
Conformational Analysis of 1,5-Diaryl-3-Oxo-1,4-Pentadiene Derivatives: A Nuclear Overhauser Effect Spectroscopy Investigation





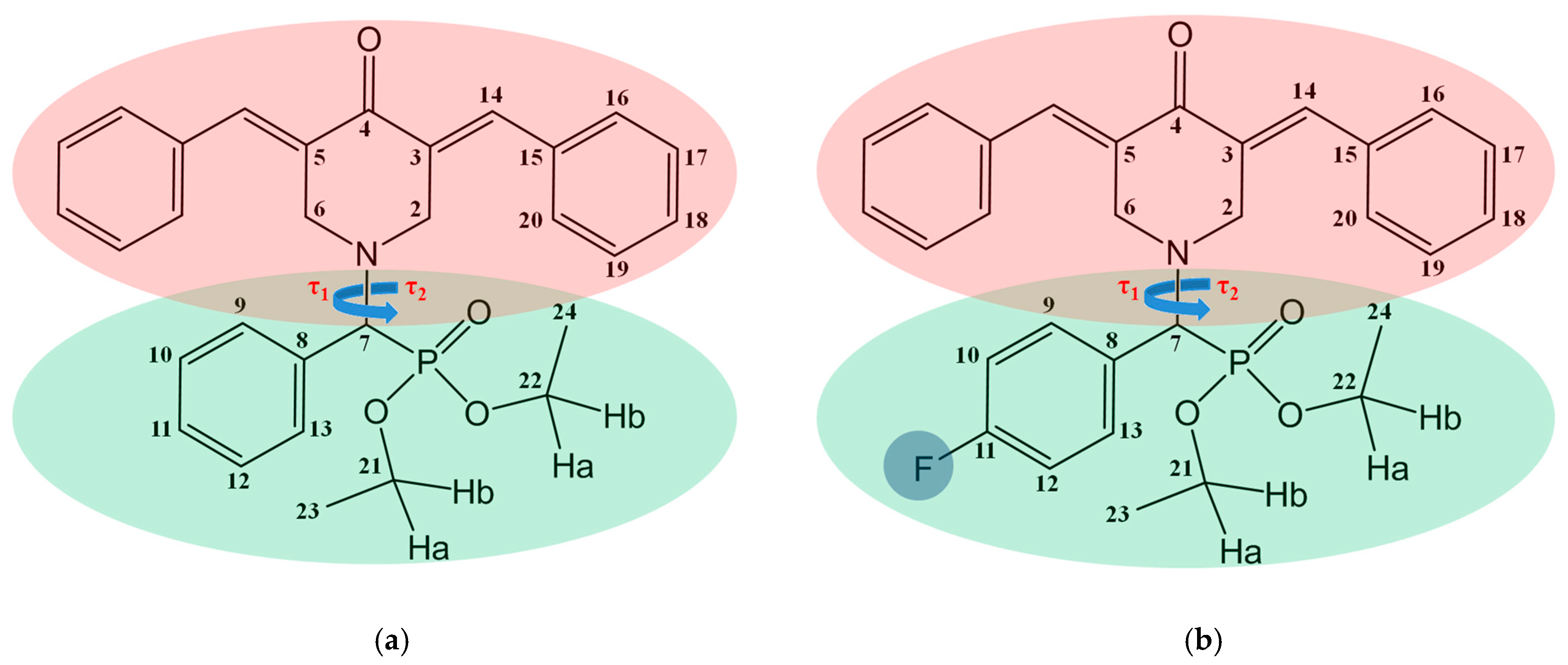{kind=link}
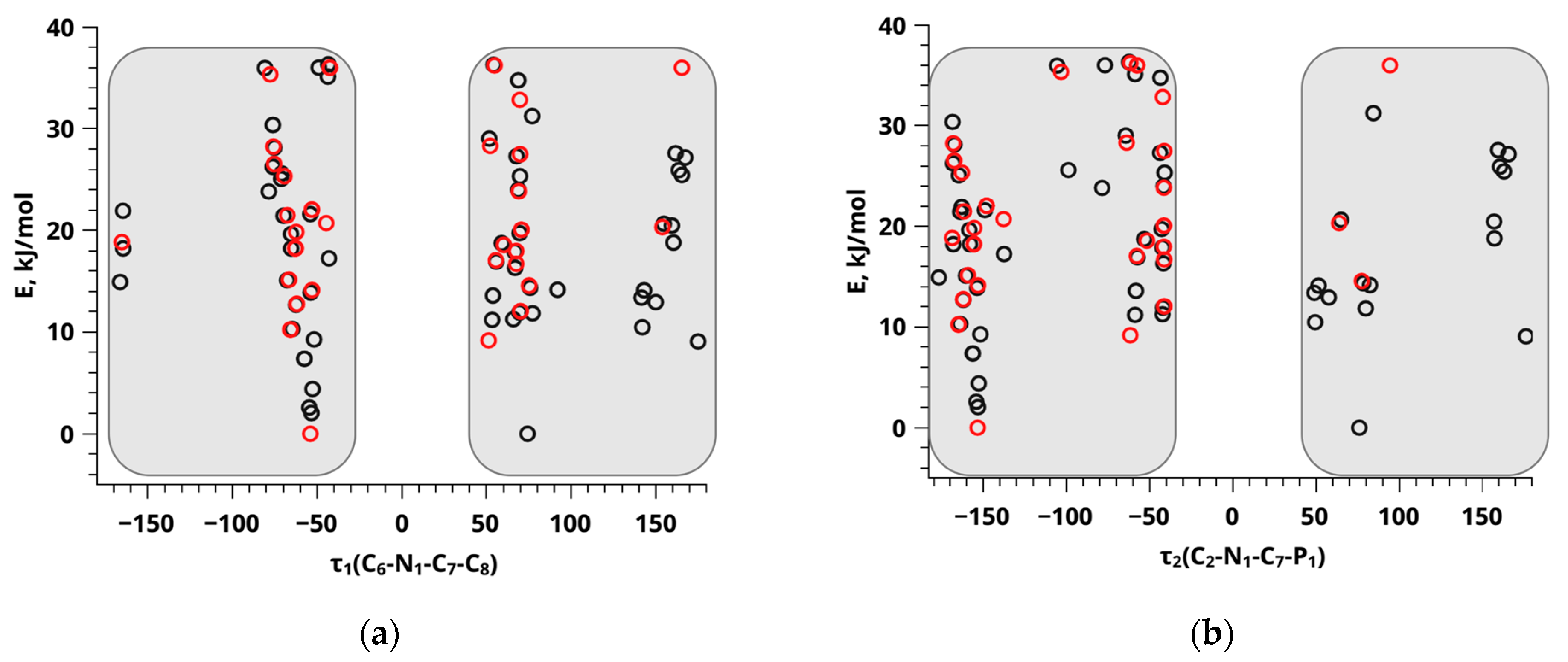{kind=link}
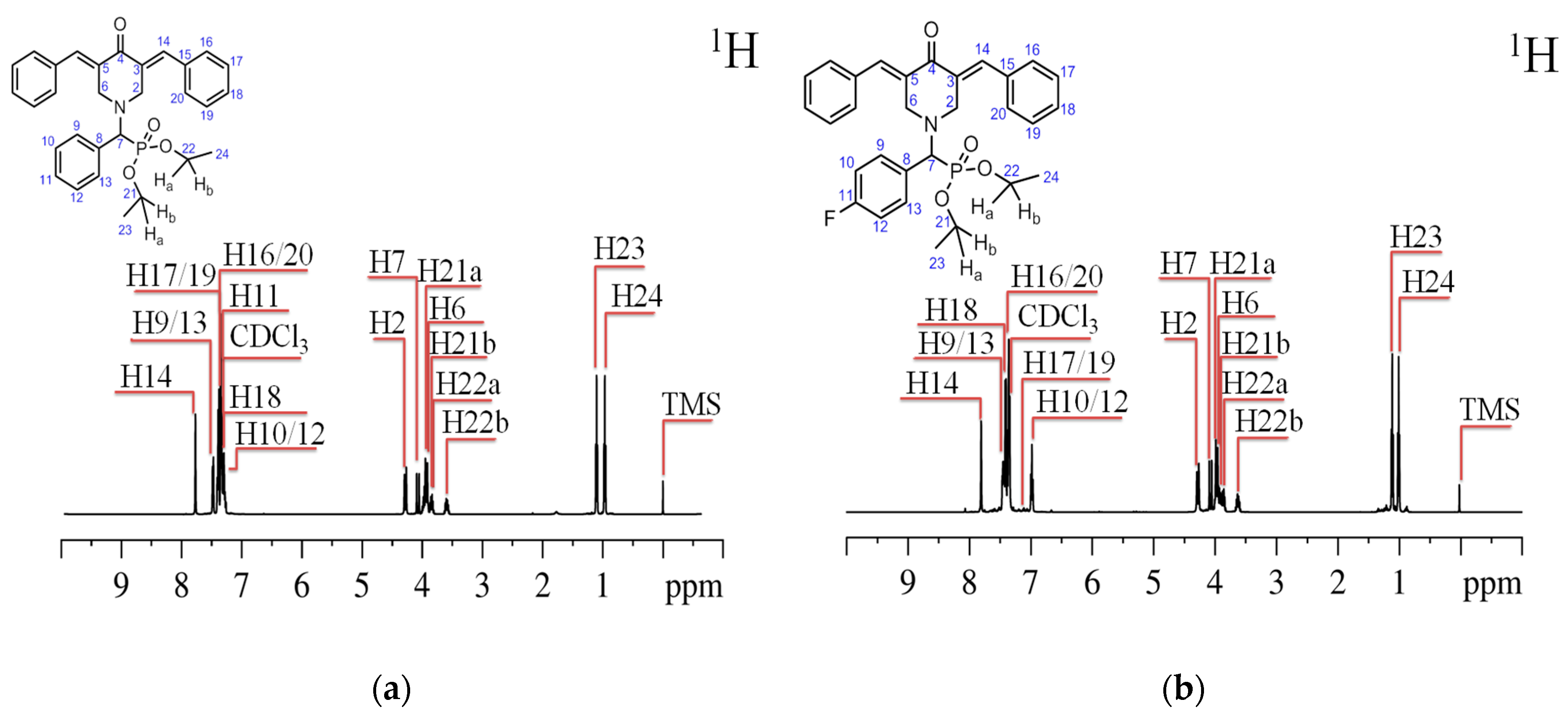{kind=link}
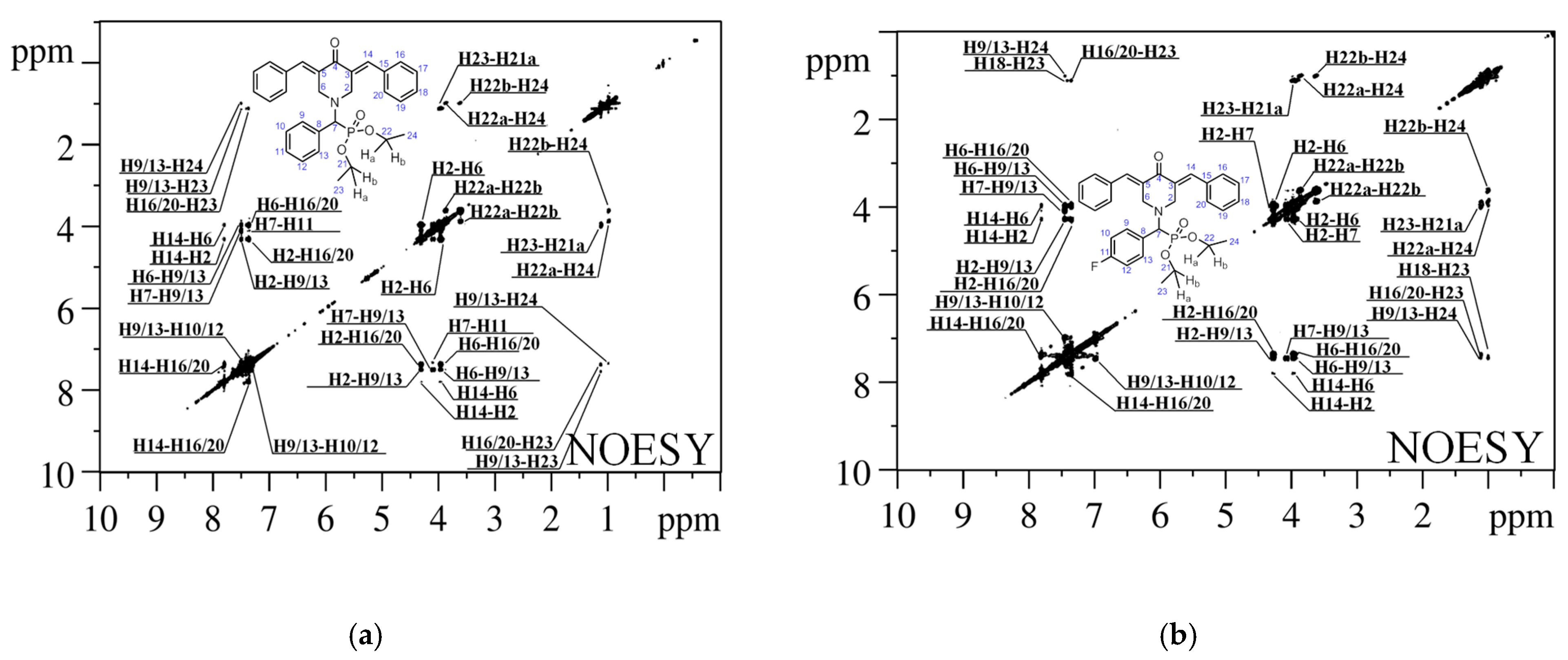{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
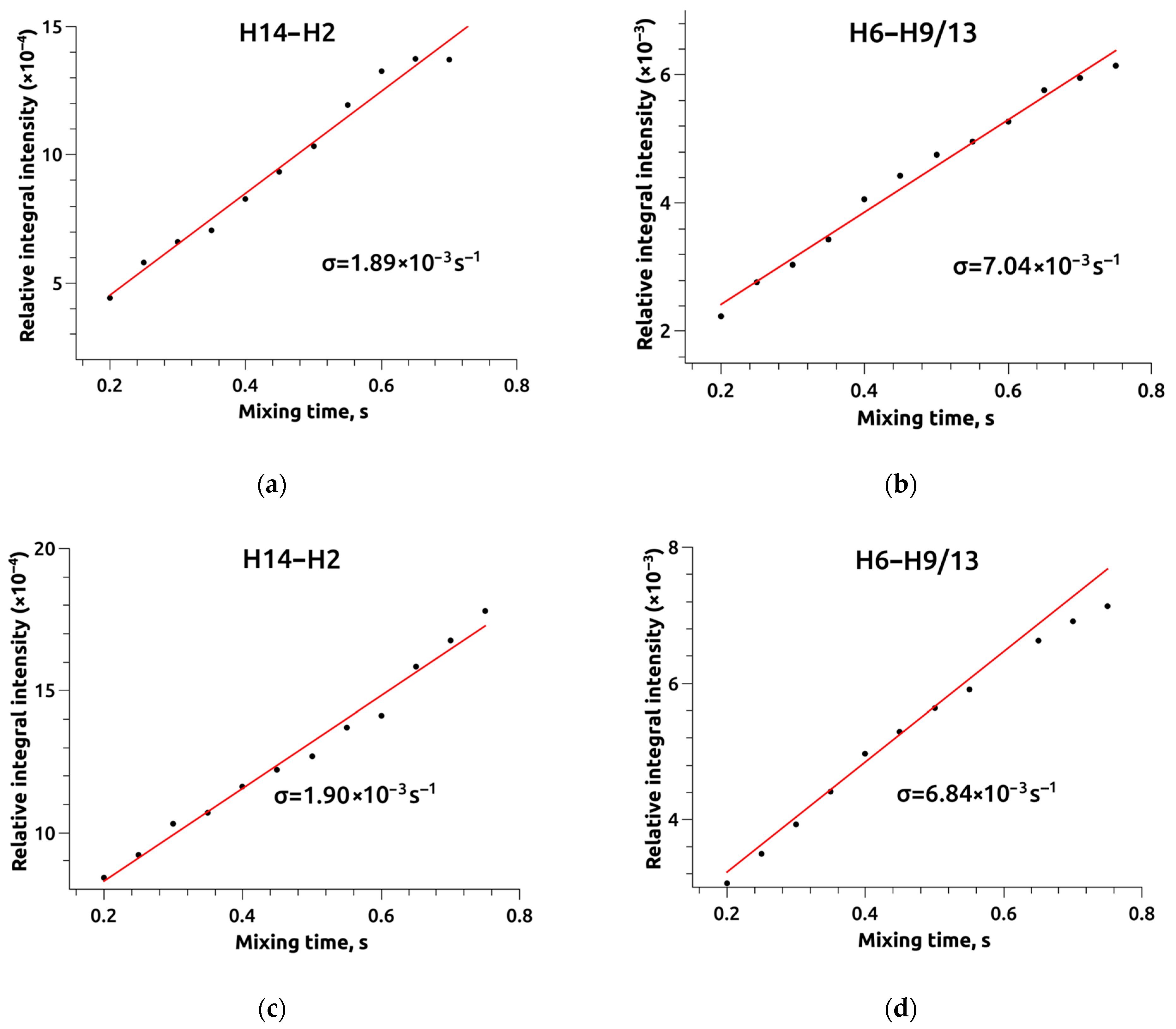{kind=link}
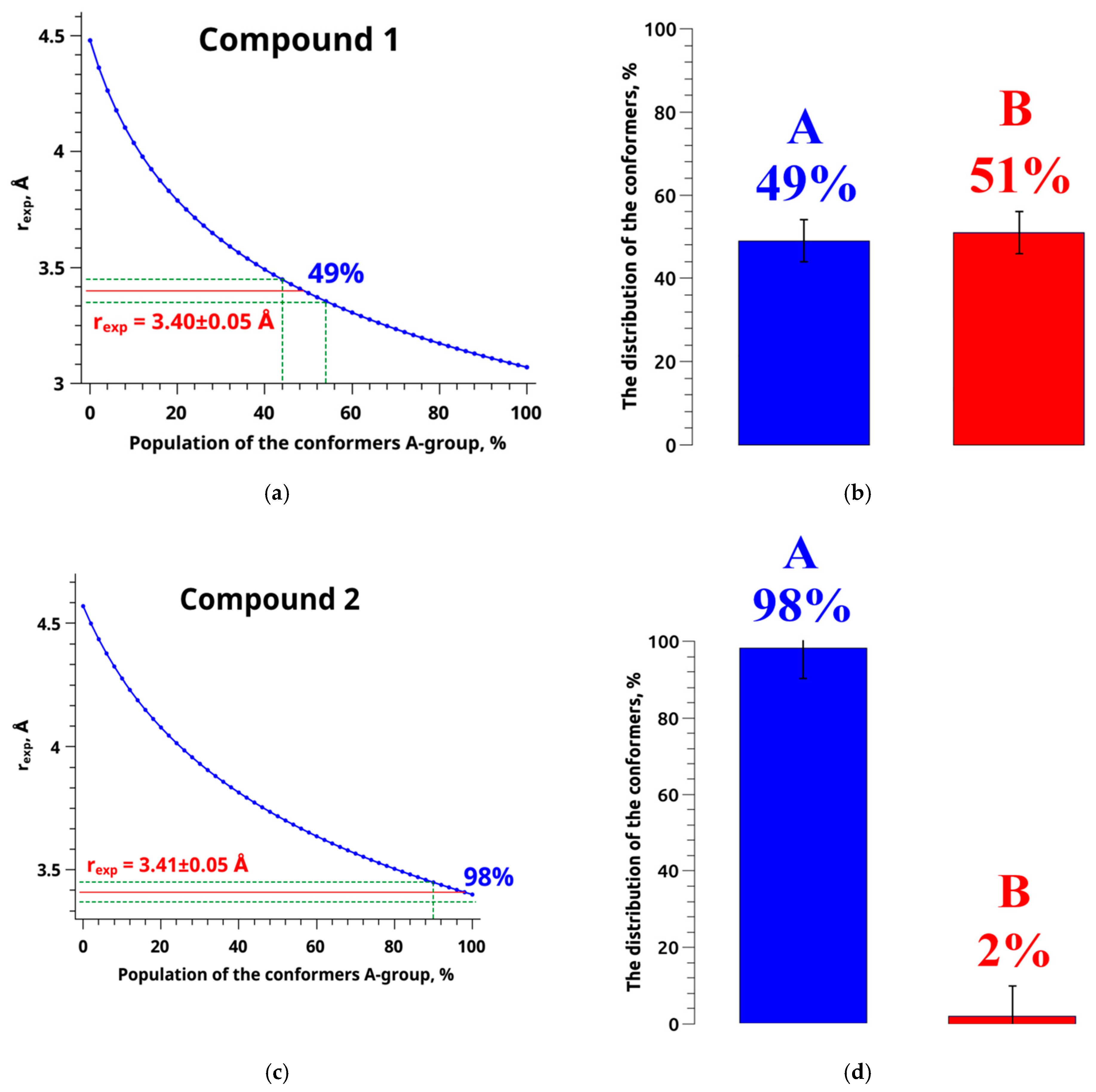{kind=link}
Abstract
:1. Introduction
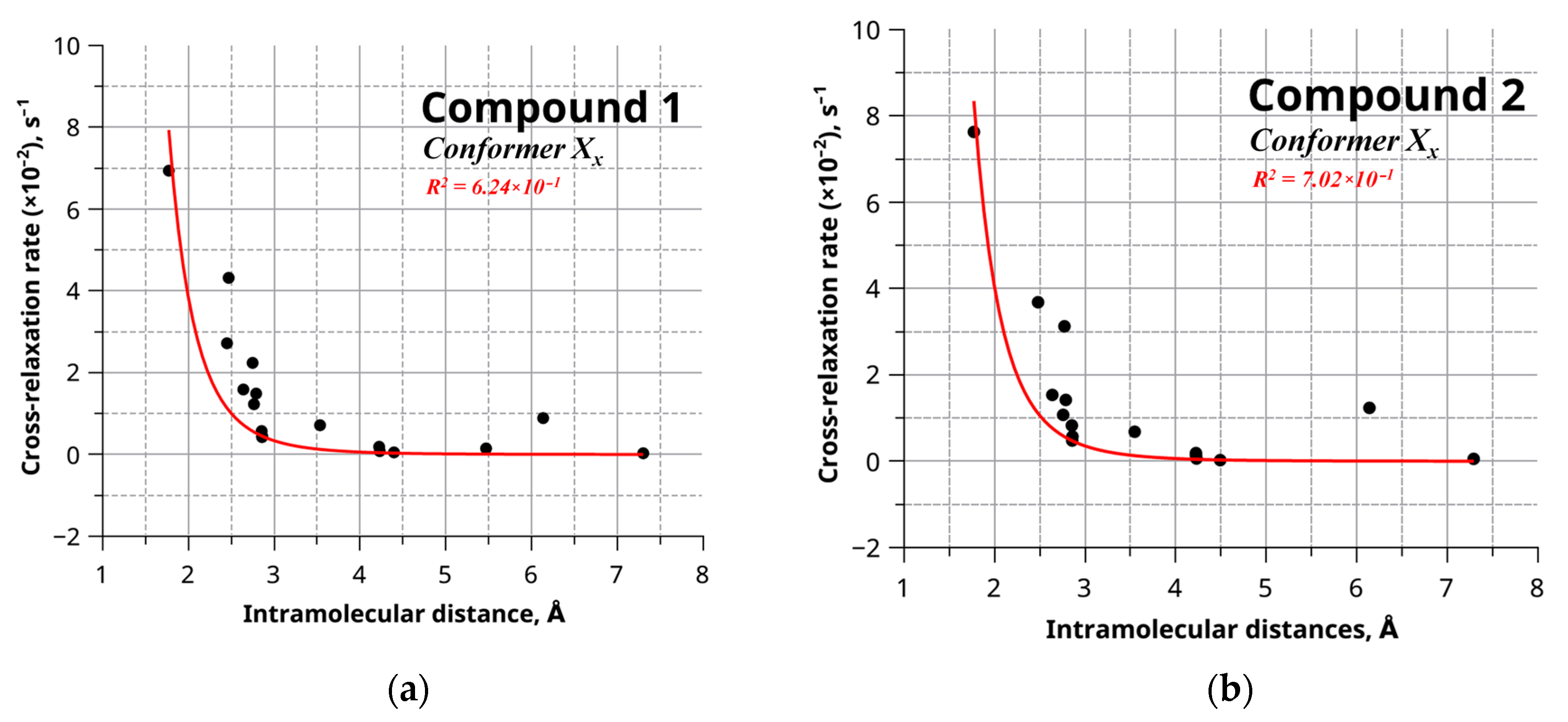
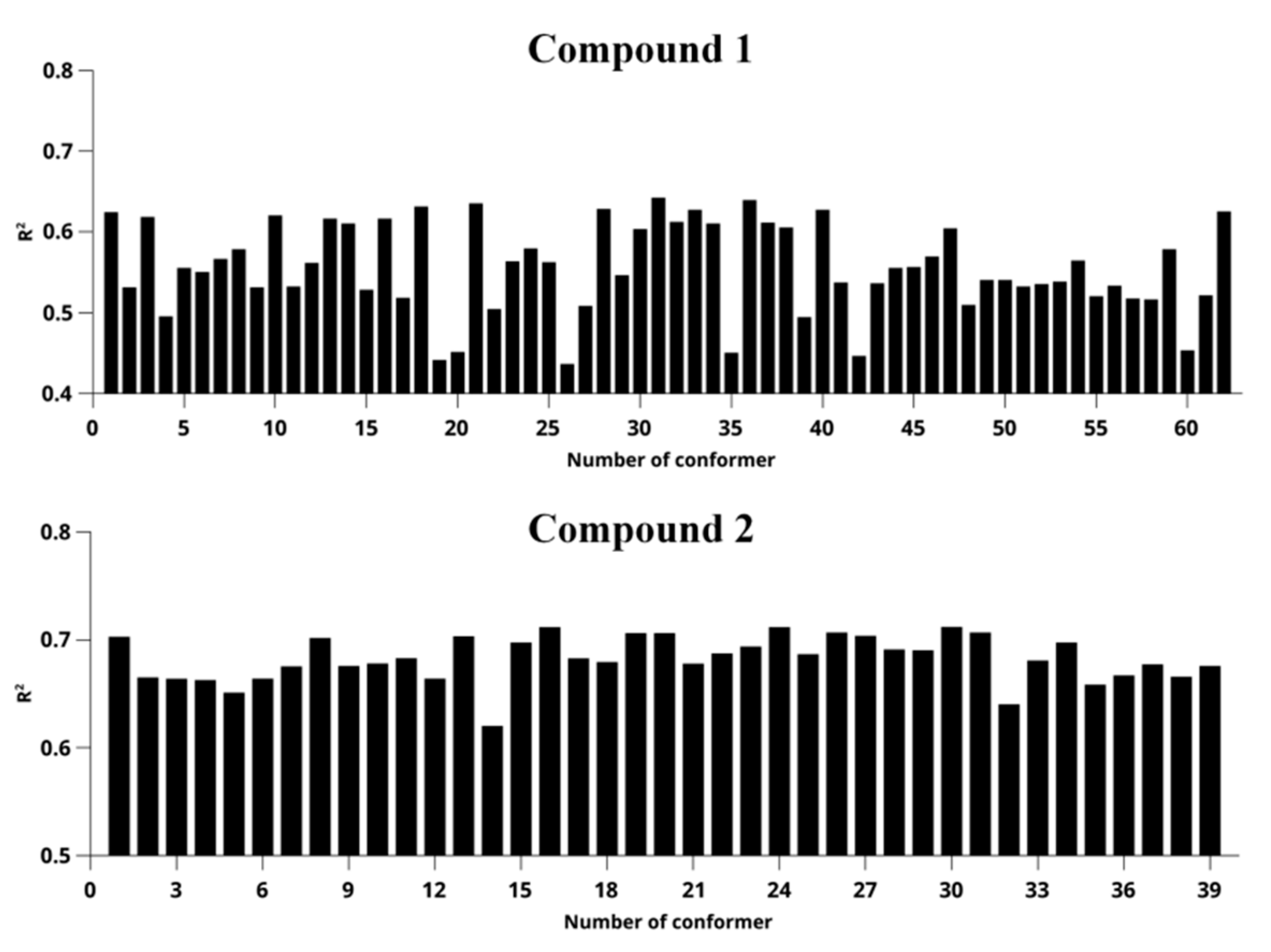
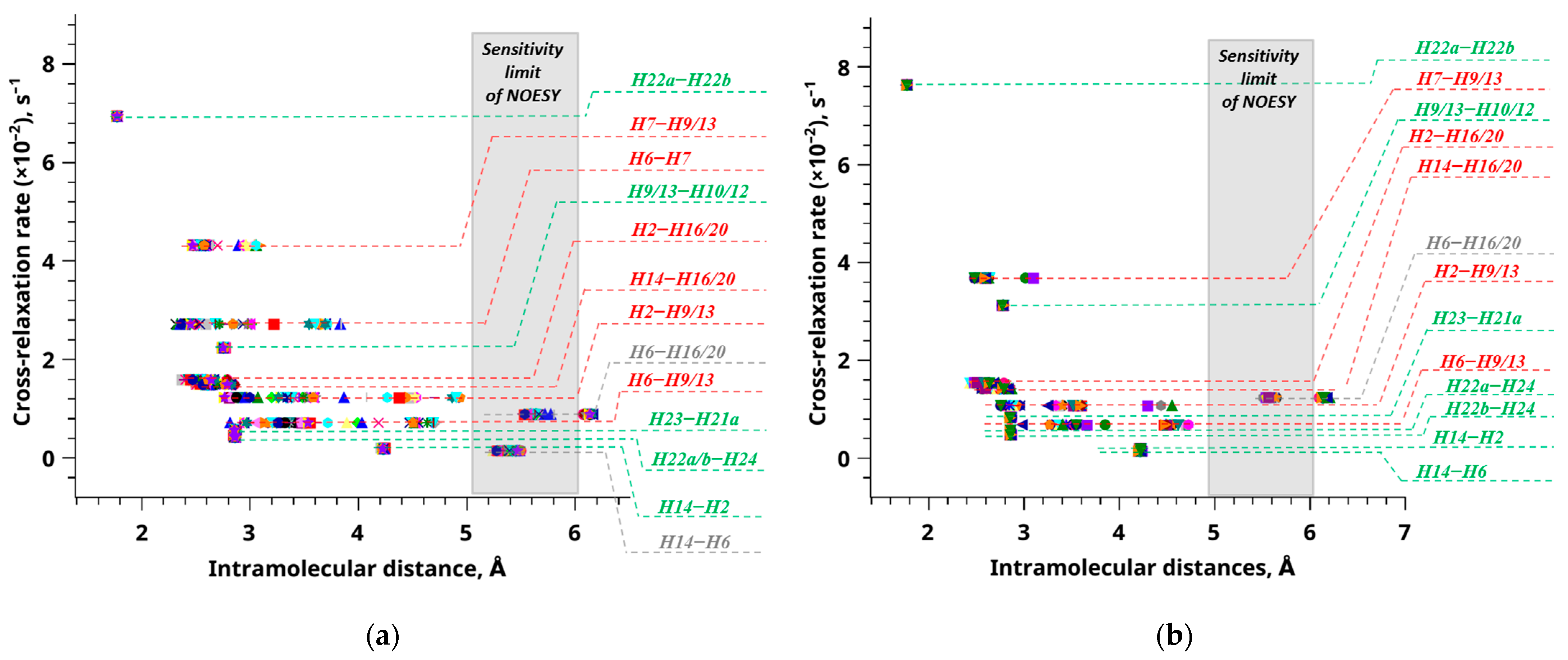
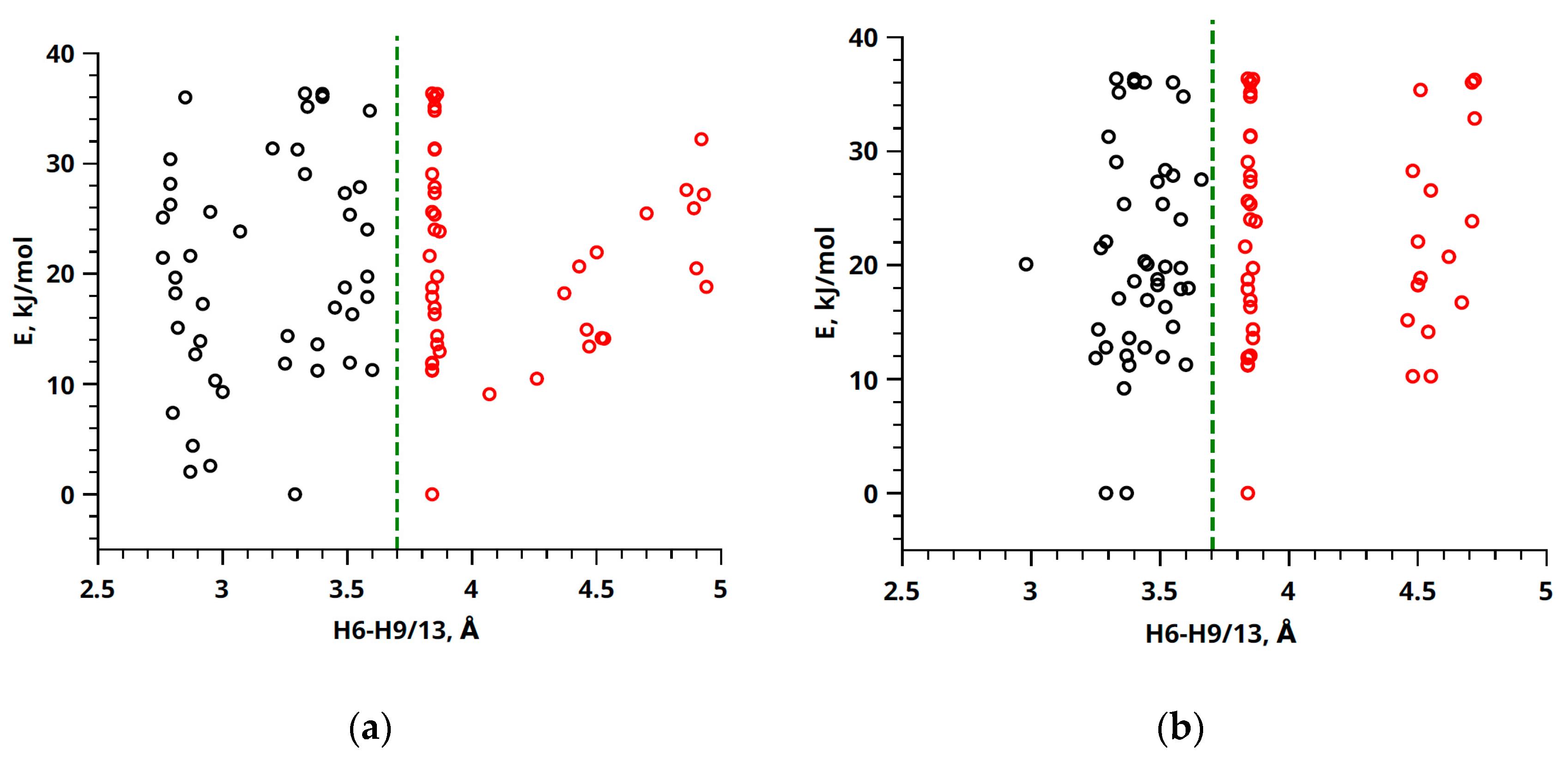
2. Results and Discussion
3. Materials and Methods
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Frigo, L.; Duque da Fonseca, G.A.M.; Marino Favero, G.; Augusto Maria, D. Managing Issues: Tumor Lysis, Extravasation, Adverse Effects, and Others. In The Golden Guide to Oncologic Pharmacy; Springer: Cham, Switzerland, 2022; pp. 371–398. [Google Scholar] [CrossRef]
- Thakur, R.; Bajaj, J.K.; Dutta, A.; Sidhu, S. Retrospective Observational Study to Evaluate Causality, Preventability and Severity of Adverse Drug Reaction Associated with Anticancer Drugs in a Tertiary Care Hospital in Northern India. Curr. Drug Saf. 2022, 17, 335–343. [Google Scholar] [CrossRef] [PubMed]
- Tewari, D.; Rawat, P.; Singh, P.K. Adverse Drug Reactions of Anticancer Drugs Derived from Natural Sources. Food Chem. Toxicol. 2019, 123, 522–535. [Google Scholar] [CrossRef] [PubMed]
- Basak, D.; Arrighi, S.; Darwiche, Y.; Deb, S. Comparison of Anticancer Drug Toxicities: Paradigm Shift in Adverse Effect Profile. Life 2022, 12, 48. [Google Scholar] [CrossRef] [PubMed]
- Moreau Bachelard, C.; Coquan, E.; du Rusquec, P.; Paoletti, X.; Le Tourneau, C. Risks and Benefits of Anticancer Drugs in Advanced Cancer Patients: A Systematic Review and Meta-Analysis. EClinicalMedicine 2021, 40, 101130. [Google Scholar] [CrossRef] [PubMed]
- Schirrmacher, V. From Chemotherapy to Biological Therapy: A Review of Novel Concepts to Reduce the Side Effects of Systemic Cancer Treatment (Review). Int. J. Oncol. 2019, 54, 407–419. [Google Scholar] [CrossRef] [PubMed]
