
Ion Channels of the Sarcolemma and Intracellular Organelles in Duchenne Muscular Dystrophy: A Role in the Dysregulation of Ion Homeostasis and a Possible Target for Therapy



Abstract
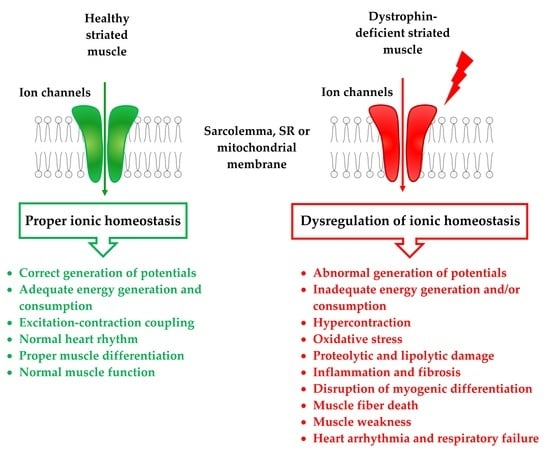
:
1. Introduction
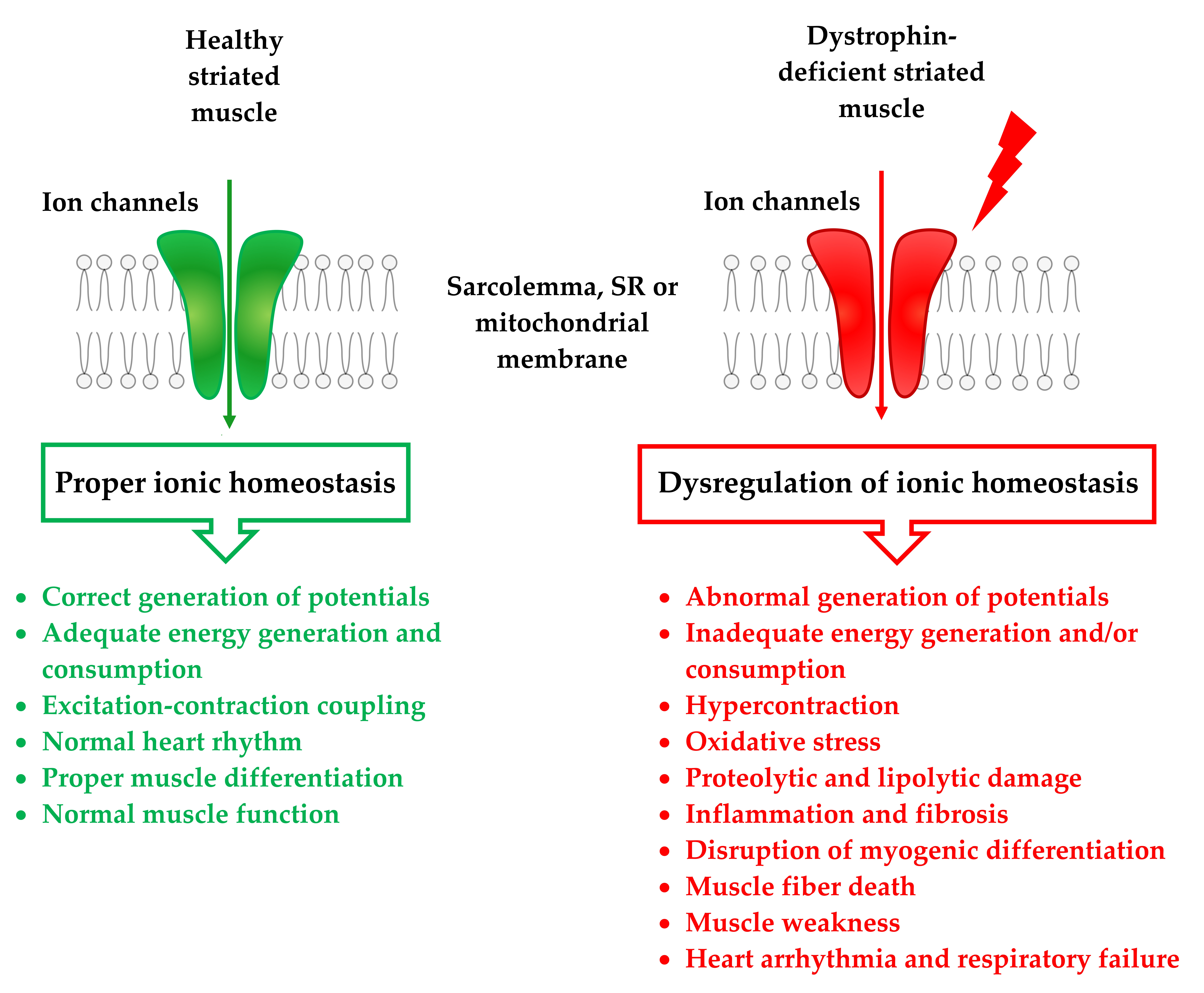
2. Dystrophin and DAPC and Their Role in the Regulation of Sarcolemmal Ion Channels
2.1. Dysregulation of Sarcolemmal Calcium Channels in DMD
2.2. Dysregulation of Sarcolemmal Sodium Channels in DMD
2.3. Dysregulation of Sarcolemmal Potassium Channels in DMD
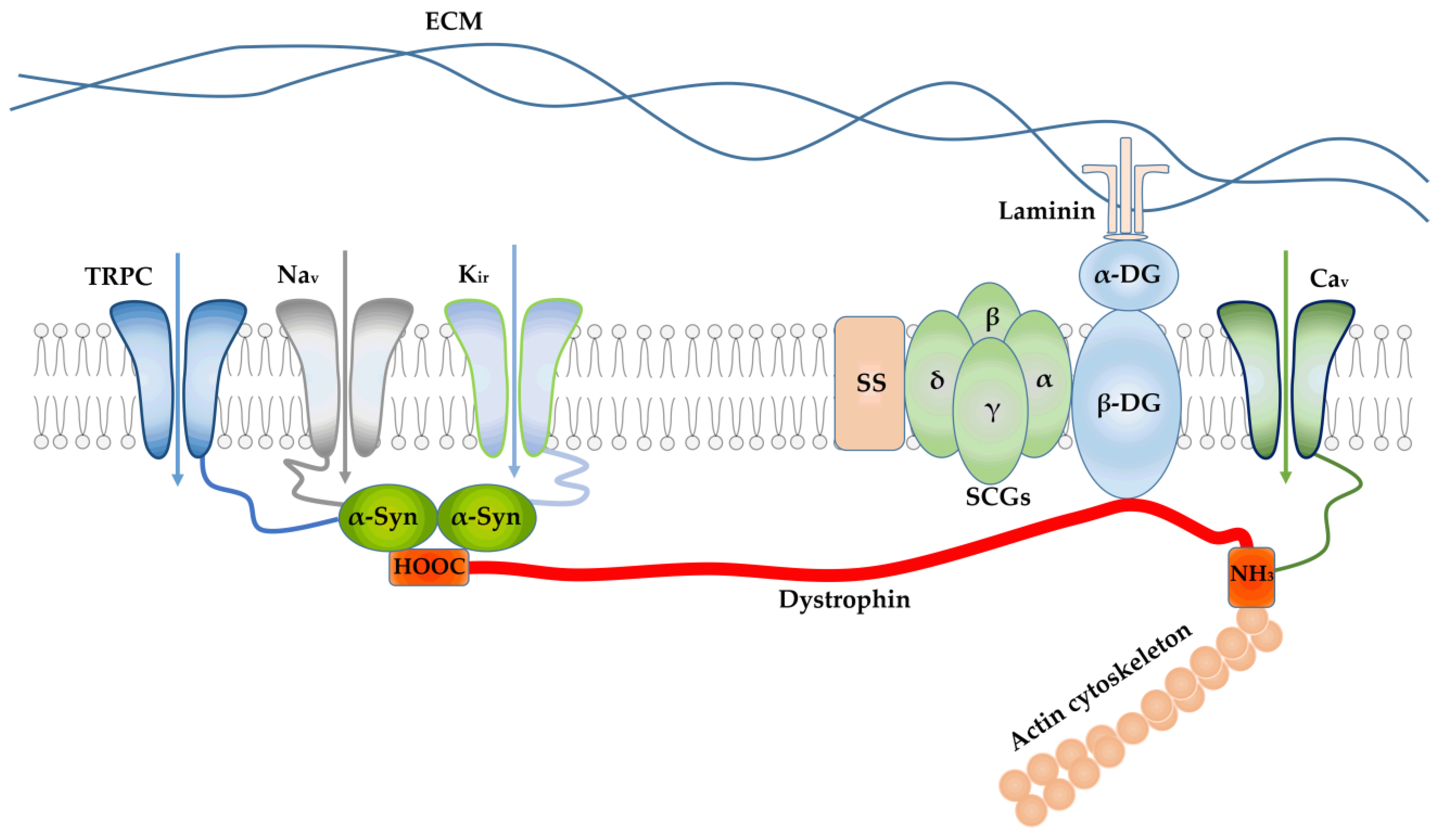
3. Consequences of Impaired Sarcolemmal Ion Permeability in DMD
4. The Role of Ion Channels of Intracellular Organelles in the Development of DMD
4.1. SR Channel Abnormalities in DMD
4.2. Mitochondrial Ion Channels in DMD
4.3. SR–Mitochondria Axis and MAM Contacts in DMD
5. Contribution of SR and Mitochondrial Channels to the Development of DMD
6. The Summary
7. Clinical Implications
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| ANT | Adenine nucleotide translocator |
| BKCa | High conductance Ca2+-activated K+ channel |
| CaV | Voltage-gated calcium channel |
| CypD | Cyclophilin D |
| DAPC | Dystrophin-associated protein complex |
| DMD | Duchenne muscular dystrophy |
| DWORF | Dwarf open reading frame |
| GRP75 | Glucose-regulated protein 75 |
| IP3R | Inositol 1,4,5-trisphosphate receptor |
| KATP | ATP-sensitive potassium channel |
| Kir | Inward-rectifier potassium channel |
| MAM | Mitochondria-associated membranes |
| MCUC | Mitochondrial calcium uniporter complex |
| MLN | Myoregulin |
| MPT | Mitochondrial permeability transition |
| NaV | Voltage-gated sodium channel |
| NCLX | Na+-Ca2+-Li+ exchanger |
| NCX | Na+/Ca2+ exchanger |
| NHE | Na+–H+ exchanger |
| NOS | Nitric oxide synthase |
| PLN | Phospholamban |
| RNS | Reactive nitrogen species |
| ROS | Reactive oxygen species |
| RyR | Ryanodine receptor |
| SERCA | Sarco/endoplasmic reticulum Ca2+-ATPase |
| SLN | Sarcolipin |
| SOCE | Store-operated Ca2+ entry |
| SR | Sarcoplasmic reticulum |
| STIM1 | Stromal interaction molecule |
| TRIC | Trimeric intracellular cation channel |
| TRPC | Transient receptor potential channel |
| TRPV2 | Transient receptor potential vanilloid type 2 |
| UPR | Unfolded protein response |
| VDAC | Voltage-dependent anion channel |
References
- Monaco, A.P.; Neve, R.L.; Colletti-Feener, C.; Bertelson, C.J.; Kurnit, D.M.; Kunkel, L.M. Isolation of candidate cDNAs for portions of the Duchenne muscular dystrophy gene. Nature 1986, 323, 646–650. [Google Scholar] [CrossRef] [PubMed]
- Emery, A.E. Population frequencies of inherited neuromuscular diseases—A world survey. Neuromuscul. Disord. 1991, 1, 19–29. [Google Scholar] [CrossRef] [PubMed]
- Mendell, J.R.; Lloyd-Puryear, M. Report of MDA muscle disease symposium on newborn screening for Duchenne muscular dystrophy. Muscle Nerve 2013, 48, 21–26. [Google Scholar] [CrossRef] [PubMed]
- Duan, D.; Goemans, N.; Takeda, S.; Mercuri, E.; Aartsma-Rus, A. Duchenne muscular dystrophy. Nat. Rev. Dis. Prim. 2021, 7, 13. [Google Scholar] [CrossRef]
- Mercuri, E.; Bonnemann, C.G.; Muntoni, F. Muscular dystrophies. Lancet 2019, 394, 2025–2038. [Google Scholar] [CrossRef]
- Bushby, K.; Finke, R.; Birnkrant, D.J.; Case, L.E.; Clemens, P.R.; Cripe, L.; Kaul, A.; Kinnett, K.; McDonald, C.; Pandya, S.; et al. DMD Care Considerations Working Group. Diagnosis and management of Duchenne muscular dystrophy, part 1: Diagnosis, and pharmacological and psychosocial management. Lancet Neurol. 2010, 9, 77–93. [Google Scholar] [CrossRef]
- Kamdar, F.; Garry, D.J. Dystrophin-Deficient Cardiomyopathy. J. Am. Coll. Cardiol. 2016, 67, 2533–2546. [Google Scholar] [CrossRef]
- D’Amario, D.; Amodeo, A.; Adorisio, R.; Tiziano, F.D.; Leone, A.M.; Perri, G.; Bruno, P.; Massetti, M.; Ferlini, A.; Pane, M.; et al. A current approach to heart failure in Duchenne muscular dystrophy. Heart 2017, 103, 1770–1779. [Google Scholar] [CrossRef]
- Schultz, T.I.; Raucci, F.J., Jr.; Salloum, F.N. Cardiovascular Disease in Duchenne Muscular Dystrophy: Overview and Insight into Novel Therapeutic Targets. JACC Basic Transl. Sci. 2022, 7, 608–625. [Google Scholar] [CrossRef]
- Ware, S.M. Genetics of paediatriccardiomyopathies. Curr. Opin. Pediatr. 2017, 29, 534–540. [Google Scholar] [CrossRef]
- Peter, A.K.; Cheng, H.; Ross, R.S.; Knowlton, K.U.; Chen, J. The costamere bridges sarcomeres to the sarcolemma in striated muscle. Prog. Pediatr. Cardiol. 2011, 31, 83–88. [Google Scholar] [CrossRef] [Green Version]
- Ignatieva, E.; Smolina, N.; Kostareva, A.; Dmitrieva, R. Skeletal muscle mitochondria dysfunction in genetic neuromuscular disorders with cardiac phenotype. Int. J. Mol. Sci. 2021, 22, 7349. [Google Scholar] [CrossRef] [PubMed]
- Wilson, D.G.S.; Tinker, A.; Iskratsch, T. The role of the dystrophin glycoprotein complex in muscle cell mechanotransduction. Commun. Biol. 2022, 5, 1022. [Google Scholar] [CrossRef]
- Angelini, G.; Mura, G.; Messina, G. Therapeutic approaches to preserve the musculature in Duchenne muscular dystrophy: The importance of the secondary therapies. Exp. Cell Res. 2022, 410, 112968. [Google Scholar] [CrossRef] [PubMed]
- Constantin, B. Dystrophin complex functions as a scaffold for signalling proteins. Biochim. Biophys. Acta 2014, 1838, 635–642. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leyva-Leyva, M.; Sandoval, A.; Felix, R.; González-Ramírez, R. Biochemical and functional interplay between ion channels and the components of the dystrophin-associated glycoprotein complex. J. Membr. Biol. 2018, 251, 535–550. [Google Scholar] [CrossRef] [PubMed]
- Koenig, X.; Ebner, J.; Hilber, K. Voltage-Dependent Sarcolemmal Ion Channel Abnormalities in the Dystrophin-Deficient Heart. Int. J. Mol. Sci. 2018, 19, 3296. [Google Scholar] [CrossRef] [Green Version]
- Mendell, J.R.; Sahenk, Z.; Lehman, K.; Nease, C.; Lowes, L.P.; Miller, N.F.; Iammarino, M.A.; Alfano, L.N.; Nicholl, A.; Al-Zaidy, S.; et al. Assessment of systemic delivery of rAAVrh74.MHCK7 micro-dystrophin in children with Duchenne muscular dystrophy. JAMA Neurol. 2020, 77, 1122. [Google Scholar] [CrossRef]
- HappiMbakam, C.; Lamothe, G.; Tremblay, G.; Tremblay, J.P. CRISPR-Cas9 gene therapy for Duchenne muscular dystrophy. Neurotherapeutics 2022, 19, 931–941. [Google Scholar] [CrossRef]
- Matthews, E.; Brassington, R.; Kuntzer, T.; Jichi, F.; Manzur, A.Y. Corticosteroids for the treatment of Duchenne muscular dystrophy. Cochrane Database Syst Rev. 2016, 2016, CD003725. [Google Scholar] [CrossRef] [Green Version]
- Bhat, H.F.; Mir, S.S.; Dar, K.B.; Bhat, Z.F.; Shah, R.A.; Ganai, N.A. ABC of multifaceted dystrophin glycoprotein complex (DGC). J. Cell Physiol. 2017, 233, 5142–5159. [Google Scholar] [CrossRef] [PubMed]
- Bovolenta, M.; Erriquez, D.; Valli, E.; Brioschi, S.; Scotton, C.; Neri, M.; Falzarano, M.S.; Gherardi, S.; Fabris, M.; Rimessi, P.; et al. The DMD locus harbours multiple long non-coding RNAs which orchestrate and control transcription of muscle dystrophin mRNA isoforms. PLoS ONE 2012, 7, e45328. [Google Scholar] [CrossRef] [PubMed]
- Blake, D.J.; Weir, A.; Newey, S.E.; Davies, K.E. Function and genetics of dystrophin and dystrophin-related proteins in muscle. Physiol. Rev. 2002, 82, 291–329. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tozawa, T.; Itoh, K.; Yaoi, T.; Tando, S.; Umekage, M.; Dai, H.; Hosoi, H.; Fushiki, S. The shortest isoform of dystrophin (Dp40) interacts with a group of presynaptic proteins to form a presumptive novel complex in the mouse brain. Mol. Neurobiol. 2012, 45, 287–297. [Google Scholar] [CrossRef] [Green Version]
