
Dual-Specificity Phosphatase 6 Deficiency Attenuates Arterial-Injury-Induced Intimal Hyperplasia in Mice
 , , and
, , and 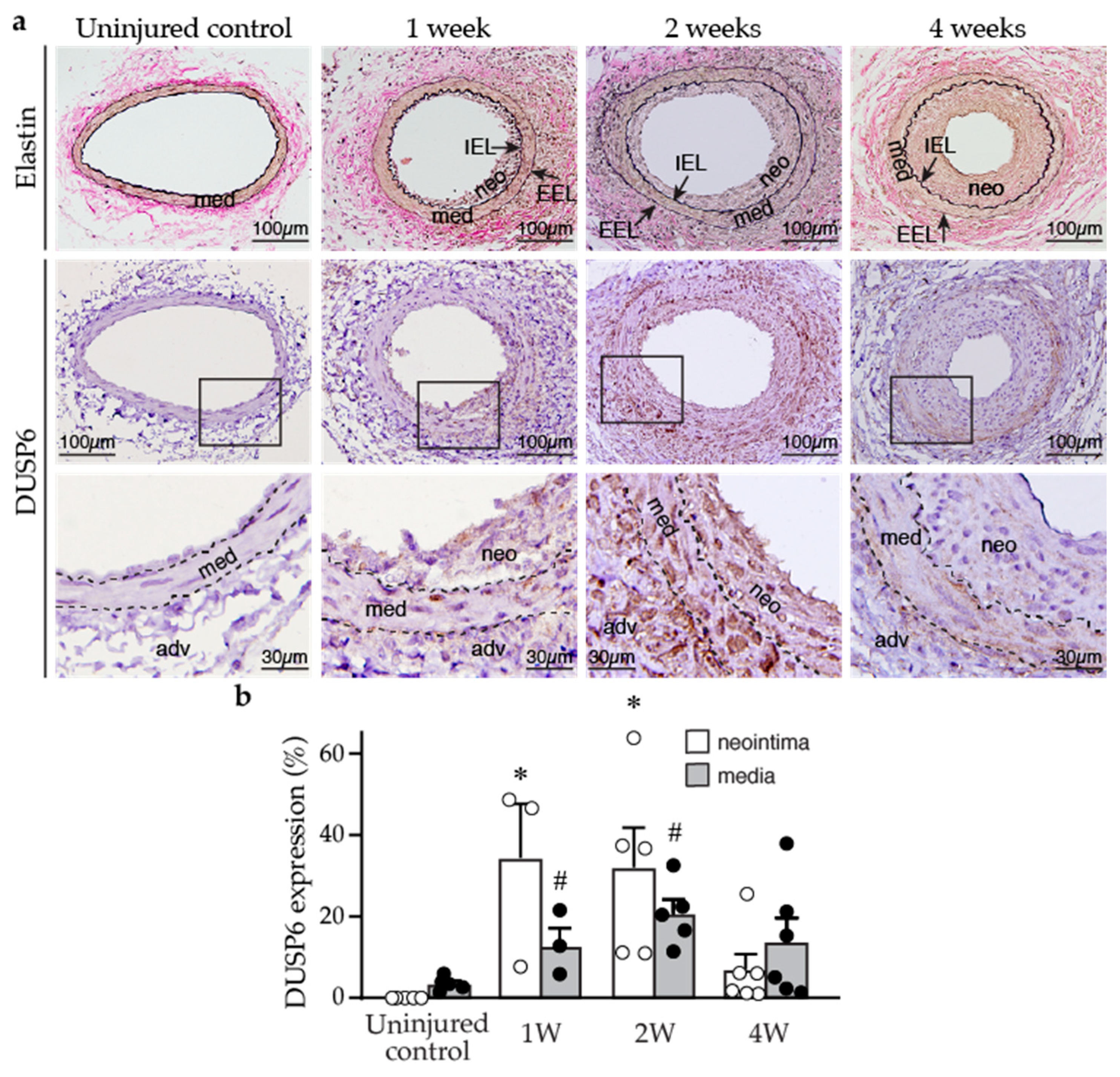{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
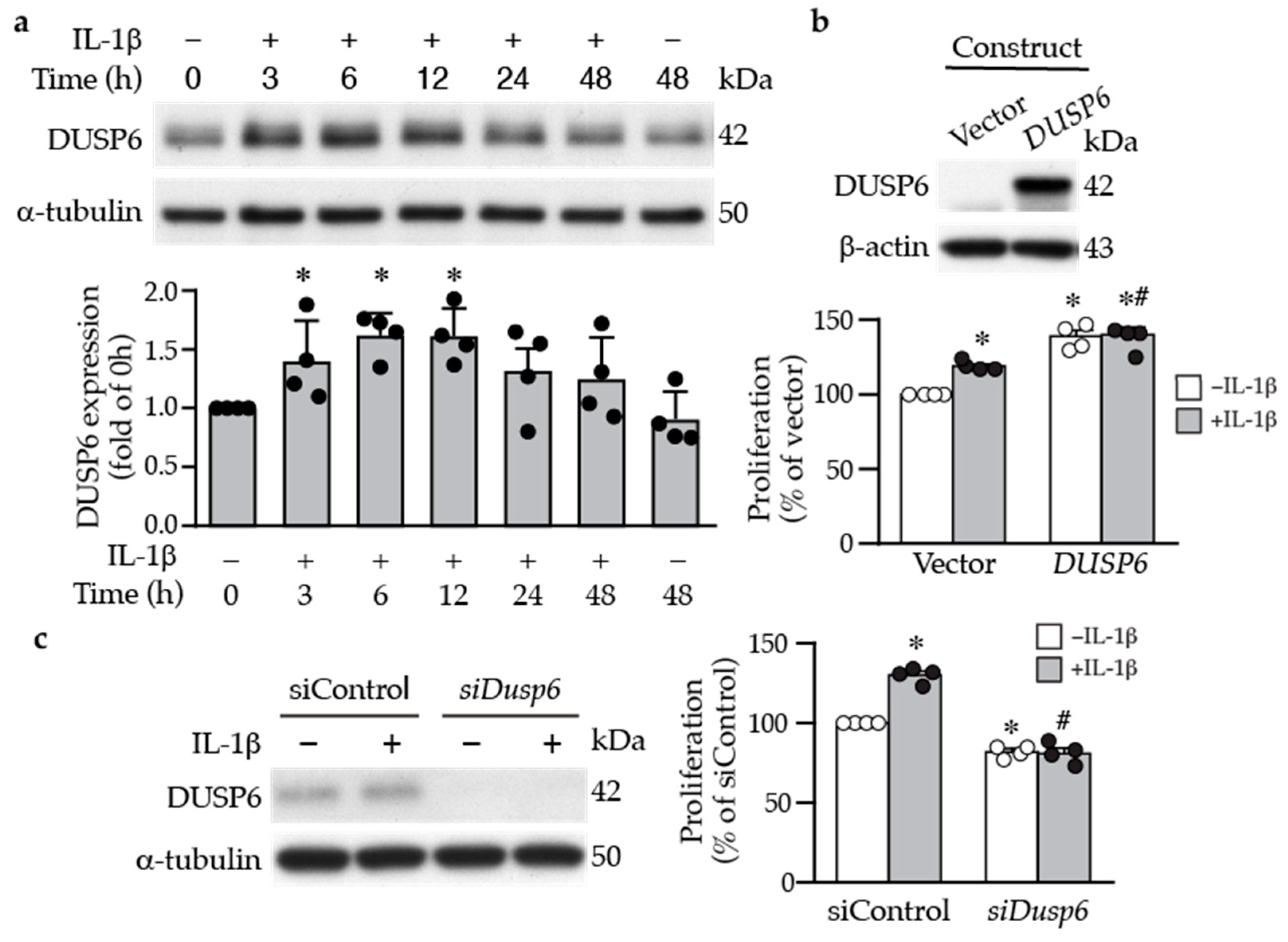
2.1. DUSP6 Is Increased in the Vessel Wall after Injury and Promotes VSMC Proliferation In Vitro
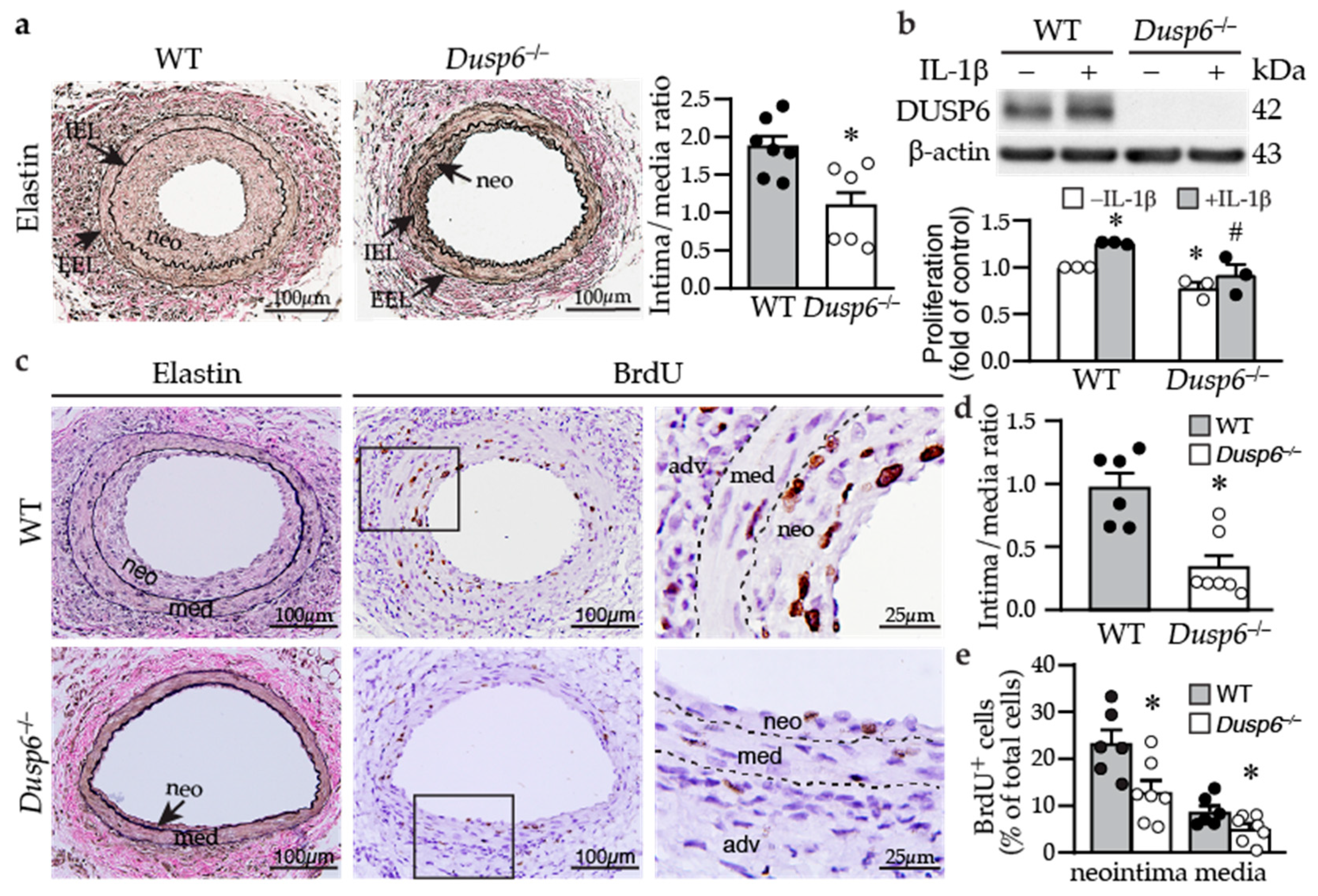
2.2. Dusp6 Deficiency Reduces Neointima Formation and Decreases VSMC Proliferation
2.3. ERK1/2 Activation Precedes DUSP6 Induction in VSMCs following IL-1β Stimulation
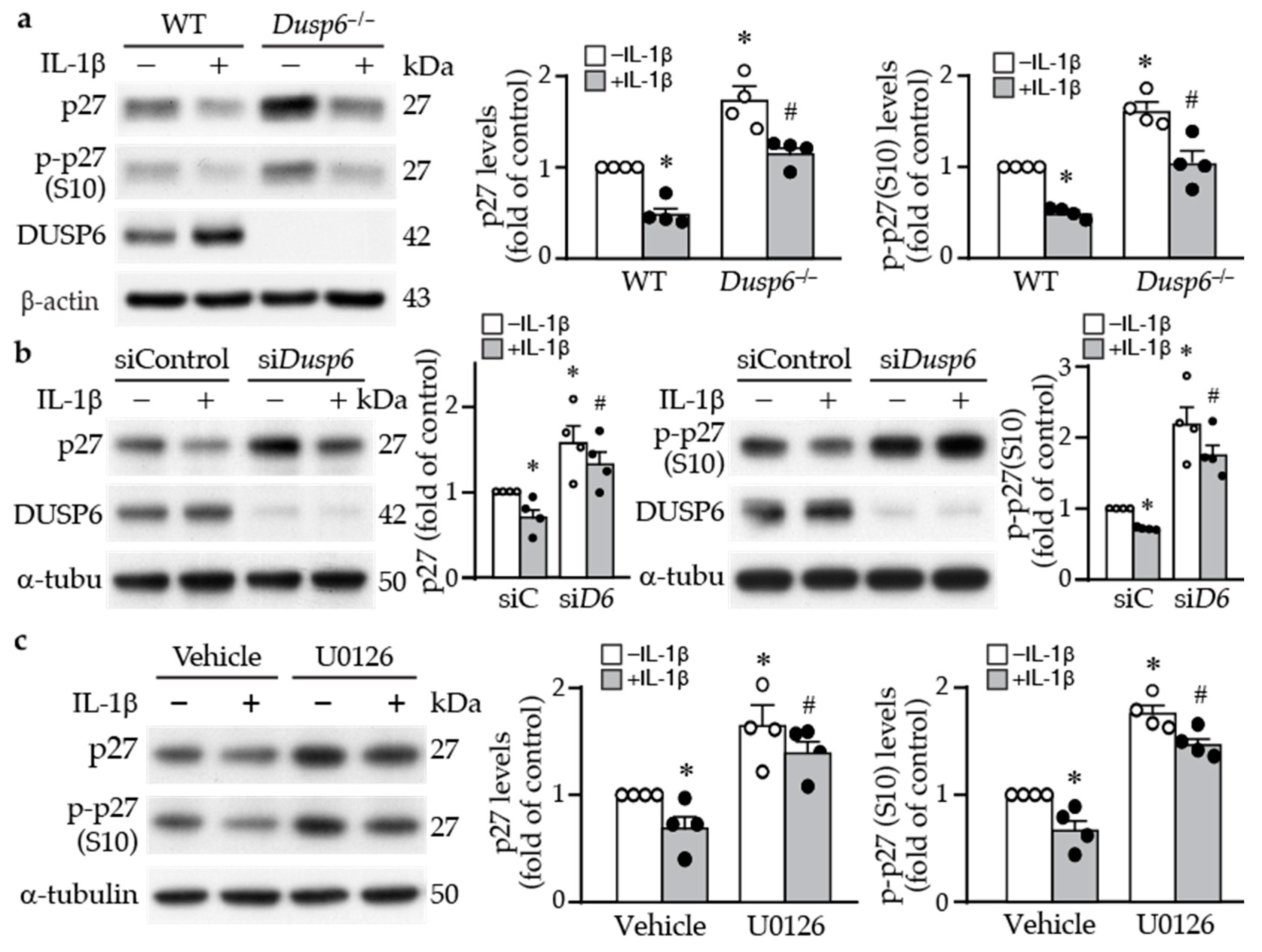
2.4. DUSP6 Regulates Cell Cycle Inhibitor p27 Levels
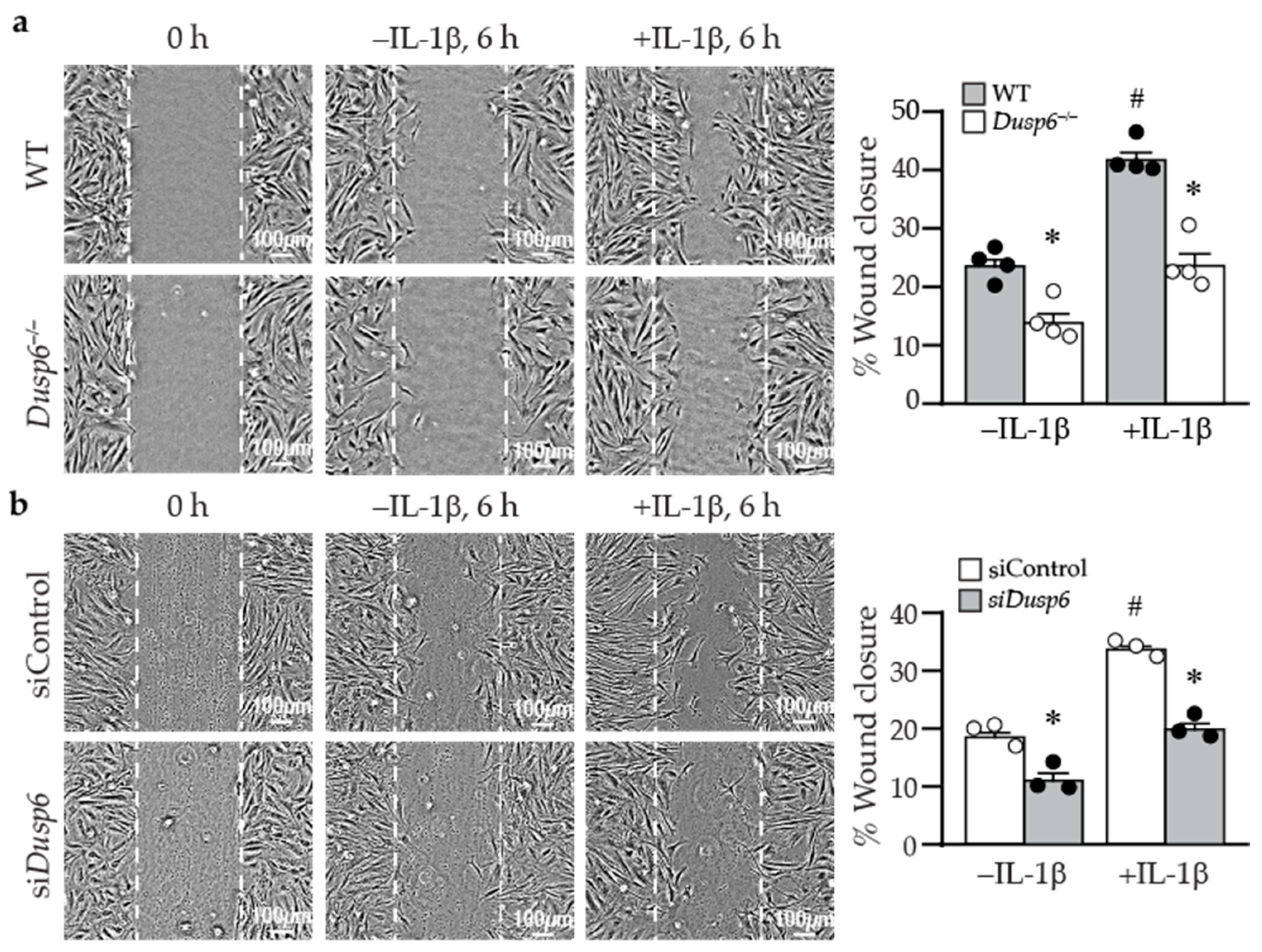
2.5. Lack of DUSP6 Reduces VSMC Migration
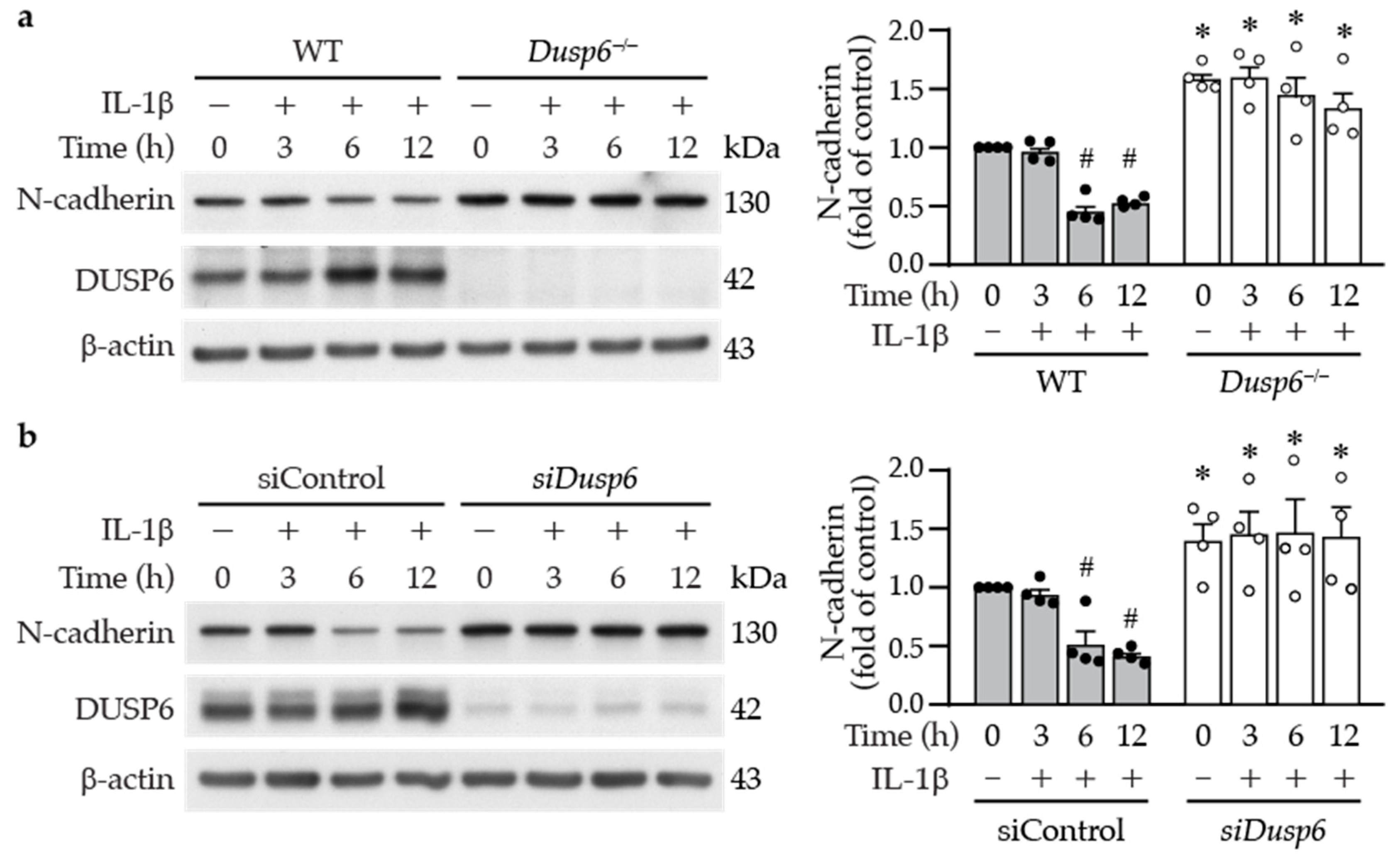
2.6. DUSP6 Deficiency Increases Cell–Cell Adhesion Molecule N-cadherin in VSMCs
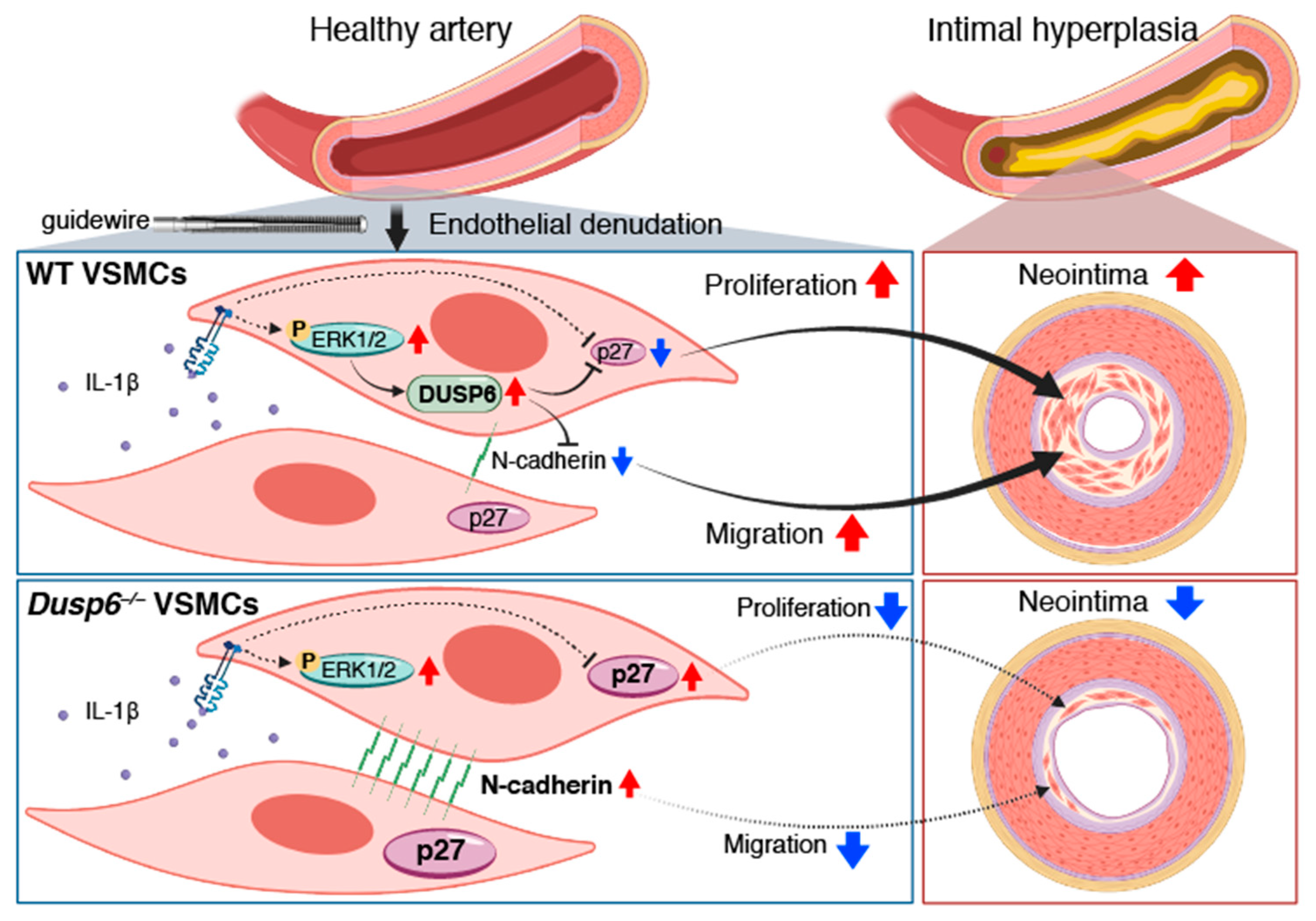
3. Discussion
4. Materials and Methods
4.1. Animal Care and Genotyping
4.2. Experimental Mouse Model of Femoral Artery Denudation Injury
4.3. Histological Analysis and Immunohistochemistry
4.4. Primary VSMC Culture, IL-1β Stimulation, and Proliferation Assays
4.5. Wound Healing Assays
4.6. Western Blot Analysis
4.7. DUSP6 Overexpression and Knockdown in VSMCs
4.8. ERK1/2 Signaling and Cell Cycle Inhibitor p27
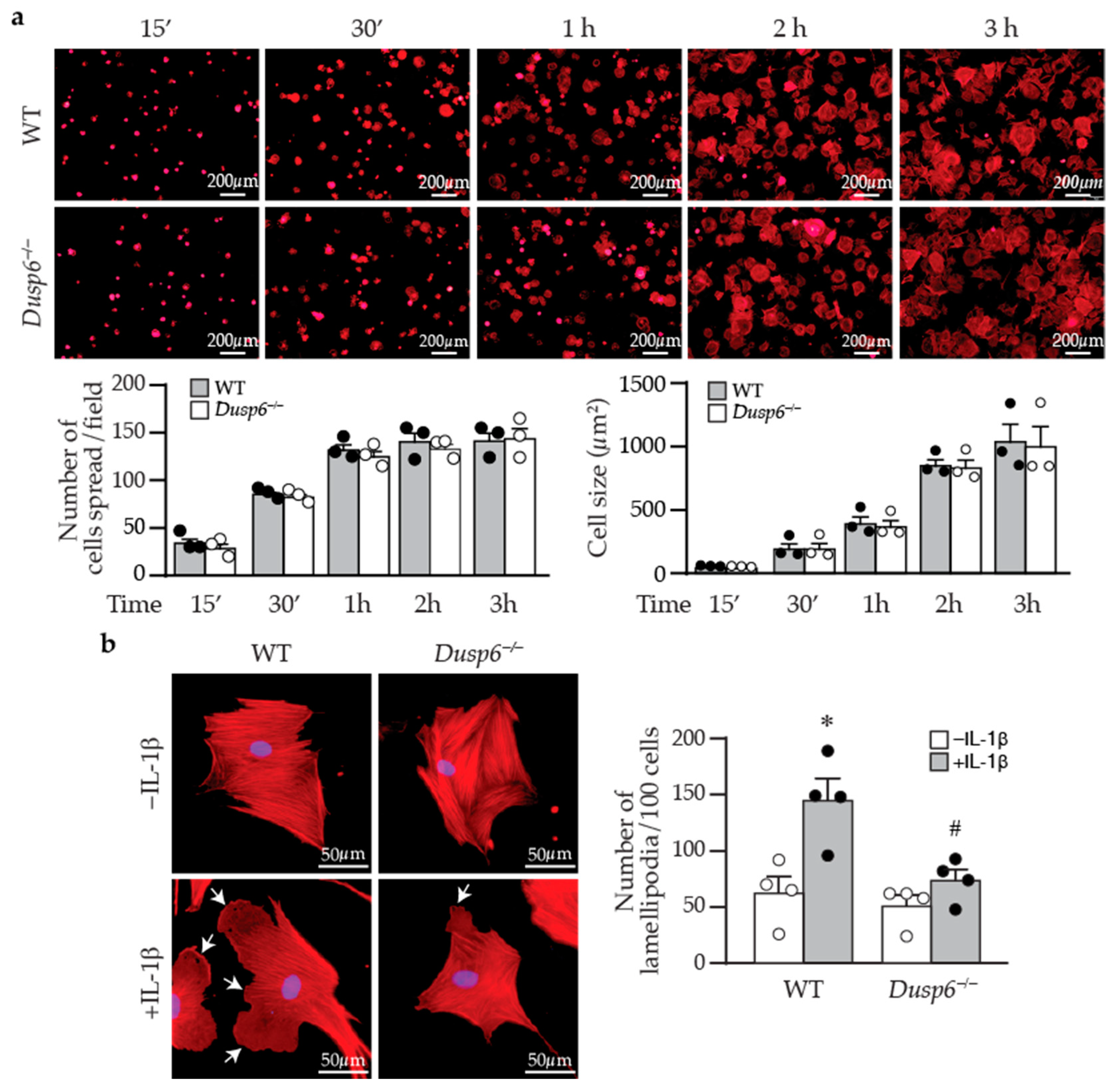
4.9. Cell Adhesion, Spreading, and Lamellipodia Formation
4.10. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Vaduganathan, M.; Mensah, G.A.; Turco, J.V.; Fuster, V.; Roth, G.A. The global burden of cardiovascular diseases and risk: A compass for future health. J. Am. Coll. Cardiol. 2022, 80, 2361–2371. [Google Scholar] [CrossRef] [PubMed]
- Tsao, C.W.; Aday, A.W.; Almarzooq, Z.I.; Anderson, C.A.M.; Arora, P.; Avery, C.L.; Baker-Smith, C.M.; Beaton, A.Z.; Boehme, A.K.; Buxton, A.E.; et al. Heart Disease and Stroke Statistics-2023 Update: A Report From the American Heart Association. Circulation 2023, 147, e93–e621. [Google Scholar] [CrossRef] [PubMed]