- Hossain, M.B.; Haldar Neer, A.H. Chemotherapy. Cancer Treat. Res. 2023, 185, 49–58. [Google Scholar] [CrossRef] [PubMed]
- Pagani, M. Antitumor/Cytostatic Drugs. In Cutaneous Drug Hypersensitivity; Springer: Cham, Switzerland, 2022; pp. 297–304. [Google Scholar] [CrossRef]
- Zeien, J.; Qiu, W.; Triay, M.; Dhaibar, H.A.; Cruz-Topete, D.; Cornett, E.M.; Urits, I.; Viswanath, O.; Kaye, A.D. Clinical Implications of Chemotherapeutic Agent Organ Toxicity on Perioperative Care. Biomed. Pharmacother. 2022, 146, 112503. [Google Scholar] [CrossRef] [PubMed]
- Restrepo, G. Chemical Space: Limits, Evolution and Modelling of an Object Bigger than Our Universal Library. Digit. Discov. 2022, 1, 568–585. [Google Scholar] [CrossRef]
- Sahoo, A.; Mandal, A.K.; Kumar, M.; Dwivedi, K.; Singh, D. Prospective Challenges for Patenting and Clinical Trials of Anticancer Compounds from Natural Products: Coherent Review. Recent Pat. Anticancer. Drug Discov. 2022, 18, 470–494. [Google Scholar] [CrossRef]
- Sofi, F.A.; Tabassum, N. Natural Product Inspired Leads in the Discovery of Anticancer Agents: An Update. J. Biomol. Struct. Dyn. 2023, 41, 8605–8628. [Google Scholar] [CrossRef]
- Goel, A.; Kunnumakkara, A.B.; Aggarwal, B.B. Curcumin as “Curecumin”: From Kitchen to Clinic. Biochem. Pharmacol. 2008, 75, 787–809. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.J.; Huang, S.Y.; Zhou, D.D.; Xiong, R.G.; Zhao, C.N.; Fang, A.P.; Zhang, Y.J.; Li, H.B.; Zhu, H.L. Effects and Mechanisms of Curcumin for the Prevention and Management of Cancers: An Updated Review. Antioxidants 2022, 11, 1481. [Google Scholar] [CrossRef]
- Sanlier, N.; Kocabas, Ş.; Erdogan, K.; Sanlier, N.T. Effects of Curcumin, Its Analogues, and Metabolites on Various Cancers: Focusing on Potential Mechanisms. Food Rev. Int. 2023, 39, 5356–5376. [Google Scholar] [CrossRef]
- Garcea, G.; Berry, D.P.; Jones, D.J.L.; Singh, R.; Dennison, A.R.; Farmer, P.B.; Sharma, R.A.; Steward, W.P.; Gescher, A.J. Consumption of the Putative Chemopreventive Agent Curcumin by Cancer Patients: Assessment of Curcumin Levels in the Colorectum and Their Pharmacodynamic Consequences. Cancer Epidemiol. Biomarkers Prev. 2005, 14, 120–125. [Google Scholar] [CrossRef] [PubMed]