- Gao, Q.Q.; McNally, E.M. The dystrophin complex: Structure, function, and implications for therapy. Compr. Physiol. 2015, 5, 1223–1239. [Google Scholar]
- Allen, D.G.; Whitehead, N.P.; Froehner, S.C. Absence of dystrophin disrupts skeletal muscle signaling: Roles of Ca2+, reactive oxygen species, and nitric oxide in the development of muscular dystrophy. Physiol. Rev. 2016, 96, 253–305. [Google Scholar] [CrossRef] [Green Version]
- Allard, B. Sarcolemmal ion channels in dystrophin-deficient skeletal muscle fibres. J. Muscle Res. Cell Motil. 2006, 27, 367–373. [Google Scholar] [CrossRef]
- Tian, L.J.; Cao, J.H.; Deng, X.Q.; Zhang, C.L.; Qian, T.; Song, X.X.; Huang, B.S. Gene expression profiling of Duchenne muscular dystrophy reveals characteristics along disease progression. Genet. Mol. Res. 2014, 13, 1402–1411. [Google Scholar] [CrossRef]
- Emery, A.E.; Burt, D. Intracellular calcium and pathogenesis and antenatal diagnosis of Duchenne muscular dystrophy. Br. Med. J. 1980, 280, 355–357. [Google Scholar] [CrossRef] [Green Version]
- Harisseh, R.; Chatelier, A.; Magaud, C.; Deliot, N.; Constantin, B. Involvement of TRPV2 and SOCE in calcium influx disorder in DMD primary human myotubes with a specific contribution of alpha1-syntrophin and PLC/PKC in SOCE regulation. Am. J. Physiol. Cell Physiol. 2013, 304, C881–C894. [Google Scholar] [CrossRef]
- Petrof, B.J.; Shrager, J.B.; Stedman, H.H.; Kelly, A.M.; Sweeney, H.L. Dystrophin protects the sarcolemma from stresses developed during muscle contraction. Proc. Natl. Acad. Sci. USA 1993, 90, 3710–3714. [Google Scholar] [CrossRef] [Green Version]
- Danialou, G.; Comtois, A.S.; Dudley, R.; Karpati, G.; Vincent, G.; Des Rosiers, C.; Petrof, B.J. Dystrophin-deficient cardiomyocytes are abnormally vulnerable to mechanical stress-induced contractile failure and injury. FASEB J. 2001, 15, 1655–1657. [Google Scholar] [CrossRef]
- Cooper, S.T.; Head, S.I. Membrane injury and repair in the muscular dystrophies. Neuroscientist 2015, 21, 653–668. [Google Scholar] [CrossRef]
- Townsend, D.; Turner, I.; Yasuda, S.; Martindale, J.; Davis, J.; Shillingford, M.; Kornegay, J.N.; Metzger, J.M. Chronic administration of membrane sealant prevents severe cardiac injury and ventricular dilatation in dystrophic dogs. J. Clin. Investig. 2010, 120, 1140–1150. [Google Scholar] [CrossRef]
- Houang, E.M.; Haman, K.J.; Filareto, A.; Perlingeiro, R.C.; Bates, F.S.; Lowe, D.A.; Metzger, J.M. Membrane-stabilizing copolymers confer marked protection to dystrophic skeletal muscle in vivo. Mol. Ther. Methods Clin. Dev. 2015, 2, 15042. [Google Scholar] [CrossRef]
- Markham, B.E.; Kernodle, S.; Nemzek, J.; Wilkinson, J.E.; Sigler, R. Chronic dosing with membrane sealant poloxamer 188 NF improves respiratory dysfunction in dystrophic Mdx and Mdx/Utrophin-/- mice. PLoS ONE 2015, 10, e0134832. [Google Scholar] [CrossRef]
- Quinlan, J.G.; Wong, B.L.; Niemeier, R.T.; McCullough, A.S.; Levin, L.; Emanuele, M. Poloxamer 188 failed to prevent exercise-induced membrane breakdown in mdx skeletal muscle fibers. Neuromuscul. Disord. 2006, 16, 855–864. [Google Scholar] [CrossRef]
- Terry, R.L.; Kaneb, H.M.; Wells, D.J. Poloxamer 188 has a deleterious effect on dystrophic skeletal muscle function. PLoS ONE 2014, 9, e91221. [Google Scholar] [CrossRef]
- Vandebrouck, A.; Sabourin, J.; Rivet, J.; Balghi, H.; Sebille, S.; Kitzis, A.; Raymond, G.; Cognard, C.; Bourmeyster, N.; Constantin, B. Regulation of capacitative calcium entries by alpha1-syntrophin: Association of TRPC1 with dystrophin complex and the PDZ domain of alpha1-syntrophin. FASEB J. 2007, 21, 608–617. [Google Scholar] [CrossRef] [Green Version]
- Gervasio, O.L.; Whitehead, N.P.; Yeung, E.W.; Phillips, W.D.; Allen, D.G. TRPC1 binds to caveolin-3 and is regulated by Src kinase—Role in Duchenne muscular dystrophy. J. Cell Sci. 2008, 121 Pt 13, 2246–2255. [Google Scholar] [CrossRef] [Green Version]
- Lopez, J.R.; Uryash, A.; Faury, G.; Estève, E.; Adams, J.A. Contribution of TRPC Channels to Intracellular Ca2+ Dyshomeostasis in Smooth Muscle From mdx Mice. Front. Physiol. 2020, 11, 126. [Google Scholar] [CrossRef] [PubMed]
- Sabourin, J.; Lamiche, C.; Vandebrouck, A.; Magaud, C.; Rivet, J.; Cognard, C.; Bourmeyster, N.; Constantin, B. Regulation of TRPC1 and TRPC4 cation channels requires an alpha1-syntrophindependent complex in skeletal mouse myotubes. J. Biol. Chem. 2009, 284, 36248–36261. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sabourin, J.; Cognard, C.; Constantin, B. Regulation by scaffolding proteins of canonical transient receptor potential channels in striated muscle. J. Muscle Res. Cell Motil. 2009, 30, 289–297. [Google Scholar] [CrossRef] [PubMed]
- Shi, J.; Geshi, N.; Takahashi, S.; Kiyonaka, S.; Ichikawa, J.; Hu, Y.; Mori, Y.; Ito, Y.; Inoue, R. Molecular determinants for cardiovascular TRPC6 channel regulation by Ca2+/calmodulin-dependent kinase II. J. Physiol. 2013, 591, 2851–2866. [Google Scholar] [CrossRef] [PubMed]
- Shen, B.; Kwan, H.Y.; Ma, X.; Wong, C.O.; Du, J.; Huang, Y.; Yao, X. cAMP activates TRPC6 channels via the phosphatidylinositol 3-kinase (PI3K)-protein kinase B (PKB)-mitogen-activated protein kinase kinase (MEK)-ERK1/2 signaling pathway. J. Biol. Chem. 2011, 286, 19439–19445. [Google Scholar] [CrossRef] [Green Version]
- Seo, K.; Rainer, P.P.; Lee, D.I.; Hao, S.; Bedja, D.; Birnbaumer, L.; Cingolani, O.H.; Kass, D.A. Hyperactive adverse mechanical stress responses in dystrophic heart are coupled to transient receptor potential canonical 6 and blocked by cGMP-protein kinase G modulation. Circ. Res. 2014, 114, 823–832. [Google Scholar] [CrossRef]
- Seo, K.; Rainer, P.P.; Shalkey Hahn, V.; Lee, D.I.; Jo, S.H.; Andersen, A.; Liu, T.; Xu, X.; Willette, R.N.; Lepore, J.J.; et al. Combined TRPC3 and TRPC6 blockade by selective small-molecule or genetic deletion inhibits pathological cardiac hypertrophy. Proc. Natl. Acad. Sci. USA 2014, 111, 1551–1556. [Google Scholar] [CrossRef] [Green Version]
- Khairallah, R.J.; Shi, G.; Sbrana, F.; Prosser, B.L.; Borroto, C.; Mazaitis, M.J.; Hoffman, E.P.; Mahurkar, A.; Sachs, F.; Sun, Y.; et al. Microtubules underlie dysfunction in Duchenne muscular dystrophy. Sci. Signal 2012, 5, ra56. [Google Scholar] [CrossRef] [Green Version]
- Hammers, D.W.; Sleeper, M.M.; Forbes, S.C.; Shima, A.; Walter, G.A.; Sweeney, H.L. Tadalafil treatment delays the onset of cardiomyopathy in dystrophin-deficient hearts. J. Am. Heart Assoc. 2016, 5, e003911. [Google Scholar] [CrossRef] [Green Version]
- Lin, B.L.; Shin, J.Y.; Jeffreys, W.P.; Wang, N.; Lukban, C.A.; Moorer, M.C.; Velarde, E.; Hanselman, O.A.; Kwon, S.; Kannan, S.; et al. Pharmacological TRPC6 inhibition improves survival and muscle function in mice with Duchenne muscular dystrophy. JCI Insight 2022, 7, e158906. [Google Scholar] [CrossRef]
- Creisméas, A.; Gazaille, C.; Bourdon, A.; Lallemand, M.A.; François, V.; Allais, M.; Ledevin, M.; Larcher, T.; Toumaniantz, G.; Lafoux, A.; et al. TRPC3, but not TRPC1, as a good therapeutic target for standalone or complementary treatment of DMD. J. Transl. Med. 2021, 19, 519. [Google Scholar] [CrossRef] [PubMed]