- Moussa, I.D.; Mohananey, D.; Saucedo, J.; Stone, G.W.; Yeh, R.W.; Kennedy, K.F.; Waksman, R.; Teirstein, P.; Moses, J.W.; Simonton, C. Trends and outcomes of restenosis after coronary stent implantation in the United States. J. Am. Coll. Cardiol. 2020, 76, 1521–1531. [Google Scholar] [CrossRef] [PubMed]
- Giustino, G.; Colombo, A.; Camaj, A.; Yasumura, K.; Mehran, R.; Stone, G.W.; Kini, A.; Sharma, S.K. Coronary In-Stent Restenosis: JACC State-of-the-Art Review. J. Am. Coll. Cardiol. 2022, 80, 348–372. [Google Scholar] [CrossRef] [PubMed]
- Shi, J.; Yang, Y.; Cheng, A.; Xu, G.; He, F. Metabolism of vascular smooth muscle cells in vascular diseases. Am. J. Physiol. Heart Circ. Physiol. 2020, 319, H613–H631. [Google Scholar] [CrossRef] [PubMed]
- Cao, G.; Xuan, X.; Hu, J.; Zhang, R.; Jin, H.; Dong, H. How vascular smooth muscle cell phenotype switching contributes to vascular disease. Cell Commun. Signal. 2022, 20, 180. [Google Scholar] [CrossRef] [PubMed]
- Tang, H.Y.; Chen, A.Q.; Zhang, H.; Gao, X.F.; Kong, X.Q.; Zhang, J.J. Vascular smooth muscle cells phenotypic switching in cardiovascular diseases. Cells 2022, 11, 4060. [Google Scholar] [CrossRef] [PubMed]
- Déglise, S.; Bechelli, C.; Allagnat, F. Vascular smooth muscle cells in intimal hyperplasia, an update. Front. Physiol. 2023, 13, 1081881. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Liu, H.T. MAPK signal pathways in the regulation of cell proliferation in mammalian cells. Cell Res. 2002, 12, 9–18. [Google Scholar] [CrossRef] [PubMed]
- Pearson, G.; Robinson, F.; Beers Gibson, T.; Xu, B.E.; Karandikar, M.; Berman, K.; Cobb, M.H. Mitogen-activated protein (MAP) kinase pathways: Regulation and physiological functions. Endocr. Rev. 2001, 22, 153–183. [Google Scholar]
- Samson, S.C.; Khan, A.M.; Mendoza, M.C. ERK signaling for cell migration and invasion. Front. Mol. Biosci. 2022, 9, 998475. [Google Scholar] [CrossRef] [PubMed]
