
Acetylcorynoline Induces Apoptosis and G2/M Phase Arrest through the c-Myc Signaling Pathway in Colon Cancer Cells

 ,
,  , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
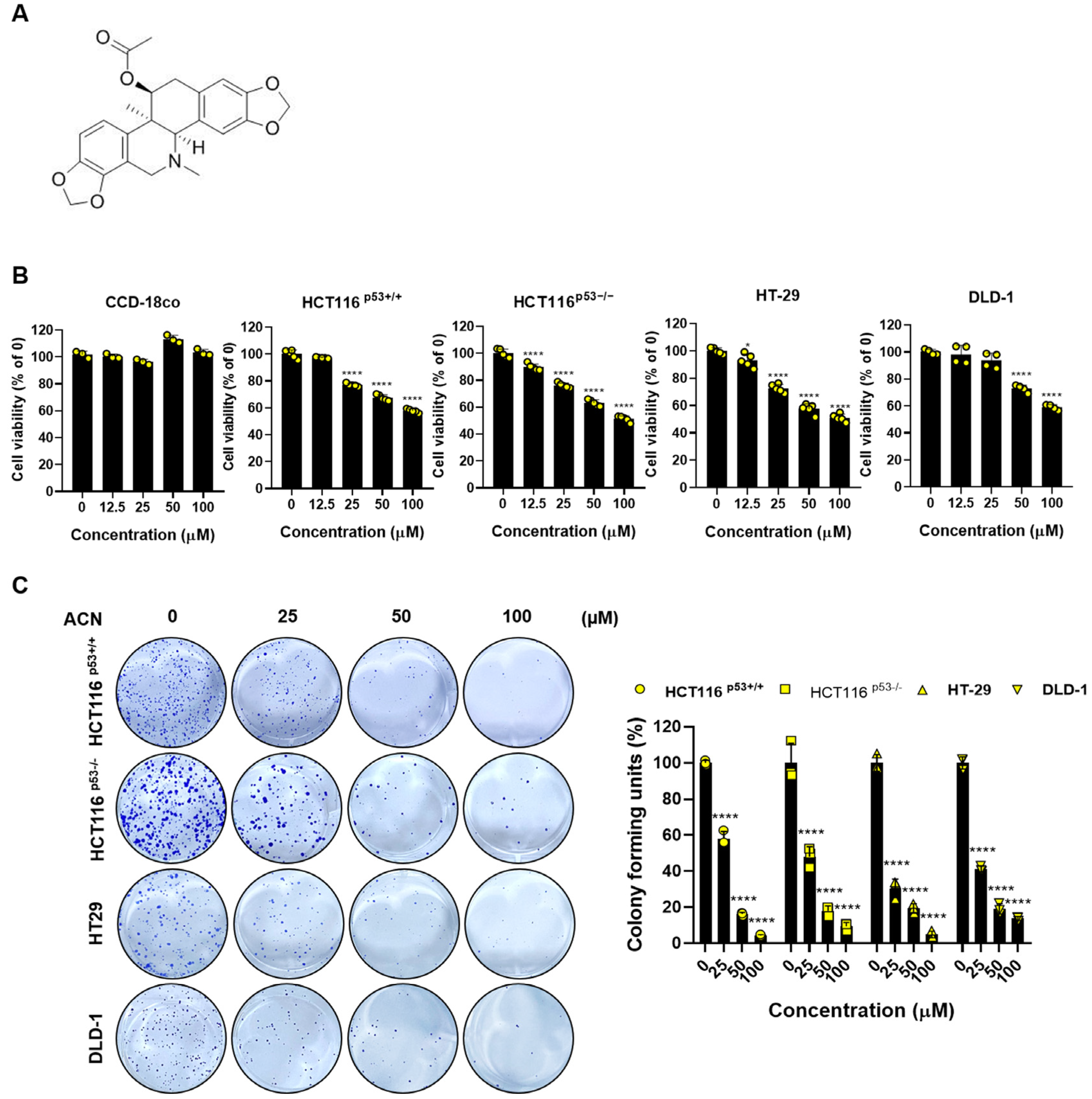
2.1. ACN Inhibits the Cell Viability and Proliferation of Colon Cancer Cells
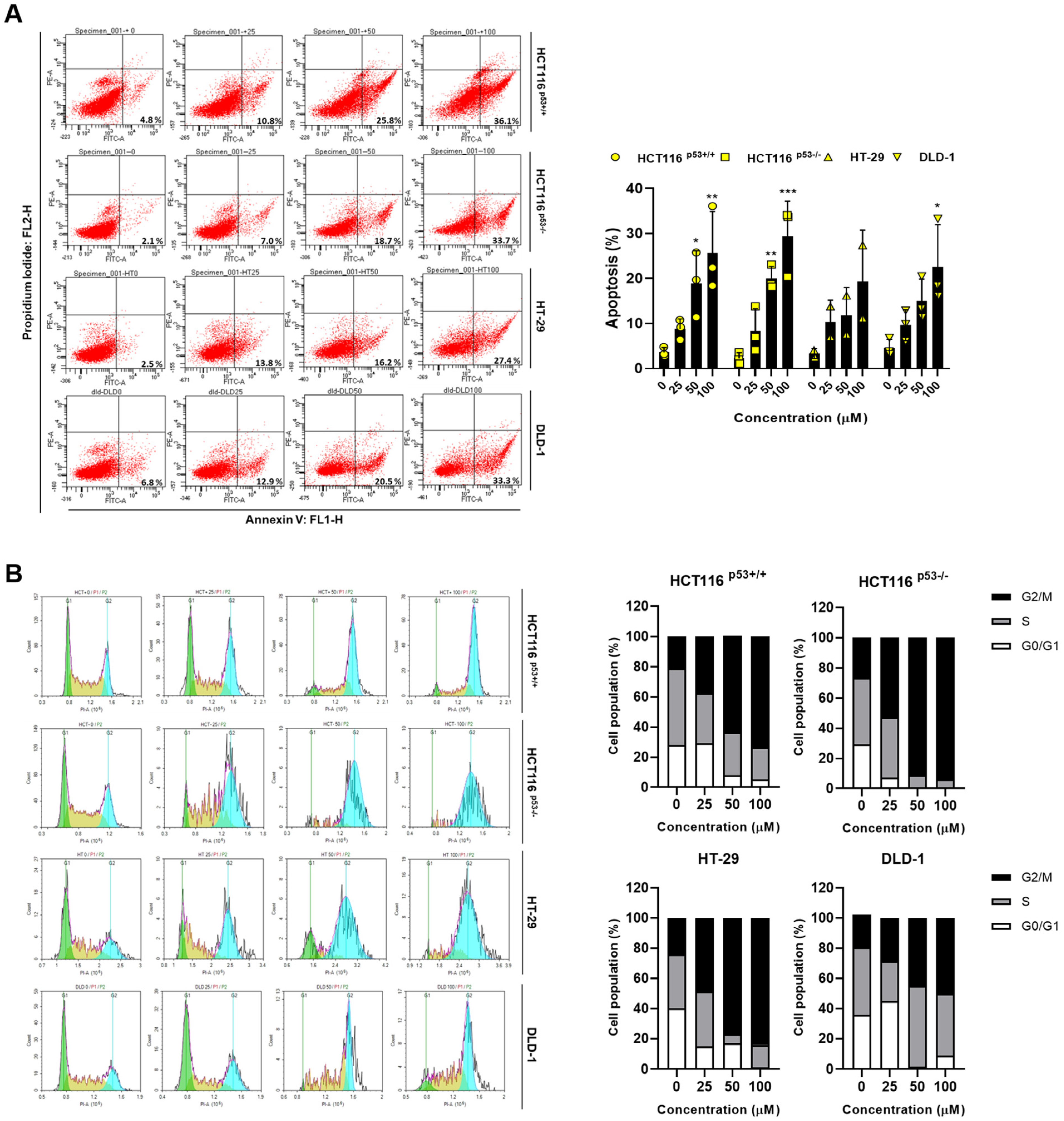
2.2. ACN Regulates Apoptosis and Induces G2/M Phase Arrest
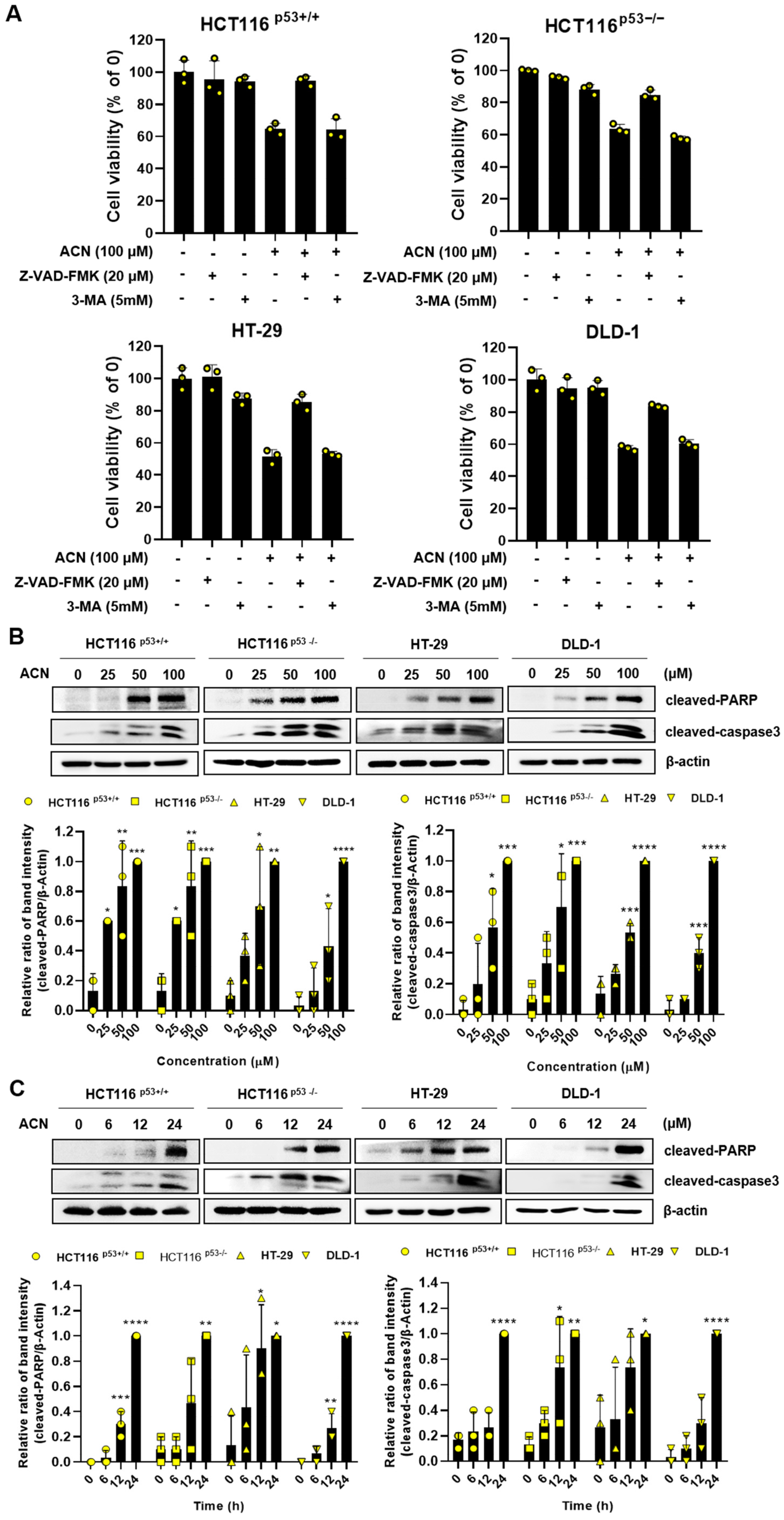
2.3. ACN Regulates the Expression of Apoptosis-Related Signaling Proteins
2.4. c-Myc Is a Potential Target for the Inhibition of Colon Cancer in ACN
2.5. ACN Attenuates c-Myc via CNOT2, MID1IP1, and Ribosomal Proteins
2.6. ACN Attenuates c-Myc Stability
2.7. ACN Regulates Serum-Induced c-Myc Stimulation
2.8. Potential Effect of ACN with 5-FU or Dox in Colon Cancer Cells
3. Discussion
4. Materials and Methods
4.1. Chemicals and Reagents
4.2. Cell Culture
4.3. Cytotoxicity Assay
4.4. Colony Formation Assay
4.5. Western Blotting
4.6. Cell Cycle Analysis via Flow Cytometry
4.7. Annexin V/Propidium Iodide (PI) Assay
4.8. Cycloheximide (CHX) Chase Assay for c-Myc Stability
4.9. Serum Stimulation for c-Myc Induction
4.10. Immunofluorescence Assay
4.11. Gene Silencing Using Small Interfering RNA (siRNA)
4.12. Real-Time Polymerase Chain Reaction (RT-PCR)
4.13. Statistical Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sung, H.; Ferlay, J.; Siegel, R.L. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2018. CA Cancer J. Clin. 2018, 68, 7–30. [Google Scholar] [CrossRef] [PubMed]