- Sharma, R.A.; Euden, S.A.; Platton, S.L.; Cooke, D.N.; Shafayat, A.; Hewitt, H.R.; Marczylo, T.H.; Morgan, B.; Hemingway, D.; Plummer, S.M.; et al. Phase I Clinical Trial of Oral CurcuminBiomarkers of Systemic Activity and Compliance. Clin. Cancer Res. 2004, 10, 6847–6854. [Google Scholar] [CrossRef] [PubMed]
- Kudo, C.; Yamakoshi, H.; Sato, A.; Nanjo, H.; Ohori, H.; Ishioka, C.; Iwabuchi, Y.; Shibata, H. Synthesis of 86 Species of 1,5-Diaryl-3-Oxo-1,4-Pentadienes Analogs of Curcumin Can Yield a Good Lead in Vivo. BMC Pharmacol. 2011, 11, 1–9. [Google Scholar] [CrossRef]
- Makarov, M.V.; Rybalkina, E.Y.; Anikina, L.V.; Pukhov, S.A.; Klochkov, S.G.; Mischenko, D.V.; Neganova, M.E.; Khrustalev, V.N.; Klemenkova, Z.S.; Brel, V.K. 1,5-Diaryl-3-Oxo-1,4-Pentadienes Based on (4-Oxopiperidin-1-Yl)(Aryl)Methyl Phosphonate Scaffold: Synthesis and Antitumor Properties. Med. Chem. Res. 2017, 26, 140–152. [Google Scholar] [CrossRef]
- Jha, A.; Mukherjee, C.; Prasad, A.K.; Parmar, V.S.; Clercq, E.D.; Balzarini, J.; Stables, J.P.; Manavathu, E.K.; Shrivastav, A.; Sharma, R.K.; et al. E,E,E-1-(4-Arylamino-4-Oxo-2-Butenoyl)-3,5-Bis(Arylidene)-4-Piperidones: A Topographical Study of Some Novel Potent Cytotoxins. Bioorg. Med. Chem. 2007, 15, 5854–5865. [Google Scholar] [CrossRef] [PubMed]
- Bazzaro, M.; Anchoori, R.K.; Mudiam, M.K.R.; Issaenko, O.; Kumar, S.; Karanam, B.; Lin, Z.; Isaksson Vogel, R.; Gavioli, R.; Destro, F.; et al. α,β-Unsaturated Carbonyl System of Chalcone-Based Derivatives Is Responsible for Broad Inhibition of Proteasomal Activity and Preferential Killing of Human Papilloma Virus (HPV) Positive Cervical Cancer Cells. J. Med. Chem. 2011, 54, 449–456. [Google Scholar] [CrossRef]
- Kálai, T.; Kuppusamy, M.L.; Balog, M.; Selvendiran, K.; Rivera, B.K.; Kuppusamy, P.; Hideg, K. Synthesis of N-Substituted 3,5-Bis(Arylidene)-4-Piperidones with High Antitumor and Antioxidant Activity. J. Med. Chem. 2011, 54, 5414–5421. [Google Scholar] [CrossRef]
- Williamson, M.P. Nuclear Magnetic Resonance Spectroscopy Nuclear Overhauser Effect. In Encyclopedia of Analytical Science, 3rd ed.; Elsevier: Amsterdam, The Netherlands, 2019; pp. 264–271. [Google Scholar] [CrossRef]
- Wieske, L.H.E.; Erdélyi, M. Non-Uniform Sampling for NOESY? A Case Study on Spiramycin. Magn. Reson. Chem. 2021, 59, 723–737. [Google Scholar] [CrossRef]