- Purcell, N.H.; Wilkins, B.J.; York, A.; Saba-El-Leil, M.K.; Meloche, S.; Robbins, J.; Molkentin, J.D. Genetic inhibition of cardiac ERK1/2 promotes stress-induced apoptosis and heart failure but has no effect on hypertrophy in vivo. Proc. Natl. Acad. Sci. USA 2007, 104, 14074–14079. [Google Scholar] [CrossRef] [PubMed]
- Xie, X.L.; Nie, X.; Wu, J.; Zhang, F.; Zhao, L.L.; Lin, Y.L.; Yin, Y.J.; Liu, H.; Shu, Y.N.; Miao, S.B.; et al. Smooth muscle 22alpha facilitates angiotensin II-induced signaling and vascular contraction. J. Mol. Med. 2015, 93, 547–558. [Google Scholar] [CrossRef] [PubMed]
- Yoshizumi, M.; Kyotani, Y.; Zhao, J.; Nakahira, K. Targeting the mitogen-activated protein kinase-mediated vascular smooth muscle cell remodeling by angiotensin II. Ann. Transl. Med. 2020, 8, 157. [Google Scholar] [CrossRef] [PubMed]
- Eun, S.Y.; Ko, Y.S.; Park, S.W.; Chang, K.C.; Kim, H.J. IL-1β enhances vascular smooth muscle cell proliferation and migration via P2Y2 receptor-mediated RAGE expression and HMGB1 release. Vasc. Pharmacol. 2015, 72, 108–117. [Google Scholar] [CrossRef] [PubMed]
- Dong, L.H.; Wen, J.K.; Liu, G.; McNutt, M.A.; Miao, S.B.; Gao, R.; Zheng, B.; Zhang, H.; Han, M. Blockade of the Ras-extracellular signal-regulated kinase 1/2 pathway is involved in smooth muscle 22α-mediated suppression of vascular smooth muscle cell proliferation and neointima hyperplasia. Arterioscler. Thromb. Vasc. Biol. 2010, 30, 683–691. [Google Scholar] [CrossRef] [PubMed]
- Keyse, S.M. Protein phosphatases and the regulation of mitogen-activated protein kinase signalling. Curr. Opin. Cell Biol. 2000, 12, 186–192. [Google Scholar] [CrossRef] [PubMed]