- Liang, B.J.; Pigula, M.; Baglo, Y.; Najafali, D.; Hasan, T.; Huang, H.-C. Breaking the selectivity-uptake trade-off of photoimmunoconjugates with nanoliposomal irinotecan for synergistic multi-tier cancer targeting. J. Nanobiotechnol. 2020, 18, 1. [Google Scholar] [CrossRef] [PubMed]
- Abd-Rabou, A.A.; Ahmed, H.H.; Shalby, A.B. Selenium Overcomes Doxorubicin Resistance in Their Nano-platforms Against Breast and Colon Cancers. Biol. Trace Elem. Res. 2020, 193, 377–389. [Google Scholar] [CrossRef] [PubMed]
- Weng, W.; Feng, J.; Qin, H.; Ma, Y. Molecular therapy of colorectal cancer: Progress and future directions. Int. J. Cancer 2015, 136, 493–502. [Google Scholar] [CrossRef] [PubMed]
- Jung, J.H.; Lee, H.-J.; Kim, J.-H.; Sim, D.Y.; Im, E.; Kim, S.; Chang, S.; Kim, S.-H. Colocalization of MID1IP1 and c-Myc is Critically Involved in Liver Cancer Growth via Regulation of Ribosomal Protein L5 and L11 and CNOT2. Cells 2020, 9, 985. [Google Scholar] [CrossRef] [PubMed]
- Al-Kuraya, K.; Novotny, H.; Bavi, P.; Siraj, A.K.; Uddin, S.; Ezzat, A.; Sanea, N.A.; Al-Dayel, F.; Al-Mana, H.; Sheikh, S.S.; et al. HER2, TOP2A, CCND1, EGFR and C-MYC oncogene amplification in colorectal cancer. J. Clin. Pathol. 2007, 60, 768–772. [Google Scholar] [CrossRef] [PubMed]