- Dietrich, A.; Mederos y Schnitzler, M.; Emmel, J.; Kalwa, H.; Hofmann, T.; Gudermann, T. N-linked protein glycosylation is a major determinant for basal TRPC3 and TRPC6 channel activity. J. Biol. Chem. 2003, 278, 47842–47852. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Millay, D.P.; Goonasekera, S.A.; Sargent, M.A.; Maillet, M.; Aronow, B.J.; Molkentin, J.D. Calcium influx is sufficient to induce muscular dystrophy through a TRPC-dependent mechanism. Proc. Natl. Acad. Sci. USA 2009, 106, 19023–19028. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vandebrouck, C.; Martin, D.; Colson-Van Schoor, M.; Debaix, H.; Gailly, P. Involvement of TRPC in the abnormal calcium influx observed in dystrophic (mdx) mouse skeletal muscle fibers. J. Cell Biol. 2002, 158, 1089–1096. [Google Scholar] [CrossRef]
- Iwata, Y.; Katanosaka, Y.; Arai, Y.; Komamura, K.; Miyatake, K.; Shigekawa, M. A novel mechanism of myocyte degeneration involving the Ca2+-permeable growth factor-regulated channel. J. Cell Biol. 2003, 161, 957–967. [Google Scholar] [CrossRef] [Green Version]
- Iwata, Y.; Katanosaka, Y.; Arai, Y.; Shigekawa, M.; Wakabayashi, S. Dominant-negative inhibition of Ca2+ influx via TRPV2 ameliorates muscular dystrophy in animal models. Hum. Mol. Genet. 2009, 18, 824–834. [Google Scholar] [CrossRef] [Green Version]
- Aguettaz, E.; Lopez, J.J.; Krzesiak, A.; Lipskaia, L.; Adnot, S.; Hajjar, R.J.; Cognard, C.; Constantin, B.; Sebille, S. Axial stretch-dependent cation entry in dystrophic cardiomyopathy: Involvement of several TRPs channels. Cell Calcium 2016, 59, 145–155. [Google Scholar] [CrossRef] [Green Version]
- Koenig, X.; Rubi, L.; Obermair, G.J.; Cervenka, R.; Dang, X.B.; Lukacs, P.; Kummer, S.; Bittner, R.E.; Kubista, H.; Todt, H.; et al. Enhanced currents through L-type calcium channels in cardiomyocytes disturb the electrophysiology of the dystrophic heart. Am. J. Physiol. Heart Circ. Physiol. 2014, 306, H564–H573. [Google Scholar] [CrossRef] [Green Version]
- Matsumura, C.Y.; Pertille, A.; Albuquerque, T.C.; Santo Neto, H.; Marques, M.J. Diltiazem and verapamil protect dystrophin-deficient muscle fibers of MDX mice from degeneration: A potential role in calcium buffering and sarcolemmal stability. Muscle Nerve 2009, 39, 167–176. [Google Scholar] [CrossRef]
- Altamirano, F.; Valladares, D.; Henriquez-Olguin, C.; Casas, M.; Lopez, J.R.; Allen, P.D.; Jaimovich, E. Nifedipine treatment reduces resting calciumconcentration, oxidative and apoptotic gene expression, and improves muscle function in dystrophic mdx mice. PLoS ONE 2013, 8, e81222. [Google Scholar] [CrossRef] [Green Version]
- Yeung, D.; Kharidia, R.; Brown, S.C.; Górecki, D.C. Enhanced expression of the P2X4 receptor in Duchenne muscular dystrophy 516 correlates with macrophage invasion. Neurobiol. Dis. 2004, 15, 212–220. [Google Scholar] [CrossRef]
- Young, C.N.; Brutkowski, W.; Lien, C.F.; Arkle, S.; Lochmuller, H.; Zablocki, K.; Górecki, D.C. P2X7 purinoceptor alterations in dystrophic mdx mouse muscles: Relationship to pathology and potential target for treatment. J. Cell. Mol. Med. 2012, 16, 1026–1037. [Google Scholar] [CrossRef]
- Sinadinos, A.; Young, C.N.; Al-Khalidi, R.; Teti, A.; Kalinski, P.; Mohamad, S.; Floriot, L.; Henry, T.; Tozzi, G.; Jiang, T.; et al. P2RX7 purinoceptor: A therapeutic target for ameliorating the symptoms of Duchenne muscular dystrophy. PLoS Med. 2015, 12, e1001888. [Google Scholar] [CrossRef]
- Gazzerro, E.; Baldassari, S.; Assereto, S.; Fruscione, F.; Pistorio, A.; Panicucci, C.; Volpi, S.; Perruzza, L.; Fiorillo, C.; Minetti, C.; et al. Enhancement of Muscle T Regulatory Cells and Improvement of Muscular Dystrophic Process in mdx Mice by Blockade of Extracellular ATP/P2X Axis. Am. J. Pathol. 2015, 185, 3349–3360. [Google Scholar] [CrossRef] [Green Version]
- Taniguti, A.P.; Pertille, A.; Matsumura, C.Y.; Santo Neto, H.; Marques, M.J. Prevention of muscle fibrosis and myonecrosis in mdx mice by suramin, a TGF-β1 blocker. Muscle Nerve 2011, 43, 82–87. [Google Scholar] [CrossRef]
- Sztretye, M.; Geyer, N.; Vincze, J.; Al-Gaadi, D.; Olah, T.; Szentesi, P.; Kis, G.; Antal, M.; Balatoni, I.; Csernoch, L.; et al. SOCE Is Important for Maintaining Sarcoplasmic Calcium Content and Release in Skeletal Muscle Fibers. Biophys. J. 2017, 113, 2496–2507. [Google Scholar] [CrossRef] [Green Version]
- Ben-Kasus Nissim, T.; Zhang, X.; Elazar, A.; Roy, S.; Stolwijk, J.A.; Zhou, Y.; Motiani, R.K.; Gueguinou, M.; Hempel, N.; Hershfinkel, M.; et al. Mitochondria control store-operated Ca2+ entry through Na+ and redox signals. EMBO J. 2017, 36, 797–815. [Google Scholar] [CrossRef] [Green Version]
- Edwards, J.N.; Friedrich, O.; Cully, T.R.; von Wegner, F.; Murphy, R.M.; Launikonis, B.S. Upregulation of store-operated Ca2+ entry in dystrophic mdx mouse muscle. Am. J. Physiol. Cell Physiol. 2010, 299, 42–50. [Google Scholar] [CrossRef] [Green Version]
- Roos, J.; DiGregorio, P.J.; Yeromin, A.V.; Ohlsen, K.; Lioudyno, M.; Zhang, S.; Safrina, O.; Kozak, J.A.; Wagner, S.L.; Cahalan, M.D.; et al. STIM1, an essential and conserved component of store-operated Ca2+ channel function. J. Cell Biol. 2005, 169, 435–445. [Google Scholar] [CrossRef] [Green Version]
- Liou, J.; Fivaz, M.; Inoue, T.; Meyer, T. Live-cell imaging reveals sequential oligomerization and local plasma membrane targeting of stromal interaction molecule 1 after Ca2+ store depletion. Proc. Natl. Acad. Sci. USA 2007, 104, 9301–9306. [Google Scholar] [CrossRef] [Green Version]
- Zhang, S.L.; Yeromin, A.V.; Zhang, X.H.; Yu, Y.; Safrina, O.; Penna, A.; Roos, J.; Stauderman, K.A.; Cahalan, M.D. Genome-wide RNAi screen of Ca2+ influx identifies genes that regulate Ca2+ release-activated Ca2+ channel activity. Proc. Natl. Acad. Sci. USA 2006, 103, 9357–9362. [Google Scholar] [CrossRef] [Green Version]
- Wei-LaPierre, L.; Carrell, E.M.; Boncompagni, S.; Protasi, F.; Dirksen, R.T. Orai1-dependent calcium entry promotes skeletal muscle growth and limits fatigue. Nat. Commun. 2013, 4, 2805. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, X.; Moloughney, J.G.; Zhang, S.; Komazaki, S.; Weisleder, N. Orai1 mediates exacerbated Ca2+ entry in dystrophic skeletal muscle. PLoS ONE 2012, 7, e49862. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goonasekera, S.A.; Davis, J.; Kwong, J.Q.; Accornero, F.; Wei-LaPierre, L.; Sargent, M.A.; Dirksen, R.T.; Molkentin, J.D. Enhanced Ca2+ influx from STIM1-Orai1 induces muscle pathology in mouse models of muscular dystrophy. Hum. Mol. Genet. 2014, 23, 3706–3715. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uchimura, T.; Sakurai, H. Orai1-STIM1 Regulates Increased Ca2+ Mobilization, Leading to Contractile Duchenne Muscular Dystrophy Phenotypes in Patient-Derived Induced Pluripotent Stem Cells. Biomedicines 2021, 9, 1589. [Google Scholar] [CrossRef]
- García-Castañeda, M.; Michelucci, A.; Zhao, N.; Malik, S.; Dirksen, R.T. Postdevelopmental knockout of Orai1 improves muscle pathology in a mouse model of Duchenne muscular dystrophy. J. Gen. Physiol. 2022, 154, e202213081. [Google Scholar] [CrossRef]
- Lopez, J.J.; Salido, G.M.; Pariente, J.A.; Rosado, J.A. Interaction of STIM1 with endogenously expressed human canonical TRP1 upon depletion of intracellular Ca2+ stores. J. Biol. Chem. 2006, 281, 28254–28264. [Google Scholar] [CrossRef] [Green Version]
- Yuan, J.P.; Zeng, W.; Huang, G.N.; Worley, P.F.; Muallem, S. STIM1 heteromultimerizes TRPC channels to determine their function as store-operated channels. Nat. Cell Biol. 2007, 9, 636–645. [Google Scholar] [CrossRef] [Green Version]
- Vandebrouck, C.; Duport, G.; Cognard, C.; Raymond, G. Cationic channels in normal and dystrophic human myotubes. Neuromuscul. Disord. 2001, 11, 72–79. [Google Scholar] [CrossRef]
- Tutdibi, O.; Brinkmeier, H.; Rudel, R.; Fohr, K.J. Increased calcium entry into dystrophin-deficient muscle fibres of MDX and ADR-MDX mice is reduced by ion channel blockers. J. Physiol. 1999, 515 Pt 3, 859–868. [Google Scholar] [CrossRef]