- Patterson, K.I.; Brummer, T.; O’Brien, P.M.; Daly, R.J. Dual-specificity phosphatases: Critical regulators with diverse cellular targets. Biochem. J. 2009, 418, 475–489. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.S.; Asmis, R. Mitogen-activated protein kinase phosphatase 1 (MKP-1) in macrophage biology and cardiovascular disease. A redox-regulated master controller of monocyte function and macrophage phenotype. Free Radic. Biol. Med. 2017, 109, 75–83. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Sun, J.P.; Zhou, B.; Zhang, Z.Y. Structural basis of docking interactions between ERK2 and MAP kinase phosphatase 3. Proc. Natl. Acad. Sci. USA 2006, 103, 5326–5331. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, M.K.; Abdollah, N.A.; Shafie, N.H.; Yusof, N.M.; Razak, S.R.A. Dual-specificity phosphatase 6 (DUSP6): A review of its molecular characteristics and clinical relevance in cancer. Cancer Biol. Med. 2018, 15, 14–28. [Google Scholar] [PubMed]
- Ruan, J.W.; Statt, S.; Huang, C.T.; Tsai, Y.T.; Kuo, C.C.; Chan, H.L.; Liao, Y.C.; Tan, T.H.; Kao, C.Y. Dual-specificity phosphatase 6 deficiency regulates gut microbiome and transcriptome response against diet-induced obesity in mice. Nat. Microbiol. 2016, 2, 16220. [Google Scholar] [CrossRef] [PubMed]
- Beaudry, K.; Langlois, M.J.; Montagne, A.; Cagnol, S.; Carrier, J.C.; Rivard, N. Dual-specificity phosphatase 6 deletion protects the colonic epithelium against inflammation and promotes both proliferation and tumorigenesis. J. Cell. Physiol. 2019, 234, 6731–6745. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.S.; Liao, Y.C.; Huang, C.T.; Lin, C.M.; Cheung, C.H.Y.; Ruan, J.W.; Yu, W.H.; Tsai, Y.T.; Lin, I.J.; Huang, C.H.; et al. Identification of a gut microbiota member that ameliorates DSS-induced colitis in intestinal barrier enhanced Dusp6-deficient mice. Cell Rep. 2021, 37, 110016. [Google Scholar] [CrossRef] [PubMed]
- Reis, E.D.; Roqué, M.; Cordon-Cardo, C.; Drobnjak, M.; Fuster, V.; Badimon, J.J. Apoptosis, proliferation, and p27 expression during vessel wall healing: Time course study in a mouse model of transluminal femoral artery injury. J. Vasc. Surg. 2000, 32, 1022–1029. [Google Scholar] [CrossRef] [PubMed]