- Belov, K.V.; Dyshin, A.A.; Krestyaninov, M.A.; Efimov, S.V.; Khodov, I.A.; Kiselev, M.G. Conformational Preferences of Tolfenamic Acid in DMSO-CO2 Solvent System by 2D NOESY. J. Mol. Liq. 2022, 367, 120481. [Google Scholar] [CrossRef]
- Khodov, I.A.; Belov, K.V.; Krestyaninov, M.A.; Dyshin, A.A.; Kiselev, M.G.; Krestov, G.A. Investigation of the Spatial Structure of Flufenamic Acid in Supercritical Carbon Dioxide Media via 2D NOESY. Materials 2023, 16, 1524. [Google Scholar] [CrossRef] [PubMed]
- Eventova, V.A.; Belov, K.V.; Efimov, S.V.; Khodov, I.A. Conformational Screening of Arbidol Solvates: Investigation via 2D NOESY. Pharmaceutics 2023, 15, 226. [Google Scholar] [CrossRef] [PubMed]
- Khodov, I.A.; Belov, K.V.; Huster, D.; Scheidt, H.A. Conformational State of Fenamates at the Membrane Interface: A MAS NOESY Study. Membranes 2023, 13, 607. [Google Scholar] [CrossRef] [PubMed]
- Baishya, B.; Verma, R.; Parihar, R. Spatially Encoded Polarization Transfer for Improving the Quantitative Aspect of 1H–13C HSQC. J. Magn. Reson. Open 2022, 12–13, 100063. [Google Scholar] [CrossRef]
- Williamson, R.T.; Sørensen, O.W. Origin and Remedy for HSQC Artifacts in Proton-Detected INADEQUATE Spectra. Phys. Chem. Chem. Phys. 2023, 25, 11080–11084. [Google Scholar] [CrossRef]
- Bigler, P.; Furrer, J. Measurement of Long-Range Heteronuclear Coupling Constants Using the Peak Intensity in Classical 1D HMBC Spectra. Magn. Reson. Chem. 2018, 56, 329–337. [Google Scholar] [CrossRef] [PubMed]
- Timári, I.; Bagi, P.; Keglevich, G.; Kövér, K.E. Ultrahigh-Resolution Homo- and Heterodecoupled 1H and TOCSY NMR Experiments. ACS Omega 2022, 7, 43283–43289. [Google Scholar] [CrossRef]
- Nolis, P.; Parella, T. Multiplicity-Edited 1 H- 1 H TOCSY Experiment. Magn. Reson. Chem. 2018, 56, 976–982. [Google Scholar] [CrossRef]
- Nolis, P.; Parella, T. Practical Aspects of the Simultaneous Collection of COSY and TOCSY Spectra. Magn. Reson. Chem. 2019, 57, S85–S94. [Google Scholar] [CrossRef]
- Szalai, Z.; Keglevich, G.; Micheletti, G.; Trofimov, B.A. Tetraalkyl Hydroxymethylene-Bisphosphonate and Dialkyl 1-Diphenylphosphinoyl-1-Hydroxy-Ethylphosphonate Derivatives by the Pudovik Reaction and Their Rearranged Products. Molecules 2021, 26, 7575. [Google Scholar] [CrossRef]