- Maroto, R.; Raso, A.; Wood, T.G.; Kurosky, A.; Martinac, B.; Hamill, O.P. TRPC1 forms the stretch-activated cation channel in vertebrate cells. Nat. Cell Biol. 2005, 7, 179–185. [Google Scholar] [CrossRef]
- Gee, S.H.; Madhavan, R.; Levinson, S.R.; Caldwell, J.H.; Sealock, R.; Froehner, S.C. Interaction of muscle and brain sodium channels with multiple members of the syntrophin family of dystrophin associated proteins. J. Neurosci. 1998, 18, 128–137. [Google Scholar] [CrossRef] [Green Version]
- Hirn, C.; Shapovalov, G.; Petermann, O.; Roulet, E.; Ruegg, U.T. Nav1.4 deregulation in dystrophic skeletal muscle leads to Na+ overload and enhanced cell death. J. Gen. Physiol. 2008, 132, 199–208. [Google Scholar] [CrossRef] [Green Version]
- Gavillet, B.; Rougier, J.S.; Domenighetti, A.A. Cardiac sodium channel Nav1.5 is regulated by a multiprotein complex composed of syntrophins and dystrophin. Circ. Res. 2006, 99, 407–414. [Google Scholar] [CrossRef] [Green Version]
- Iwata, Y.; Katanosaka, Y.; Hisamitsu, T.; Wakabayashi, S. Enhanced Na+/H+ exchange activity contributes to the pathogenesis of muscular dystrophy via involvement of P2 receptors. Am. J. Pathol. 2007, 171, 1576–1587. [Google Scholar] [CrossRef] [Green Version]
- Burr, A.R.; Molkentin, J.D. Genetic evidence in the mouse solidifies the calcium hypothesis of myofiber death in muscular dystrophy. Cell Death Differ. 2015, 22, 1402–1412. [Google Scholar] [CrossRef] [Green Version]
- Previtali, S.C.; Gidaro, T.; Díaz-Manera, J.; Zambon, A.; Carnesecchi, S.; Roux-Lombard, P.; Spitali, P.; Signorelli, M.; Szigyarto, C.A.-K.; Johansson, C.; et al. Rimeporide as a first- in-class NHE-1 inhibitor: Results of a phase Ib trial in young patients with Duchenne Muscular Dystrophy. Pharmacol Res. 2020, 159, 104999. [Google Scholar] [CrossRef]
- Ghaleh, B.; Barthélemy, I.; Wojcik, J.; Sambin, L.; Bizé, A.; Hittinger, L.; Tran, T.D.; Thomé, F.P.; Blot, S.; Su, J.B. Protective effects of rimeporide on left ventricular function in golden retriever muscular dystrophy dogs. Int. J. Cardiol. 2020, 312, 89–95. [Google Scholar] [CrossRef]
- Deval, E.; Levitsky, D.O.; Marchand, E.; Cantereau, A.; Raymond, G.; Cognard, C. Na(+)/Ca(2+) exchange in human myotubes: Intracellular calcium rises in response to external sodium depletion are enhanced in DMD. Neuromuscul. Disord. 2002, 12, 665–673. [Google Scholar] [CrossRef]
- Alloatti, G.; Gallo, M.P.; Penna, C.; Levi, R.C. Properties of Cardiac Cells from Dystrophic Mouse. J. Mol. Cell. Cardiol. 1995, 27, 1775–1779. [Google Scholar] [CrossRef]
- Pacioretty, L.M.; Cooper, B.J.; Gilmour, R.F. Reduction of the Transient Outward Potassium Current in Canine X-Linked Muscular Dystrophy. Circulation 1994, 90, 1350–1356. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rubi, L.; Koenig, X.; Kubista, H.; Todt, H.; Hilber, K. Decreased Inward Rectifier Potassium Current IK1 in Dystrophin-Deficient Ventricular Cardiomyocytes. Channels 2017, 11, 101–108. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Cai, H.; Dang, Y.; Qiu, C.; Wang, J. Adenosine triphosphate-sensitive potassium channels and cardiomyopathies (Review). Mol. Med. Rep. 2016, 13, 1447–1454. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Graciotti, L.; Becker, J.; Granata, A.L.; Procopio, A.D.; Tessarollo, L.; Fulgenzi, G. Dystrophin is required for the normal function of the cardio-protective K(ATP) channel in cardiomyocytes. PLoS ONE 2011, 6, e27034. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bienengraeber, M.; Olson, T.M.; Selivanov, V.A.; Kathmann, E.C.; O’Cochlain, F.; Gao, F.; Karger, A.B.; Ballew, J.D.; Hodgson, D.M.; Zingman, L.V.; et al. ABCC9 mutations identified in human dilated cardiomyopathy disrupt catalytic KATP channel gating. Nat. Genet. 2004, 36, 382–387. [Google Scholar] [CrossRef] [Green Version]
- Farid, T.A.; Nair, K.; Massé, S.; Azam, M.A.; Maguy, A.; Lai, P.F.; Umapathy, K.; Dorian, P.; Chauhan, V.; Varró, A.; et al. Role of KATP channels in the maintenance of ventricular fibrillation in cardiomyopathic human hearts. Circ. Res. 2011, 109, 1309–1318. [Google Scholar] [CrossRef] [Green Version]
- Allard, B.; Rougier, O. Similarity of ATP-dependent K+ channels in skeletal muscle fibres from normal and mutant mdx mice. J. Physiol. 1997, 498, 319–325. [Google Scholar] [CrossRef] [Green Version]
- Mallouk, N.; Jacquemond, V.; Allard, B. Elevated subsarcolemmal Ca2+ in mdx mouse skeletal muscle fibers detected with Ca2+-activated K+ channels. Proc. Natl. Acad. Sci. USA 2000, 97, 4950–4955. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.W.; Saifee, O.; Nonet, M.L.; Salkoff, L. SLO-1 potassium channels control quantal content of neurotransmitter release at the C. elegans neuromuscular junction. Neuron 2001, 32, 867–881. [Google Scholar] [CrossRef] [Green Version]
- Yuan, A.; Dourado, M.; Butler, A.; Walton, N.; Wei, A.; Salkoff, L. SLO-2, a K+ channel with an unusual Cl-dependence. Nat. Neurosci. 2000, 3, 771–779. [Google Scholar] [CrossRef]
- Kim, H.; Pierce-Shimomura, J.T.; Oh, H.J.; Johnson, B.E.; Goodman, M.B.; McIntire, S.L. The dystrophin complex controls bk channel localization and muscle activity in Caenorhabditis elegans. PLoS Genet. 2009, 5, e1000780. [Google Scholar] [CrossRef] [Green Version]
- Eisner, D.A.; Caldwell, J.L.; Kistamás, K.; Trafford, A.W. Calcium and Excitation-Contraction Coupling in the Heart. Circ Res. 2017, 121, 181–195. [Google Scholar] [CrossRef]
- Bolaños, P.; Calderón, J.C. Excitation-contraction coupling in mammalian skeletal muscle: Blending old and last-decade research. Front. Physiol. 2022, 13, 989796. [Google Scholar] [CrossRef] [PubMed]
- Hara, H.; Nolan, P.M.; Scott, M.O.; Bucan, M.; Wakayama, Y.; Fischbeck, K.H. Running endurance abnormality in mdx mice. Muscle Nerve 2002, 25, 207–211. [Google Scholar] [CrossRef]
- Ravens, U.; Cerbai, E. Role of potassium currents in cardiac arrhythmias. Europace 2008, 10, 1133–1137. [Google Scholar] [CrossRef]
- Burg, S.; Attali, B. Targeting of Potassium Channels in Cardiac Arrhythmias. Trends Pharmacol. Sci. 2021, 42, 491–506. [Google Scholar] [CrossRef]
- Niranjan, N.; Mareedu, S.; Tian, Y.; Kodippili, K.; Fefelova, N.; Voit, A.; Xie, L.-H.; Duan, N.; Babu, G.J. Sarcolipin overexpression impairs myogenic differentiation in Duchenne muscular dystrophy. Am. J. Physiol. Cell Physiol. 2019, 317, C813–C824. [Google Scholar] [CrossRef]
- Gailly, P.; De Backer, F.; Van Schoor, M.; Gillis, J.M. In situ measurements of calpain activity in isolated muscle fibres from normal and dystrophin-lacking mdx mice. J. Physiol. 2007, 582 Pt 3, 1261–1275. [Google Scholar] [CrossRef] [PubMed]
- Voit, A.; Patel, V.; Pachon, R.; Shah, V.; Bakhutma, M.; Kohlbrenner, E.; McArdle, J.J.; Dell’Italia, L.J.; Mendell, J.R.; Xie, L.-H.; et al. Reducing sarcolipin expression mitigates Duchenne muscular dystrophy and associated cardiomyopathy in mice. Nat. Commun. 2017, 8, 1068. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lindahl, M.; Backman, E.; Henriksson, K.G.; Gorospe, J.R.; Hoffman, E.P. Phospholipase A2 activity in dystrophinopathies. Neuromuscul. Disord. 1995, 5, 193–199. [Google Scholar] [CrossRef] [PubMed]
- Kikkawa, N.; Ohno, T.; Nagata, Y.; Shiozuka, M.; Kogure, T.; Matsuda, R. Ectopic calcification is caused by elevated levels of serum inorganic phosphate in mdx mice. Cell Struct. Funct. 2009, 34, 77–88. [Google Scholar] [CrossRef] [Green Version]
- Young, C.N.J.; Gosselin, M.R.F.; Rumney, R.; Oksiejuk, A.; Chira, N.; Bozycki, L.; Matryba, P.; Łukasiewicz, K.; Kao, A.P.; Dunlop, J.; et al. Total Absence of Dystrophin Expression Exacerbates Ectopic Myofiber Calcification and Fibrosis and Alters Macrophage Infiltration Patterns. Am. J. Pathol. 2020, 190, 190–205. [Google Scholar] [CrossRef]