- Sprague, A.H.; Khalil, R.A. Inflammatory cytokines in vascular dysfunction and vascular disease. Biochem. Pharmacol. 2009, 78, 539–552. [Google Scholar] [CrossRef] [PubMed]
- Ishida, N.; Kitagawa, M.; Hatakeyama, S.; Nakayama, K. Phosphorylation at serine 10, a major phosphorylation site of p27(Kip1), increases its protein stability. J. Biol. Chem. 2000, 275, 25146–25154. [Google Scholar] [CrossRef]
- Ilic, D.; Furuta, Y.; Kanazawa, S.; Takeda, N.; Sobue, K.; Nakatsuji, N.; Nomura, S.; Fujimoto, J.; Okada, M.; Yamamoto, T. Reduced cell motility and enhanced focal adhesion contact formation in cells from FAK-deficient mice. Nature 1995, 377, 539–544. [Google Scholar]
- Sun, Z.; Parrish, A.R.; Hill, M.A.; Meininger, G.A. N-cadherin, a vascular smooth muscle cell-cell adhesion molecule: Function and signaling for vasomotor control. Microcirculation 2014, 21, 208–218. [Google Scholar] [CrossRef]
- Jones, M.; Sabatini, P.J.; Lee, F.S.; Bendeck, M.P.; Langille, B.L. N-cadherin upregulation and function in response of smooth muscle cells to arterial injury. Arterioscler. Thromb. Vasc. Biol. 2002, 22, 1972–1977. [Google Scholar] [CrossRef]
- Lyon, C.; Mill, C.; Tsaousi, A.; Williams, H.; George, S. Regulation of VSMC behavior by the cadherin-catenin complex. Front. Biosci. (Landmark Ed) 2011, 16, 644–657. [Google Scholar] [CrossRef] [PubMed]
- Skopelja-Gardner, S.; Saha, M.; Alvarado-Vazquez, P.A.; Liponis, B.S.; Martinez, E.; Romero-Sandoval, E.A. Mitogen-activated protein kinase phosphatase-3 (MKP-3) in the surgical wound is necessary for the resolution of postoperative pain in mice. J. Pain Res. 2017, 10, 763–774. [Google Scholar] [CrossRef] [PubMed]
- Lake, D.; Correa, S.A. Negative feedback regulation of the ERK1/2 MAPK pathway. Cell. Mol. Life Sci. 2016, 73, 4397–4413. [Google Scholar] [CrossRef] [PubMed]