- Kress, T.R.; Sabò, A.; Amati, B. MYC: Connecting selective transcriptional control to global RNA production. Nat. Rev. Cancer 2015, 15, 593–607. [Google Scholar] [CrossRef]
- Hoffman, B.; Liebermann, D.A. Apoptotic signaling by c-MYC. Oncogene 2008, 27, 6462–6472. [Google Scholar] [CrossRef]
- Dang, C.V. c-Myc target genes involved in cell growth, apoptosis, and metabolism. Mol. Cell. Biol. 1999, 19, 1–11. [Google Scholar] [CrossRef]
- Sohn, E.J.; Jung, D.-B.; Lee, H.; Han, I.; Lee, J.; Lee, H.; Kim, S.-H. CNOT2 promotes proliferation and angiogenesis via VEGF signaling in MDA-MB-231 breast cancer cells. Cancer Lett. 2018, 412, 88–98. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Jung, J.H.; Hwang, J.; Park, J.E.; Kim, J.-H.; Park, W.Y.; Suh, J.Y.; Kim, S.-H. CNOT2 Is Critically Involved in Atorvastatin Induced Apoptotic and Autophagic Cell Death in Non-Small Cell Lung Cancers. Cancers 2019, 11, 1470. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.J.; Sim, D.Y.; Lee, H.M.; Lee, H.-J.; Kim, S.-H. Hypolipogenic Effect of Shikimic Acid Via Inhibition of MID1IP1 and Phosphorylation of AMPK/ACC. Int. J. Mol. Sci. 2019, 20, 582. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Huang, R.; Hua, J.; Liang, H.; Pan, Y.; Dai, L.; Liang, D.; Wang, H. Antitumor lignanamides from the aerial parts of Corydalis saxicola. Phytomed. Int. J. Phytother. Phytopharm. 2016, 23, 1599–1609. [Google Scholar] [CrossRef] [PubMed]