- Lehmann-Horn, F.; Weber, M.A.; Nagel, A.M.; Meinck, H.M.; Breitenbach, S.; Scharrer, J.; Jurkat-Rott, K. Rationale for treating oedema in Duchenne muscular dystrophy with eplerenone. Acta Myol. 2012, 31, 31–39. [Google Scholar]
- Budzinska, M.; Zimna, A.; Kurpisz, M. The role of mitochondria in Duchenne muscular dystrophy. J Physiol. Pharmacol. 2021, 72, 157–166. [Google Scholar] [CrossRef]
- Bellissimo, C.A.; Garibotti, M.C.; Perry, C.G.R. Mitochondrial stress responses in Duchenne muscular dystrophy: Metabolic dysfunction or adaptive reprogramming? Am. J. Physiol. Cell Physiol. 2022, 323, C718–C730. [Google Scholar] [CrossRef]
- Rossi, D.; Pierantozzi, E.; Amadsun, D.O.; Buonocore, S.; Rubino, E.M.; Sorrentino, V. The Sarcoplasmic Reticulum of Skeletal Muscle Cells: A Labyrinth of Membrane Contact Sites. Biomolecules 2022, 12, 488. [Google Scholar] [CrossRef]
- Periasamy, M.; Kalyanasundaram, A. SERCA pump isoforms: Their role in calcium transport and disease. Muscle Nerve 2007, 35, 430–442. [Google Scholar] [CrossRef]
- Tupling, A.R. The decay phase of Ca2+ transients in skeletal muscle: Regulation and physiology. Appl. Physiol. Nutr. Metab. 2009, 34, 373–376. [Google Scholar] [CrossRef]
- Dowling, P.; Lohan, J.; Ohlendieck, K. Comparative analysis of Dp427-deficient mdx tissues shows that the milder dystrophic phenotype of extraocular and toe muscle fibres is associated with a persistent expression of beta-dystroglycan. Eur. J. Cell Biol. 2003, 82, 222–230. [Google Scholar] [CrossRef] [Green Version]
- Ferretti, R.; Marques, M.J.; Pertille, A.; Santo Neto, H. Sarcoplasmicendoplasmic-reticulum Ca2+-ATPase and calsequestrin are overexpressed in spared intrinsic laryngeal muscles of dystrophin-deficient mdx mice. Muscle Nerve 2009, 39, 609–615. [Google Scholar] [CrossRef]
- Schneider, J.S.; Shanmugam, M.; Gonzalez, J.P.; Lopez, H.; Gordan, R.; Fraidenraich, D.; Babu, G.J. Increased sarcolipin expression and decreased sarco(endo)plasmic reticulum Ca2+ uptake in skeletal muscles of mouse models of Duchenne muscular dystrophy. J. Muscle Res. Cell Motil. 2013, 34, 349–356. [Google Scholar] [CrossRef] [PubMed]
- Cleverdon, R.E.G.; Braun, J.L.; Geromella, M.S.; Whitley, K.C.; Marko, D.M.; Hamstra, S.I.; Roy, B.D.; MacPherson, R.E.; Fajardo, V.A. Sarco(endo)plasmic reticulum Ca2+-ATPase function is impaired in skeletal and cardiac muscles from young DBA/2J mdx mice. iScience 2022, 25, 104972. [Google Scholar] [CrossRef] [PubMed]
- Goonasekera, S.A.; Lam, C.K.; Millay, D.P.; Sargent, M.A.; Hajjar, R.J.; Kranias, E.G.; Molkentin, J.D. Mitigation of muscular dystrophy in mice by SERCA overexpression in skeletal muscle. J. Clin. Investig. 2011, 121, 1044–1052. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mazala, D.A.; Pratt, S.J.P.; Chen, D.; Molkentin, J.D.; Lovering, R.M.; Chin, E.R. SERCA1 overexpression minimizes skeletal muscle damage in dystrophic mouse models. Am. J. Physiol. Cell Physiol. 2015, 308, C699–C709. [Google Scholar] [CrossRef] [Green Version]
- Shin, J.H.; Bostick, B.; Yue, Y.; Hajjar, R.; Duan, D. SERCA2a gene transfer improves electrocardiographic performance in aged mdx mice. J. Transl. Med. 2011, 9, 132. [Google Scholar] [CrossRef] [Green Version]
- Wasala, N.B.; Yue, Y.; Lostal, W.; Wasala, L.P.; Niranjan, N.; Hajjar, R.J.; Babu, G.J.; Duan, D. Single SERCA2a therapy ameliorated dilated cardiomyopathy for 18 months in a mouse model of duchenne muscular dystrophy. Mol. Ther. 2020, 28, 845–854. [Google Scholar] [CrossRef]
- Viner, R.I.; Ferrington, D.A.; Huhmer, A.F.; Bigelow, D.J.; Schöneich, C. Accumulation of nitrotyrosine on the SERCA2a isoform of SR Ca-ATPase of rat skeletal muscle during aging: A peroxynitrite-mediated process? FEBS Lett. 1996, 379, 286–290. [Google Scholar] [CrossRef] [Green Version]
- Viner, R.I.; Ferrington, D.A.; Williams, T.D.; Bigelow, D.J.; Schöneich, C. Protein modification during biological aging: Selective tyrosine nitration of the SERCA2a isoform of the sarcoplasmic reticulum Ca2+-ATPase in skeletal muscle. Biochem. J. 1999, 340 Pt 3, 657–669. [Google Scholar] [CrossRef]
- Viner, R.I.; Krainev, A.G.; Williams, T.D.; Schöneic, C.; and Bigelow, D.J. Identification of oxidation-sensitive peptides within the cytoplasmic domain of the sarcoplasmic reticulum Ca2+-ATPase. Biochemistry 1997, 36, 7706–7716. [Google Scholar] [CrossRef]
- Viner, R.I.; Williams, T.D.; Schöneic, C. Peroxynitrite modification of protein thiols: Oxidation, nitrosylation, and S-glutathiolation of functionally important cysteine residue(s) in the sarcoplasmic reticulum Ca-ATPase. Biochemistry 1999, 38, 12408–12415. [Google Scholar] [CrossRef]
- Babu, G.J.; Bhupathy, P.; Carnes, C.A.; Billman, G.E.; Periasamy, M. Differential expression of sarcolipin protein during muscle development and cardiac pathophysiology. J. Mol. Cell. Cardiol. 2007, 43, 215–222. [Google Scholar] [CrossRef] [Green Version]
- Shaikh, S.A.; Sahoo, S.K.; Periasamy, M. Phospholamban and sarcolipin: Are they functionally redundant or distinct regulators of the Sarco(Endo)Plasmic Reticulum Calcium ATPase? J. Mol. Cell. Cardiol. 2016, 91, 81–91. [Google Scholar] [CrossRef] [Green Version]
- Anderson, D.M.; Anderson, K.M.; Chang, C.L.; Makarewich, C.A.; Nelson, B.R.; McAnally, J.R.; Kasaragod, P.; Shelton, J.M.; Liou, J.; Bassel-Duby, R.; et al. A micropeptide encoded by a putative long noncoding RNA regulates muscle performance. Cell 2015, 160, 595–606. [Google Scholar] [CrossRef] [Green Version]
- Bhupathy, P.; Babu, G.J.; Periasamy, M. Sarcolipin and phospholamban as regulators of cardiac sarcoplasmic reticulum Ca2+ ATPase. J. Mol. Cell. Cardiol. 2007, 42, 903–911. [Google Scholar] [CrossRef] [Green Version]
- Pant, M.; Bal, N.C.; Periasamy, M. Sarcolipin: A Key Thermogenic and Metabolic Regulator in Skeletal Muscle. Trends Endocrinol. Metab. 2016, 27, 881–892. [Google Scholar] [CrossRef] [Green Version]
- Nelson, B.R.; Makarewich, C.A.; Anderson, D.M.; Winders, B.R.; Troupes, C.D.; Wu, F.; Reese, A.L.; McAnally, J.R.; Chen, X.; Kavalali, E.T.; et al. A peptide encoded by a transcript annotated as long noncoding RNA enhances SERCA activity in muscle. Science 2016, 351, 271–275. [Google Scholar] [CrossRef] [Green Version]
- Law, M.L.; Prins, K.W.; Olander, M.E.; Metzger, J.M. Exacerbation of dystrophic cardiomyopathy by phospholamban deficiency mediated chronically increased cardiac Ca2+ cycling in vivo. Am. J. Physiol. Heart Circ. Physiol. 2018, 315, H1544–H1552. [Google Scholar] [CrossRef]
- Balakrishnan, R.; Mareedu, S.; Babu, G.J. Reducing sarcolipin expression improves muscle metabolism in mdx mice. Am. J. Physiol. Cell Physiol. 2022, 322, 260–274. [Google Scholar] [CrossRef]
- Mareedu, S.; Pachon, R.; Thilagavathi, J.; Fefelova, N.; Balakrishnan, R.; Niranjan, N.; Xie, L.H.; Babu, G.J. Sarcolipin haploinsufficiency prevents dystrophic cardiomyopathy in mdx mice. Am. J. Physiol. Heart Circ. Physiol. 2021, 320, 200–210. [Google Scholar] [CrossRef] [PubMed]
- Tanihata, J.; Nagata, T.; Ito, N.; Saito, T.; Nakamura, A.; Minamisawa, S.; Aoki, Y.; Ruegg, U.T.; Takeda, S. Truncated dystrophin ameliorates the dystrophic phenotype of mdx mice by reducing sarcolipin-mediated SERCA inhibition. Biochem. Biophys. Res. Commun. 2018, 505, 51–59. [Google Scholar] [CrossRef] [PubMed]