- Maillet, M.; Purcell, N.H.; Sargent, M.A.; York, A.J.; Bueno, O.F.; Molkentin, J.D. DUSP6 (MKP3) null mice show enhanced ERK1/2 phosphorylation at baseline and increased myocyte proliferation in the heart affecting disease susceptibility. J. Biol. Chem. 2008, 283, 31246–31255. [Google Scholar] [CrossRef] [PubMed]
- Ekerot, M.; Stavridis, M.P.; Delavaine, L.; Mitchell, M.P.; Staples, C.; Owens, D.M.; Keenan, I.D.; Dickinson, R.J.; Storey, K.G.; Keyse, S.M. Negative-feedback regulation of FGF signalling by DUSP6/MKP-3 is driven by ERK1/2 and mediated by Ets factor binding to a conserved site within the DUSP6/MKP-3 gene promoter. Biochem. J. 2008, 412, 287–298. [Google Scholar] [CrossRef] [PubMed]
- Caunt, C.J.; Armstrong, S.P.; Rivers, C.A.; Norman, M.R.; McArdle, C.A. Spatiotemporal regulation of ERK2 by dual specificity phosphatases. J. Biol. Chem. 2008, 283, 26612–26623. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.F.; Chuang, H.C.; Tan, T.H. Regulation of dual-specificity phosphatase (DUSP) ubiquitination and protein stability. Int. J. Mol. Sci. 2019, 20, 2668. [Google Scholar] [CrossRef] [PubMed]
- Hsu, S.F.; Lee, Y.B.; Lee, Y.C.; Chung, A.L.; Apaya, M.K.; Shyur, L.F.; Cheng, C.F.; Ho, F.M.; Meng, T.C. Dual specificity phosphatase DUSP6 promotes endothelial inflammation through inducible expression of ICAM-1. FEBS J. 2018, 285, 1593–1610. [Google Scholar] [CrossRef] [PubMed]
- Besson, A.; Dowdy, S.F.; Roberts, J.M. CDK inhibitors: Cell cycle regulators and beyond. Dev. Cell 2008, 14, 159–169. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.; Krasinski, K.; Sylvester, A.; Chen, J.; Nisen, P.D.; Andrés, V. Downregulation of cyclin-dependent kinase 2 activity and cyclin A promoter activity in vascular smooth muscle cells by p27(KIP1), an inhibitor of neointima formation in the rat carotid artery. J. Clin. Investig. 1997, 99, 2334–2341. [Google Scholar] [CrossRef] [PubMed]