- Delehedde, C.; Culcasi, M.; Ricquebourg, E.; Cassien, M.; Siri, D.; Blaive, B.; Pietri, S.; Thétiot-Laurent, S. Novel Sterically Crowded and Conformationally Constrained α-Aminophosphonates with a Near-Neutral PKa as Highly Accurate 31P NMR PH Probes. Application to Subtle PH Gradients Determination in Dictyostelium Discoideum Cells. Molecules 2022, 27, 4506. [Google Scholar] [CrossRef]
- Rádai, Z.; Hodula, V.; Kiss, N.Z.; Kóti, J.; Keglevich, G. Phosphorylation of (1-Aryl-1-Hydroxymethyl)Phosphonates. Mendeleev Commun. 2019, 29, 153–154. [Google Scholar] [CrossRef]
- Kuz’mina, N.E.; Moiseev, S.V.; Severinova, E.Y.; Stepanov, E.A.; Bunyatyan, N.D. Identification and Quantification by NMR Spectroscopy of the 22R and 22S Epimers in Budesonide Pharmaceutical Forms. Molecules 2022, 27, 2262. [Google Scholar] [CrossRef]
- Jameson, C.J. Understanding NMR chemical shifts. Annu. Rev. Phys. Chem. 2003, 47, 135–169. [Google Scholar] [CrossRef]
- Abraham, R.J.; Griffiths, L.; Perez, M. 1H NMR Spectra Part 31: 1H Chemical Shifts of Amides in DMSO Solvent. Magn. Reson. Chem. 2014, 52, 395–408. [Google Scholar] [CrossRef]
- Emenike, B.U.; Farshadmand, A.; Zeller, M.; Roman, A.J.; Sevimler, A.; Shinn, D.W. Electrostatic CH−π Interactions Can Override Fluorine Gauche Effects to Exert Conformational Control. Chem.—A Eur. J. 2023, 29, e202203139. [Google Scholar] [CrossRef]
- Fuhrer, T.J.; Houck, M.; Iacono, S.T. Fluoromaticity: The Molecular Orbital Contributions of Fluorine Substituents to the π-Systems of Aromatic Rings. ACS Omega 2021, 6, 32607–32617. [Google Scholar] [CrossRef]
- Viesser, R.V.; Tormena, C.F. Counterintuitive Deshielding on the 13C NMR Chemical Shift for the Trifluoromethyl Anion. Magn. Reson. Chem. 2020, 58, 540–547. [Google Scholar] [CrossRef]
- Casabianca, L.B.; Reson, M. Calculating Nuclear Magnetic Resonance Chemical Shifts in Solvated Systems. Magn. Reson. Chem. 2020, 58, 611–624. [Google Scholar] [CrossRef]
- Scheiner, S. Influence of Internal Noncovalent Bonds on Rotational Dynamics. Inorg. Chem. 2023, 62, 13030–13037. [Google Scholar] [CrossRef]
- Stanger, A. Nucleus Independent Chemical Shift (NICS) at Small Distances from the Molecular Plane: The Effect of Electron Density. ChemPhysChem 2023, 24, e202300080. [Google Scholar] [CrossRef]