- Fajardo, V.A.; Chambers, P.J.; Juracic, E.S.; Rietze, B.A.; Gamu, D.; Bellissimo, C.; Kwon, F.; Quadrilatero, J.; Russell Tupling, A. Sarcolipin deletion in mdx mice impairs calcineurin signalling and worsens dystrophic pathology. Hum. Mol. Genet. 2018, 27, 4094–4102. [Google Scholar] [CrossRef] [PubMed]
- Chakkalakal, J.V.; Harrison, M.A.; Carbonetto, S.; Chin, E.; Michel, R.N.; Jasmin, B.J. Stimulation of calcineurin signaling attenuates the dystrophic pathology in mdx mice. Hum. Mol. Genet. 2004, 13, 379–388. [Google Scholar] [CrossRef] [Green Version]
- Stupka, N.; Schertzer, J.D.; Bassel-Duby, R.; Olson, E.N.; Lynch, G.S. Stimulation of calcineurin Aalpha activity attenuates muscle pathophysiology in mdx dystrophic mice. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2008, 294, R983–R992. [Google Scholar] [CrossRef] [Green Version]
- Makarewich, C.A.; Bezprozvannaya, S.; Gibson, A.M.; Bassel-Duby, R.; Olson, E.N. Gene therapy with the DWORF micropeptide attenuates cardiomyopathy in mice. Circ. Res. 2020, 127, 1340–1342. [Google Scholar] [CrossRef]
- Zhang, S.-S.; Zhou, S.; Crowley-McHattan, Z.J.; Wang, R.-Y.; Li, J.-P. A Review of the Role of Endo/Sarcoplasmic Reticulum-Mitochondria Ca2+ Transport in Diseases and Skeletal Muscle Function. Int. J. Environ. Res. Public Health 2021, 18, 3874. [Google Scholar] [CrossRef]
- Bellinger, A.M.; Reiken, S.; Carlson, C.; Mongillo, M.; Liu, X.; Rothman, L.; Matecki, S.; Lacampagne, A.; Marks, A.R. Hypernitrosylated ryanodine receptor calcium release channels are leaky in dystrophic muscle. Nat. Med. 2009, 15, 325–330. [Google Scholar] [CrossRef]
- Nogami, K.; Maruyama, Y.; Elhussieny, A.; Sakai-Takemura, F.; Tanihata, J.; Kira, J.-I.; Miyagoe-Suzuki, Y.; Takeda, S. iNOS is not responsible for RyR1 S-nitrosylation in mdx mice with truncated dystrophin. BMC Musculoskelet Disord. 2020, 21, 479. [Google Scholar] [CrossRef]
- Fauconnier, J.; Thireau, J.; Reiken, S.; Cassan, C.; Richard, S.; Matecki, S.; Marks, A.R.; Lacampagne, A. Leaky RyR2 trigger ventricular arrhythmias in Duchenne muscular dystrophy. Proc. Natl. Acad. Sci. USA 2010, 107, 1559–1564. [Google Scholar] [CrossRef] [Green Version]
- Kyrychenko, S.; Polakova, E.; Kang, C.; Pocsai, K.; Ullrich, N.D.; Niggli, E.; Shirokova, N. Hierarchical accumulation of RyR post-translational modifications drives disease progression in dystrophic cardiomyopathy. Cardiovasc Res. 2013, 97, 666–675. [Google Scholar] [CrossRef] [Green Version]
- Wang, Q.; Wang, W.; Wang, G.; Rodney, G.G.; Wehrens, X.H. Crosstalk between RyR2 oxidation and phosphorylation contributes to cardiac dysfunction in mice with Duchenne muscular dystrophy. J. Mol. Cell Cardiol. 2015, 89 Pt B, 177–184. [Google Scholar] [CrossRef] [Green Version]
- Capogrosso, R.F.; Mantuano, P.; Uaesoontrachoon, K.; Cozzoli, A.; Giustino, A.; Dow, T.; Srinivassane, S.; Filipovic, M.; Bell, C.; Vandermeulen, J.; et al. Ryanodine channel complex stabilizer compound S48168/ARM210 as a disease modifier in dystrophin-deficient mdx mice: Proof-of-concept study and independent validation of efficacy. FASEB J. 2018, 32, 1025–1043. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barthelemy, F.; Wang, R.T.; Hsu, C.; Douine, E.D.; Marcantonio, E.E.; Nelson, S.F.; Miceli, M.C. Targeting RyR activity boosts antisense exon 44 and 45 skipping in human DMD skeletal or cardiac muscle culture models. Mol. Ther. Nucleic Acids 2019, 18, 580–589. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mareedu, S.; Million, E.D.; Duan, D.; Babu, G.J. Abnormal Calcium Handling in Duchenne Muscular Dystrophy: Mechanisms and Potential Therapies. Front Physiol. 2021, 12, 647010. [Google Scholar] [CrossRef]
- Takeshima, H.; Venturi, E.; Sitsapesan, R. New and notable ion-channels in the sarcoplasmic/endoplasmic reticulum: Do they support the process of intracellular Ca²⁺ release? J. Physiol. 2015, 593, 3241–3251. [Google Scholar] [CrossRef] [Green Version]
- Kuum, M.; Veksler, V.; Kaasik, A. Potassium fluxes across the endoplasmic reticulum and their role in endoplasmic reticulum calcium homeostasis. Cell Calcium 2015, 58, 79–85. [Google Scholar] [CrossRef]
- Gillespie, D.; Fill, M. Intracellular calcium release channels mediate their own countercurrent: The ryanodine receptor case study. Biophys. J. 2008, 95, 3706–3714. [Google Scholar] [CrossRef] [Green Version]
- Yazawa, M.; Ferrante, C.; Feng, J.; Mio, K.; Ogura, T.; Zhang, M.; Lin, P.H.; Pan, Z.; Komazaki, S.; Kato, K.; et al. TRIC channels are essential for Ca2+ handling in intracellular stores. Nature 2007, 448, 78–82. [Google Scholar] [CrossRef]
- Mado, K.; Chekulayev, V.; Shevchuk, I.; Puurand, M.; Tepp, K.; Kaambre, T. On the role of tubulin, plectin, desmin, and vimentin in the regulation of mitochondrial energy fluxes in muscle cells. Am. J. Physiol. Cell Physiol. 2019, 316, C657–C667. [Google Scholar] [CrossRef]
- Ramos, S.V.; Hughes, M.C.; Delfinis, L.J.; Bellissimo, C.A.; Perry, C.G. Mitochondrial bioenergetic dysfunction in the D2.mdx model of Duchenne muscular dystrophy is associated with microtubule disorganization in skeletal muscle. PLoS ONE 2020, 15, e0237138. [Google Scholar] [CrossRef]
- Viola, H.M.; Adams, A.M.; Davies, S.M.; Fletcher, S.; Filipovska, A.; Hool, L.C. Impaired functional communication between the L-type calcium channel and mitochondria contributes to metabolic inhibition in the mdx heart. Proc. Natl. Acad. Sci. USA 2014, 111, E2905–E2914. [Google Scholar] [CrossRef] [Green Version]
- De Stefani, D.; Raffaello, A.; Teardo, E.; Szabo, I.; Rizzuto, R. A forty-kilodalton protein of the inner membrane is the mitochondrial calcium uniporter. Nature 2011, 476, 336–340. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baughman, J.M.; Perocchi, F.; Girgis, H.S.; Plovanich, M.; Belcher-Timme, C.A.; Sancak, Y.; Bao, X.R.; Strittmatter, L.; Goldberger, O.; Bogorad, R.L.; et al. Integrative genomics identifies MCU as an essential component of the mitochondrial calcium uniporter. Nature 2011, 476, 341–345. [Google Scholar] [CrossRef] [Green Version]
- Palty, R.; Silverman, W.F.; Hershfinkel, M.; Caporale, T.; Sensi, S.L.; Parnis, J.; Nolte, C.; Fishman, D.; Shoshan Barmatz, V.; Herrmann, S.; et al. NCLX is an essential component of mitochondrial Na+/Ca2+ exchange. Proc. Natl. Acad. Sci. USA 2010, 107, 436–441. [Google Scholar] [CrossRef] [Green Version]
- Jiang, D.; Zhao, L.; Clapham, D.E. Genome-wide RNAi screen identifies Letm1 as a mitochondrial Ca2+/H+ antiporter. Science 2009, 326, 144–147. [Google Scholar] [CrossRef] [Green Version]
- Belosludtsev, K.N.; Dubinin, M.; Belosludtseva, N.; Mironova, G.D. Mitochondrial Ca2+ Transport: Mechanisms, Molecular Structures, and Role in Cells. Biochemistry 2019, 84, 593–607. [Google Scholar] [CrossRef]
- Bonora, M.; Giorgi, C.; Pinton, P. Molecular mechanisms and consequences of mitochondrial permeability transition. Nat. Rev. Mol. Cell Biol. 2022, 23, 266–285. [Google Scholar] [CrossRef]
- Dubinin, M.V.; Talanov, E.Y.; Tenkov, K.S.; Starinets, V.S.; Mikheeva, I.B.; Sharapov, M.G.; Belosludtsev, K.N. Duchenne muscular dystrophy is associated with the inhibition of calcium uniport in mitochondria and an increased sensitivity of the organelles to the calcium-induced permeability transition. Biochim. Biophys. Acta Mol. Basis Dis. 2020, 1866, 165674. [Google Scholar] [CrossRef] [PubMed]
- Dubinin, M.V.; Talanov, E.Y.; Tenkov, K.S.; Starinets, V.S.; Belosludtseva, N.V.; Belosludtsev, K.N. The Effect of Deflazacort Treatment on the Functioning of Skeletal Muscle Mitochondria in Duchenne Muscular Dystrophy. Int. J. Mol. Sci. 2020, 21, 8763. [Google Scholar] [CrossRef]