- Fasciano, S.; Patel, R.C.; Handy, I.; Patel, C.V. Regulation of vascular smooth muscle proliferation by heparin: Inhibition of cyclin-dependent kinase 2 activity by p27(kip1). J. Biol. Chem. 2005, 280, 15682–15689. [Google Scholar] [CrossRef]
- Park, K.W.; Kim, D.H.; You, H.J.; Sir, J.J.; Jeon, S.I.; Youn, S.W.; Yang, H.M.; Skurk, C.; Park, Y.B.; Walsh, K.; et al. Activated forkhead transcription factor inhibits neointimal hyperplasia after angioplasty through induction of p27. Arterioscler. Thromb. Vasc. Biol. 2005, 25, 742–747. [Google Scholar] [CrossRef]
- Dong, L.H.; Wen, J.K.; Miao, S.B.; Jia, Z.; Hu, H.J.; Sun, R.H.; Wu, Y.; Han, M. Baicalin inhibits PDGF-BB-stimulated vascular smooth muscle cell proliferation through suppressing PDGFRβ-ERK signaling and increase in p27 accumulation and prevents injury-induced neointimal hyperplasia. Cell Res. 2010, 20, 1252–1262. [Google Scholar] [CrossRef]
- Blindt, R.; Bosserhoff, A.K.; Dammers, J.; Krott, N.; Demircan, L.; Hoffmann, R.; Hanrath, P.; Weber, C.; Vogt, F. Downregulation of N-cadherin in the neointima stimulates migration of smooth muscle cells by RhoA deactivation. Cardiovasc. Res. 2004, 62, 212–222. [Google Scholar] [CrossRef] [PubMed]
- Sabatini, P.J.; Zhang, M.; Silverman-Gavrila, R.; Bendeck, M.P.; Langille, B.L. Homotypic and endothelial cell adhesions via N-cadherin determine polarity and regulate migration of vascular smooth muscle cells. Circ. Res. 2008, 103, 405–412. [Google Scholar] [CrossRef] [PubMed]
- Lyon, C.A.; Koutsouki, E.; Aguilera, C.M.; Blaschuk, O.W.; George, S.J. Inhibition of N-cadherin retards smooth muscle cell migration and intimal thickening via induction of apoptosis. J. Vasc. Surg. 2010, 52, 1301–1309. [Google Scholar] [CrossRef] [PubMed]
- Roque, M.; Fallon, J.T.; Badimon, J.J.; Zhang, W.X.; Taubman, M.B.; Reis, E.D. Mouse model of femoral artery denudation injury associated with the rapid accumulation of adhesion molecules on the luminal surface and recruitment of neutrophils. Arterioscler. Thromb. Vasc. Biol. 2000, 20, 335–342. [Google Scholar] [CrossRef] [PubMed]