- Wang, S.; Krummenacher, K.; Landrum, G.A.; Sellers, B.D.; Di Lello, P.; Robinson, S.J.; Martin, B.; Holden, J.K.; Tom, J.Y.K.; Murthy, A.C.; et al. Incorporating NOE-Derived Distances in Conformer Generation of Cyclic Peptides with Distance Geometry. J. Chem. Inf. Model. 2022, 62, 472–485. [Google Scholar] [CrossRef]
- Koos, M.R.M.; Schulz, K.H.G.; Gil, R.R. Reference-Free NOE NMR Analysis. Chem. Sci. 2020, 11, 9930–9936. [Google Scholar] [CrossRef]
- Lee, W.; Krishna, N.R. Influence of Conformational Exchange on the 2D NOESY Spectra of Biomolecules Existing in Multiple Conformations. J. Magn. Reson. 1992, 98, 36–48. [Google Scholar] [CrossRef]
- Macur, S.; Farmer, B.T.; Brown, L.R. An Improved Method for the Determination of Cross-Relaxation Rates from NOE Data. J. Magn. Reson. 1986, 70, 493–499. [Google Scholar] [CrossRef]
- Wieske, L.H.E.; Peintner, S.; Erdélyi, M. Ensemble Determination by NMR Data Deconvolution. Nat. Rev. Chem. 2023, 7, 511–524. [Google Scholar] [CrossRef]
- Macura, S.; Huang, Y.; Suter, D.; Ernst, R.R. Two-Dimensional Chemical Exchange and Cross-Relaxation Spectroscopy of Coupled Nuclear Spins. J. Magn. Reson. 1981, 43, 259–281. [Google Scholar] [CrossRef]
- Gadiev, T.A.; Khairutdinov, B.I.; Shaikhutdinov, R.A.; Karatayeva, F.K.; Aganov, A.V.; Klochkov, V.V. Spatial Structure of Dimeric Capsules of Tetraurea Calix[4]Arenes in Solutions According to 2-D NMR (NOESY) Spectroscopy. Appl. Magn. Reson. 2003, 25, 347–352. [Google Scholar] [CrossRef]
- Hu, H.; Krishnamurthy, K. Revisiting the Initial Rate Approximation in Kinetic NOE Measurements. J. Magn. Reson. 2006, 182, 173–177. [Google Scholar] [CrossRef]
- Gadiev, T.A.; Khairutdinov, B.I.; Antipin, I.S.; Klochkov, V.V. Analysis of the Spatial Structure of Calixarenes in Solutions by 2-D NMR (NOESY) Spectroscopy. Appl. Magn. Reson. 2006, 30, 165–173. [Google Scholar] [CrossRef]
- Khodov, I.A.; Belov, K.V.; Krestyaninov, M.A.; Sobornova, V.V.; Dyshin, A.A.; Kiselev, M.G. Does DMSO Affect the Conformational Changes of Drug Molecules in Supercritical CO2 Media? J. Mol. Liq. 2023, 384, 122230. [Google Scholar] [CrossRef]
- Canet, D. (Ed.) Cross-Relaxation and Cross-Correlation Parameters in NMR: Molecular Approaches; New Developments in NMR; Royal Society of Chemistry: Cambridge, UK, 2017; ISBN 978-1-84973-913-9. [Google Scholar]