- Raffaello, A.; De Stefani, D.; Sabbadin, D.; Teardo, E.; Merli, G.; Picard, A.; Checchetto, V.; Moro, S.; Szabo, I.; Rizzuto, R. The mitochondrial calcium uniporter is a multimer that can include a dominant-negative pore-forming subunit. EMBO J. 2013, 32, 2362–2376. [Google Scholar] [CrossRef] [Green Version]
- Fieni, F.; Lee, S.B.; Jan, Y.N.; Kirichok, Y. Activity of the mitochondrial calcium uniporter varies greatly between tissues. Nat. Commun. 2012, 3, 1317. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dubinin, M.V.; Talanov, E.Y.; Tenkov, K.S.; Starinets, V.S.; Mikheeva, I.B.; Belosludtsev, K.N. Transport of Ca2+ and Ca2+-dependent permeability transition in heart mitochondria in the early stages of Duchenne muscular dystrophy. Biochim. Biophys. Acta Bioenerg. 2020, 1861, 148250. [Google Scholar] [CrossRef]
- Angebault, C.; Panel, M.; Lacôte, M.; Rieusset, J.; Lacampagne, A.; Fauconnier, J. Metformin Reverses the Enhanced Myocardial SR/ER-Mitochondria Interaction and Impaired Complex I-Driven Respiration in Dystrophin-Deficient Mice. Front. Cell Dev. Biol. 2021, 8, 609493. [Google Scholar] [CrossRef]
- Ascah, A.; Khairallah, M.; Daussin, F.; Bourcier-Lucas, C.; Godin, R.; Allen, B.G.; Petrof, B.J.; Rosiers, C.D.; Burelle, Y. Stress-induced opening of the permeability transition pore in the dystrophin-deficient heart is attenuated by acute treatment with sildenafil. Am. J. Physiol. Heart Circ. Physiol. 2011, 300, H144–H153. [Google Scholar] [CrossRef]
- Jung, C.; Martins, A.S.; Niggli, E.; Shirokova, N. Dystrophic cardiomyopathy: Amplification of cellular damage by Ca2+ signalling and reactive oxygen species-generating pathways. Cardiovasc. Res. 2008, 77, 766–773. [Google Scholar] [CrossRef]
- Dubinin, M.V.; Starinets, V.S.; Talanov, E.Y.; Mikheeva, I.B.; Belosludtseva, N.V.; Serov, D.A.; Tenkov, K.S.; Belosludtseva, E.V.; Belosludtsev, K.N. Effect of the Non-Immunosuppressive MPT Pore Inhibitor Alisporivir on the Functioning of Heart Mitochondria in Dystrophin-Deficient mdx Mice. Biomedicines 2021, 9, 1232. [Google Scholar] [CrossRef]
- Willi, L.; Abramovich, I.; Fernandez-Garcia, J.; Agranovich, B.; Shulman, M.; Milman, H.; Baskin, P.; Eisen, B.; Michele, D.E.; Arad, M.; et al. Bioenergetic and Metabolic Impairments in Induced Pluripotent Stem Cell-Derived Cardiomyocytes Generated from Duchenne Muscular Dystrophy Patients. Int. J. Mol. Sci. 2022, 23, 9808. [Google Scholar] [CrossRef]
- Rybalka, E.; Timpani, C.A.; Cooke, M.B.; Williams, A.D.; Hayes, A. Defects in mitochondrial ATP synthesis in dystrophindeficient Mdx skeletal muscles may be caused by complex I insufficiency. PLoS ONE 2014, 9, e115763. [Google Scholar] [CrossRef] [Green Version]
- Hughes, M.C.; Ramos, S.V.; Turnbull, P.C.; Rebalka, I.A.; Cao, A.; Monaco, C.M.F.; Varah, N.E.; Edgett, B.A.; Huber, J.S.; Tadi, P.; et al. Early myopathy in Duchenne muscular dystrophy is associated with elevated mitochondrial H2O2 emission during impaired oxidative phosphorylation. J. Cachexia Sarcopenia Muscl. 2019, 10, 643–661. [Google Scholar] [CrossRef] [Green Version]
- Kyrychenko, V.; Polakova, E.; Janicek, R.; Shirokova, N. Mitochondrial dysfunctions during progression of dystrophic cardiomyopathy. Cell Calcium 2015, 58, 186–195. [Google Scholar] [CrossRef] [Green Version]
- Stepien, G.; Torroni, A.; Chung, A.B.; Hodge, J.A.; Wallace, D.C. Differential expression of adenine nucleotide translocator isoforms in mammalian tissues and during muscle cell differentiation. J. Biol. Chem. 1992, 267, 14592–14597. [Google Scholar] [CrossRef]
- Dubinin, M.V.; Starinets, V.S.; Talanov, E.Y.; Mikheeva, I.B.; Belosludtseva, N.V.; Belosludtsev, K.N. Alisporivir Improves Mitochondrial Function in Skeletal Muscle of mdx Mice but Suppresses Mitochondrial Dynamics and Biogenesis. Int. J. Mol. Sci. 2021, 22, 9780. [Google Scholar] [CrossRef] [PubMed]
- Millay, D.P.; Sargent, M.A.; Osinska, H.; Baines, C.; Barton, E.R.; Vuagniaux, G.; Sweeney, H.L.; Robbins, J.; Molkentin, J.D. Genetic and pharmacologic inhibition of mitochondrial-dependent necrosis attenuates muscular dystrophy. Nat. Med. 2008, 14, 442–447. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schiavone, M.; Zulian, A.; Menazza, S.; Petronilli, V.; Argenton, F.; Merlini, L.; Sabatelli, P.; Bernardi, P. Alisporivir rescues defective mitochondrial respiration in Duchenne muscular dystrophy. Pharmacol. Res. 2017, 125, 122–131. [Google Scholar] [CrossRef]
- Reutenauer, J.; Dorchies, O.M.; Patthey-Vuadens, O.; Vuagniaux, G.; Ruegg, U.T. Investigation of Debio 025, a cyclophilin inhibitor, in the dystrophic mdx mouse, a model for Duchenne muscular dystrophy. Br. J. Pharmacol. 2008, 155, 574–584. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wissing, E.R.; Millay, D.P.; Vuagniaux, G.; Molkentin, J.D. Debio-025 is more effective than prednisone in reducing muscular pathology in mdx mice. Neuromuscul. Disord. 2010, 20, 753–760. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stocco, A.; Smolina, N.; Sabatelli, P.; Šileikytė, J.; Artusi, E.; Mouly, V.; Cohen, M.; Forte, M.; Schiavone, M.; Bernardi, P. Treatment with a triazole inhibitor of the mitochondrial permeability transition pore fully corrects the pathology of sapje zebrafish lacking dystrophin. Pharmacol. Res. 2021, 165, 105421. [Google Scholar] [CrossRef]
- Dubinin, M.V.; Starinets, V.S.; Mikheeva, I.B.; Belosludtsev, K.N. Effect of Alisporivir on Calcium Ion Transport and Mitophagy in Skeletal Muscle and Heart Mitochondria in Dystrophin-Deficient Mice. Bull. Exp. Biol. Med. 2022, 172, 695–700. [Google Scholar] [CrossRef]
- Colell, A.; Garcia-Ruiz, C.; Lluis, M.; Coll, O.; Mari, M.; Fernandez-Chaca, J.C. Cholesterol impairs the adenine nucleotide translocator-mediated mitochondrial permeability transition through altered membrane fluidity. J. Biol. Chem. 2003, 278, 33928–33935. [Google Scholar] [CrossRef] [Green Version]
- Dubinin, M.V.; Starinets, V.S.; Belosludtseva, N.V.; Mikheeva, I.B.; Chelyadnikova, Y.A.; Igoshkina, A.D.; Vafina, A.B.; Vedernikov, A.A.; Belosludtsev, K.N. BKCa Activator NS1619 Improves the Structure and Function of Skeletal Muscle Mitochondria in Duchenne Dystrophy. Pharmaceutics 2022, 14, 2336. [Google Scholar] [CrossRef]
- Dubinin, M.V.; Starinets, V.S.; Belosludtseva, N.V.; Mikheeva, I.B.; Chelyadnikova, Y.A.; Penkina, D.K.; Vedernikov, A.A.; Belosludtsev, K.N. The Effect of Uridine on the State of Skeletal Muscles and the Functioning of Mitochondria in Duchenne Dystrophy. Int. J. Mol. Sci. 2022, 23, 10660. [Google Scholar] [CrossRef]
- Mironova, G.D.; Negoda, A.E.; Marinov, B.S.; Paucek, P.; Costa, A.D.; Grigoriev, S.M.; Skarga, Y.Y.; Garlid, K.D. Functional distinctions between the mitochondrial ATP-dependent K+ channel (mitoKATP) and its inward rectifier subunit (mitoKIR). J. Biol. Chem. 2004, 279, 32562–32568. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Guo, S.; Xie, C.; Fang, J. Uridine metabolism and its role in glucose, lipid, and amino acid homeostasis. Biomed. Res. Int. 2020, 2020, 7091718. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simoes, I.C.M.; Morciano, G.; Lebiedzinska-Arciszewska, M.; Aguiari, G.; Pinton, P.; Potes, Y.; Wieckowski, M.R. The mystery of mitochondria-ER contact sites in physiology and pathology: A cancer perspective. Biochim. Biophys. Acta Mol. Basis Dis. 2020, 1866, 165834. [Google Scholar] [CrossRef] [PubMed]
- Poston, C.N.; Krishnan, S.C.; Bazemore-Walker, C.R. In depth proteomic analysis of mammalian mitochondria-associated membranes (MAM). J. Proteom. 2013, 79, 219–230. [Google Scholar] [CrossRef] [PubMed]
