206 | 0.239 | 0.32 | 0.82 |
| Chr3 | 0.101 | 0.114 | 0.234 | 0.267 | 0.60 | 1.83 |
| Chr4 | 0.09 | 0.101 | 0.203 | 0.242 | 0.25 | 0.69 |
| Chr5 | 0.104 | 0.112 | 0.239 | 0.264 | 0.54 | 1.20 |
| Chr6 | 0.136 | 0.159 | 0.35 | 0.42 | 1.05 | 3.19 |
| Chr7 | 0.086 | 0.099 | 0.191 | 0.23 | 0.24 | 0.78 |
| Whole genome | 0.103 | 0.118 | 0.236 | 0.28 | 0.48 | 1.38 |
| Clusters | Membership a | Average Dist. b | Fst |
|---|---|---|---|
| K = 3 | |||
| 1 | 28.16 | 0.2130 | 0.2268 |
| 2 | 56.83 | 0.2262 | 0.3154 |
| 3 | 15.01 | 0.1514 | 0.4315 |
| K = 6 | |||
| 1 | 14.25 | 0.1836 | 0.3316 |
| 2 | 21.08 | 0.2141 | 0.3504 |
| 3 | 21.39 | 0.1670 | 0.4147 |
| 4 | 21.35 | 0.1667 | 0.4688 |
| 5 | 12.57 | 0.1424 | 0.4975 |
| 6 | 9.37 | 0.1305 | 0.5404 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rispail, N.; Wohor, O.Z.; Osuna-Caballero, S.; Barilli, E.; Rubiales, D. Genetic Diversity and Population Structure of a Wide Pisum spp. Core Collection. Int. J. Mol. Sci. 2023, 24, 2470. https://doi.org/10.3390/ijms24032470
Rispail N, Wohor OZ, Osuna-Caballero S, Barilli E, Rubiales D. Genetic Diversity and Population Structure of a Wide Pisum spp. Core Collection. International Journal of Molecular Sciences. 2023; 24(3):2470. https://doi.org/10.3390/ijms24032470
Chicago/Turabian StyleRispail, Nicolas, Osman Zakaria Wohor, Salvador Osuna-Caballero, Eleonora Barilli, and Diego Rubiales. 2023. "Genetic Diversity and Population Structure of a Wide Pisum spp. Core Collection" International Journal of Molecular Sciences 24, no. 3: 2470. https://doi.org/10.3390/ijms24032470