- Duchardt, E.; Richter, C.; Ohlenschläger, O.; Görlach, M.; Wöhnert, J.; Schwalbe, H. Determination of the Glycosidic Bond Angle χ in RNA from Cross-Correlated Relaxation of CH Dipolar Coupling and N Chemical Shift Anisotropy. J. Am. Chem. Soc. 2004, 126, 1962–1970. [Google Scholar] [CrossRef]
- Ferrage, F.; Eykyn, T.R.; Bodenhausen, G. Coherence Transfer by Single-Transition Cross-Polarization: Quantitation of Cross-Correlation Effects in Nuclear Magnetic Resonance. J. Chem. Phys. 2000, 113, 1081. [Google Scholar] [CrossRef]
- Majumdar, A.; Hosur, R.V. Simulation of 2D NMR Spectra for Determination of Solution Conformations of Nucleic Acids. Prog. Nucl. Magn. Reson. Spectrosc. 1992, 24, 109–158. [Google Scholar] [CrossRef]
- Khodov, I.A.; Belov, K.V.; Dyshin, A.A.; Krestyaninov, M.A.; Kiselev, M.G. Pressure Effect on Lidocaine Conformational Equilibria in ScCO2: A Study by 2D NOESY. J. Mol. Liq. 2022, 367, 120525. [Google Scholar] [CrossRef]
- Koning, T.M.G.M.G.; Boelens, R.; Kaptein, R. Calculation of the Nuclear Overhauser Effect and the Determination of Proton-Proton Distances in the Presence of Internal Motions. J. Magn. Reson. 1990, 90, 111–123. [Google Scholar] [CrossRef]
- Kolmer, A.; Edwards, L.J.; Kuprov, I.; Thiele, C.M. Conformational Analysis of Small Organic Molecules Using NOE and RDC Data: A Discussion of Strychnine and α-Methylene-γ-Butyrolactone. J. Magn. Reson. 2015, 261, 101–109. [Google Scholar] [CrossRef] [PubMed]
- Lai, X.; Chen, C.; Andersen, N.H. The DISCON Algorithm, an Accurate and Robust Alternative to an Eigenvalue Solution for Extracting Experimental Distances from NOESY Data. J. Magn. Reson. Ser. B 1993, 101, 271–288. [Google Scholar] [CrossRef]
- Khodov, I.A.; Efimov, S.V.; Klochkov, V.V.; Batista De Carvalho, L.A.E.; Kiselev, M.G. The Importance of Suppressing Spin Diffusion Effects in the Accurate Determination of the Spatial Structure of a Flexible Molecule by Nuclear Overhauser Effect Spectroscopy. J. Mol. Struct. 2016, 1106, 373–381. [Google Scholar] [CrossRef]