- Szabadkai, G.; Bianchi, K.; Varnai, P.; De Stefani, D.; Wieckowski, M.R.; Cavagna, D.; Nagy, A.I.; Balla, T.; Rizzuto, R. Chaperone-mediated coupling of endoplasmic reticulum and mitochondrial Ca2+ channels. J. Cell Biol. 2006, 175, 901–911. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cárdenas, C.; Juretić, N.; Bevilacqua, J.A.; García, I.E.; Figueroa, R.; Hartley, R.; Taratuto, A.L.; Gejman, R.; Riveros, N.; Molgó, J.; et al. Abnormal distribution of inositol 1,4,5-trisphosphate receptors in human muscle can be related to altered calcium signals and gene expression in Duchenne dystrophy-derived cells. FASEB J. 2010, 24, 3210–3221. [Google Scholar] [CrossRef] [Green Version]
- Farini, A.; Sitzia, C.; Cassinelli, L.; Colleoni, F.; Parolini, D.; Giovanella, U.; Maciotta, S.; Colombo, A.; Meregalli, M.; Torrente, Y. Inositol 1,4,5-trisphosphate (IP3)-dependent Ca2+ signaling mediates delayed myogenesis in Duchenne muscular dystrophy fetal muscle. Development 2016, 143, 658–669. [Google Scholar] [CrossRef] [Green Version]
- Liberona, J.L.; Powell, J.A.; Shenoi, S.; Petherbridge, L.; Caviedes, R.; Jaimovich, E. Differences in both inositol 1,4,5-trisphosphate mass and inositol 1,4,5-trisphosphate receptors between normal and dystrophic skeletal muscle cell lines. Muscle Nerve 1998, 21, 902–909. [Google Scholar] [CrossRef]
- Pauly, M.; Angebault-Prouteau, C.; Dridi, H.; Notarnicola, C.; Scheuermann, V.; Lacampagne, A.; Matecki, S.; Fauconnier, J. ER stress disturbs SR/ER-mitochondria Ca2+ transfer: Implications in Duchenne muscular dystrophy. Biochim. Biophys. Acta Mol. Basis Dis. 2017, 1863, 2229–2239. [Google Scholar] [CrossRef]
- Altamirano, F.; López, J.R.; Henríquez, C.; Molinski, T.; Allen, P.D.; Jaimovich, E. Increased resting intracellular calcium modulates NF-κB-dependent inducible nitric-oxide synthase gene expression in dystrophic mdx skeletal myotubes. J. Biol. Chem. 2012, 287, 20876–208787. [Google Scholar] [CrossRef] [Green Version]
- Mijares, A.; Altamirano, F.; Kolster, J.; Adams, J.A.; López, J.R. Age-dependent changes in diastolic Ca2+ and Na+ concentrations in dystrophic cardiomyopathy: Role of Ca2+ entry and IP3. Biochem. Biophys. Res. Commun. 2014, 452, 1054–1059. [Google Scholar] [CrossRef] [Green Version]
- Valladares, D.; Utreras-Mendoza, Y.; Campos, C.; Morales, C.; Diaz-Vegas, A.; Contreras-Ferrat, A.; Westermeier, F.; Jaimovich, E.; Marchi, S.; Pinton, P.; et al. IP3 receptor blockade restores autophagy and mitochondrial function in skeletal muscle fibers of dystrophic mice. Biochim. Biophys. Acta 2018, 1864, 3685–3695. [Google Scholar] [CrossRef]
- Meyer, P.; Notarnicola, C.; Meli, A.C.; Matecki, S.; Hugon, G.; Salvador, J.; Khalil, M.; Féasson, L.; Cances, C.; Cottalorda, J.; et al. Skeletal Ryanodine Receptors Are Involved in Impaired Myogenic Differentiation in Duchenne Muscular Dystrophy Patients. Int. J. Mol. Sci. 2021, 22, 12985. [Google Scholar] [CrossRef]
- Hulmi, J.J.; Hentilä, J.; DeRuisseau, K.C.; Oliveira, B.M.; Papaioannou, K.G.; Autio, R.; Kujala, U.M.; Ritvos, O.; Kainulainen, H.; Korkmaz, A.; et al. Effects of muscular dystrophy, exercise and blocking activin receptor IIB ligands on the unfolded protein response and oxidative stress. Free Radic. Biol. Med. 2016, 99, 308–322. [Google Scholar] [CrossRef] [Green Version]
- Kuznetsov, A.V.; Winkler, K.; Wiedemann, F.; Von Bossanyi, P.; Dietzmann, K.; Kunz, W.S. Impaired mitochondrial oxidative phosphorylation in skeletal muscle of the dystrophin-deficient mdx mouse. Mol. Cell Biochem. 1998, 183, 87–96. [Google Scholar] [CrossRef]
- Giovarelli, M.; Zecchini, S.; Catarinella, G.; Moscheni, C.; Sartori, P.; Barbieri, C.; Roux-Biejat, P.; Napoli, A.; Vantaggiato, C.; Cervia, D.; et al. Givinostat as metabolic enhancer reverting mitochondrial biogenesis deficit in Duchenne muscular dystrophy. Pharmacol. Res. 2021, 170, 105751. [Google Scholar] [CrossRef]
- Pant, M.; Sopariwala, D.H.; Bal, N.C.; Lowe, J.; Delfín, D.A.; Rafael-Fortney, J.; Periasamy, M. Metabolic dysfunction and altered mitochondrial dynamics in the utrophin-dystrophin deficient mouse model of Duchenne muscular dystrophy. PLoS ONE 2015, 10, e0123875. [Google Scholar] [CrossRef] [Green Version]



{kind=link}
{kind=link}
{kind=link}
| Target | Drug | Mechanism of Action | Models Used | References |
|---|---|---|---|---|
| Membrane microtears | Poloxamer 188 | Seals microtears in the sarcolemma | Canine model, mdx, and mdx/utrn–/– mice | Townsend et al. [34] Houang et al. [35] Markham et al. [36] |
| TRPC3 | Pyr10 | Prevents calcium overload | DMD rats | Creisméas et al. [51] |
| TRPC6 | BI 749327 | Prevents calcium overload | mdx/utrn–/– mice | Lin et al. [50] |
| L-type CaV | Diltiazem, verapamil, nifedipine | Prevent calcium overload | mdx mice | Matsumura et al. [59] Altamirano et al. [60] |
| P2X7 | Coomassie Brilliant Blue G 250, oxidized ATP, suramin | Prevent calcium overload | mdx mice, BIO14.6 hamsters | Sinadinos et al. [63] Gazzerro et al. [64] Taniguti et al. [65] Iwata et al. [85] |
| STIM1-Orai1 | AnCoA4, CM4620, and GSK7975A | Reduce Orai1-induced Ca2+ influx into the myoplasm | DMD-patient-derived myotubes | Uchimura et al. [75] |
| NaV1.4 | Tetrodotoxin | Prevents sodium overload | mdx mice | Hirn et al. [83] |
| NHE | Cariporide, 5-(N-ethyl-N-isopropyl)-amiloride, rimeporide | Prevent sodium overload | mdx mice, BIO14.6 hamsters, DMD patients | Iwata et al. [85] Previtali et al. [87] |
| SERCA1 or SERCA2a | AAV.SERCA | Overexpresses SERKA and improves calcium uptake by SR | mdx and mdx/utrn–/– mice | Goonasekera et al. [123] Mazala et al. [124] Wasala et al. [126] |
| SLN | AAV.SLN | Downregulates SLN and activates SERKA | mdx/utrn–/– mice | Viner et al. [109] |
| RyR | Rycals | Improve binding of calstabin to RyR and prevent RyR Ca2+ leak | mdx mice, DMD-patient-derived myotubes and cardiomyocytes | Capogrosso et al. [151] Barthelemy et al. [152] Meyer et al. [203] |
| VDAC | VDAC peptide | Prevents the opening of VDAC and rescues mitochondrial membrane potential | mdx mice | Viola et al. [160] |
| MPT pore | CsA, alisporivir, isoxazoles | Desensitize MPT pore to activation by calcium | mdx mice, Zebrafish model, DMD-patient-derived myotubes | Dubinin et al. [181] Millay et al. [182] Schiavone et al. [183] Reutenauer et al. [184] Wissing et al. [185] Stocco et al. [186] |
| mitoBKCa | NS1619 | Activates potassium transport into mitochondria | mdx mice | Dubinin et al. [189] |
| mitoKATP | Uridine (precursor of uridine 5’-diphosphate (UDP) | Activates potassium transport into mitochondria | mdx mice | Dubinin et al. [190] |
| IP3R | Xestospongin | Inhibits IP3R and prevents calcium overload | mdx mice | Altamirano et al. [200] Mijares et al. [201] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dubinin, M.V.; Belosludtsev, K.N. Ion Channels of the Sarcolemma and Intracellular Organelles in Duchenne Muscular Dystrophy: A Role in the Dysregulation of Ion Homeostasis and a Possible Target for Therapy. Int. J. Mol. Sci. 2023, 24, 2229. https://doi.org/10.3390/ijms24032229
Dubinin MV, Belosludtsev KN. Ion Channels of the Sarcolemma and Intracellular Organelles in Duchenne Muscular Dystrophy: A Role in the Dysregulation of Ion Homeostasis and a Possible Target for Therapy. International Journal of Molecular Sciences. 2023; 24(3):2229. https://doi.org/10.3390/ijms24032229
Chicago/Turabian StyleDubinin, Mikhail V., and Konstantin N. Belosludtsev. 2023. "Ion Channels of the Sarcolemma and Intracellular Organelles in Duchenne Muscular Dystrophy: A Role in the Dysregulation of Ion Homeostasis and a Possible Target for Therapy" International Journal of Molecular Sciences 24, no. 3: 2229. https://doi.org/10.3390/ijms24032229