- Qin, F.; Chen, Y.; Wang, F.-F.; Tang, S.-Q.; Fang, Y.-L. Corydalis saxicola Bunting: A Review of Its Traditional Uses, Phytochemistry, Pharmacology, and Clinical Applications. Int. J. Mol. Sci. 2023, 24, 1626. [Google Scholar] [CrossRef] [PubMed]
- Fu, R.-H.; Wang, Y.-C.; Chen, C.-S.; Tsai, R.-T.; Liu, S.-P.; Chang, W.-L.; Lin, H.-L.; Lu, C.-H.; Wei, J.-R.; Wang, Z.-W.; et al. Acetylcorynoline attenuates dopaminergic neuron degeneration and α-synuclein aggregation in animal models of Parkinson’s disease. Neuropharmacology 2014, 82, 108–120. [Google Scholar] [CrossRef] [PubMed]
- Wei, H.L.; Liu, G.T. Protective action of corynoline, acetylcorynoline and protopine against experimental liver injury in mice. Yao Xue Xue Bao 1997, 32, 331–336. [Google Scholar]
- Fu, R.H.; Wang, Y.C.; Liu, S.P.; Chu, C.L.; Tsai, R.T.; Ho, Y.C.; Chang, W.L.; Chiu, S.C.; Harn, H.J.; Shyu, W.C.; et al. Acetylcorynoline impairs the maturation of mouse bone marrow-derived dendritic cells via suppression of IκB kinase and mitogen-activated protein kinase activities. PLoS ONE 2013, 8, e58398. [Google Scholar] [CrossRef]
- Li, J.; Yuan, J. Caspases in apoptosis and beyond. Oncogene 2008, 27, 6194–6206. [Google Scholar] [CrossRef]
- Kaufmann, S.H.; Desnoyers, S.; Ottaviano, Y.; Davidson, N.E.; Poirier, G.G. Specific proteolytic cleavage of poly(ADP-ribose) polymerase: An early marker of chemotherapy-induced apoptosis. Cancer Res. 1993, 53, 3976–3985. [Google Scholar]