- Ho, Y.C.; Wu, M.L.; Su, C.H.; Chen, C.H.; Ho, H.H.; Lee, G.L.; Lin, W.S.; Lin, W.Y.; Hsu, Y.J.; Kuo, C.C.; et al. A novel protective function of 5-methoxytryptophan in vascular injury. Sci. Rep. 2016, 6, 25374. [Google Scholar] [CrossRef]
- Chen, C.H.; Ho, Y.C.; Ho, H.H.; Chang, I.C.; Kirsch, K.H.; Chuang, Y.J.; Layne, M.D.; Yet, S.F. Cysteine-rich protein 2 alters p130Cas localization and inhibits vascular smooth muscle cell migration. Cardiovasc. Res. 2013, 100, 461–471. [Google Scholar] [CrossRef]
- Lin, D.W.; Chang, I.C.; Tseng, A.; Wu, M.L.; Chen, C.H.; Patenaude, C.A.; Layne, M.D.; Yet, S.F. Transforming growth factor β up-regulates cysteine-rich protein 2 in vascular smooth muscle cells via activating transcription factor 2. J. Biol. Chem. 2008, 283, 15003–15014. [Google Scholar] [CrossRef]









Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hamdin, C.D.; Wu, M.-L.; Chen, C.-M.; Ho, Y.-C.; Jiang, W.-C.; Gung, P.-Y.; Ho, H.-H.; Chuang, H.-C.; Tan, T.-H.; Yet, S.-F. Dual-Specificity Phosphatase 6 Deficiency Attenuates Arterial-Injury-Induced Intimal Hyperplasia in Mice. Int. J. Mol. Sci. 2023, 24, 17136. https://doi.org/10.3390/ijms242417136
Hamdin CD, Wu M-L, Chen C-M, Ho Y-C, Jiang W-C, Gung P-Y, Ho H-H, Chuang H-C, Tan T-H, Yet S-F. Dual-Specificity Phosphatase 6 Deficiency Attenuates Arterial-Injury-Induced Intimal Hyperplasia in Mice. International Journal of Molecular Sciences. 2023; 24(24):17136. https://doi.org/10.3390/ijms242417136
Chicago/Turabian StyleHamdin, Candra D., Meng-Ling Wu, Chen-Mei Chen, Yen-Chun Ho, Wei-Cheng Jiang, Pei-Yu Gung, Hua-Hui Ho, Huai-Chia Chuang, Tse-Hua Tan, and Shaw-Fang Yet. 2023. "Dual-Specificity Phosphatase 6 Deficiency Attenuates Arterial-Injury-Induced Intimal Hyperplasia in Mice" International Journal of Molecular Sciences 24, no. 24: 17136. https://doi.org/10.3390/ijms242417136