- Jung, J.H.; Liao, J.-M.; Zhang, Q.; Zeng, S.; Nguyen, D.; Hao, Q.; Zhou, X.; Cao, B.; Kim, S.-H.; Lu, H. Inauhzin(c) Inactivates c-Myc Independently of p53. Cancer Biol. Ther. 2015, 16, 412–419. [Google Scholar] [CrossRef] [PubMed]
- Stine, Z.E.; Walton, Z.E.; Altman, B.J.; Hsieh, A.L.; Dang, C.V. MYC, Metabolism, and Cancer. Cancer Discov. 2015, 5, 1024–1039. [Google Scholar] [CrossRef] [PubMed]
- Jung, J.H.; Lee, D.; Ko, H.M.; Jang, H.-J. Inhibition of CNOT2 Induces Apoptosis via MID1IP1 in Colorectal Cancer Cells by Activating p53. Biomolecules 2021, 11, 1492. [Google Scholar] [CrossRef] [PubMed]
- Xianjun, F.; Xirui, X.; Jie, T.; Huiwen, M.; Shaojun, Z.; Qiaoyun, L.; Yunxin, L.; Xuqun, S. Momordin Ic induces G0/1 phase arrest and apoptosis in colon cancer cells by suppressing SENP1/c-MYC signaling pathway. J. Pharmacol. Sci. 2021, 146, 249–258. [Google Scholar] [CrossRef]
- Teng, T.; Mercer, C.A.; Hexley, P.; Thomas, G.; Fumagalli, S. Loss of tumor suppressor RPL5/RPL11 does not induce cell cycle arrest but impedes proliferation due to reduced ribosome content and translation capacity. Mol. Cell. Biol. 2013, 33, 4660–4671. [Google Scholar] [CrossRef]
- Dang, C.V. MYC on the Path to Cancer. Cell 2012, 149, 22–35. [Google Scholar] [CrossRef]
- Green, A.R.; Aleskandarany, M.A.; Agarwal, D.; Elsheikh, S.; Nolan, C.C.; Diez-Rodriguez, M.; Macmillan, R.D.; Ball, G.R.; Caldas, C.; Madhusudan, S.; et al. MYC functions are specific in biological subtypes of breast cancer and confers resistance to endocrine therapy in luminal tumours. Br. J. Cancer 2016, 114, 917–928. [Google Scholar] [CrossRef]
- Duffy, M.J.; O’Grady, S.; Tang, M.; Crown, J. MYC as a target for cancer treatment. Cancer Treat. Rev. 2021, 94, 102154. [Google Scholar] [CrossRef]
- Park, Y.R.; Jee, W. Viscum album Induces Apoptosis by Regulating STAT3 Signaling Pathway in Breast Cancer Cells. Int. J. Mol. Sci. 2023, 24, 11988. [Google Scholar] [CrossRef]










Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Park, Y.-R.; Jee, W.; Park, S.-M.; Kim, S.-W.; Jung, J.-H.; Kim, H.; Kim, K.-I.; Jang, H.-J. Acetylcorynoline Induces Apoptosis and G2/M Phase Arrest through the c-Myc Signaling Pathway in Colon Cancer Cells. Int. J. Mol. Sci. 2023, 24, 17589. https://doi.org/10.3390/ijms242417589
Park Y-R, Jee W, Park S-M, Kim S-W, Jung J-H, Kim H, Kim K-I, Jang H-J. Acetylcorynoline Induces Apoptosis and G2/M Phase Arrest through the c-Myc Signaling Pathway in Colon Cancer Cells. International Journal of Molecular Sciences. 2023; 24(24):17589. https://doi.org/10.3390/ijms242417589
Chicago/Turabian StylePark, Ye-Rin, Wona Jee, So-Mi Park, Seok-Woo Kim, Ji-Hoon Jung, Hyungsuk Kim, Kwan-Il Kim, and Hyeung-Jin Jang. 2023. "Acetylcorynoline Induces Apoptosis and G2/M Phase Arrest through the c-Myc Signaling Pathway in Colon Cancer Cells" International Journal of Molecular Sciences 24, no. 24: 17589. https://doi.org/10.3390/ijms242417589