- Pronina, Y.A.; Stepakov, A.V.; Popova, E.A.; Boitsov, V.M.; Baichurin, R.I.; Selivanov, S.I. Diastereomers of Cyclopropa[a]Pyrrolizine Spiro-Fused with a Benzo[4,5]Imidazo[1,2-a]Indole Fragment: Structure Determinations Using NMR Methods. Appl. Magn. Reson. 2023, 54, 999–1014. [Google Scholar] [CrossRef]
- Böhm, H.J.; Banner, D.; Bendels, S.; Kansy, M.; Kuhn, B.; Müller, K.; Obst-Sander, U.; Stahl, M. Fluorine in Medicinal Chemistry. ChemBioChem 2004, 5, 637–643. [Google Scholar] [CrossRef]
- Galván-Hernández, A.; Kobayashi, N.; Hernández-Cobos, J.; Antillón, A.; Nakabayashi, S.; Ortega-Blake, I. Morphology and Dynamics of Domains in Ergosterol or Cholesterol Containing Membranes. Biochim. Biophys. Acta—Biomembr. 2020, 1862, 183101. [Google Scholar] [CrossRef]
- Fonseca-Ornelas, L.; Eisbach, S.E.; Paulat, M.; Giller, K.; Fernández, C.O.; Outeiro, T.F.; Becker, S.; Zweckstetter, M. Small Molecule-Mediated Stabilization of Vesicle-Associated Helical α-Synuclein Inhibits Pathogenic Misfolding and Aggregation. Nat. Commun. 2014, 5, 1–11. [Google Scholar] [CrossRef]
- Liu, M.; Mao, X.A.X.-A.; Ye, C.; Huang, H.H.; Nicholson, J.K.J.K.; Lindon, J.C.J.C. Improved Watergate Pulse Sequences for Solvent Suppression in NMR Spectroscopy. J. Magn. Reson. 1998, 132, 125–129. [Google Scholar] [CrossRef]
- Guzman, A.L.; Hoye, T.R. TMS Is Superior to Residual CHCl3 for Use as the Internal Reference for Routine 1H NMR Spectra Recorded in CDCl3. J. Org. Chem. 2022, 87, 905–909. [Google Scholar] [CrossRef]
- Makulski, W.; Jackowski, K. 1H, 13C and 29Si Magnetic Shielding in Gaseous and Liquid Tetramethylsilane. J. Magn. Reson. 2020, 313, 106716. [Google Scholar] [CrossRef]
- Sakhaii, P.; Bohorc, B.; Schliedermann, U.; Bermel, W. Boosting the Resolution of Multidimensional NMR Spectra by Complete Removal of Proton Spin Multiplicities. Sci. Rep. 2021, 11, 1–9. [Google Scholar] [CrossRef]
- Bigler, P.; Furrer, J. Why Is HMBC Superior to LR-HSQC? Influence of Homonuclear Couplings JHH′ on the Intensity of Long-Range Correlations. Magn. Reson. Chem. 2018, 56, 1101–1116. [Google Scholar] [CrossRef] [PubMed]
- Kalmykov, P.A.A.; Khodov, I.A.A.; Klochkov, V.V.V.; Klyuev, M.V.V. Theoretical and Experimental Study of Imine-Enamine Tautomerism of Condensation Products of Propanal with 4-Aminobenzoic Acid in Ethanol. Russ. Chem. Bull. 2017, 66, 70–75. [Google Scholar] [CrossRef]
- Wang, Y.; Fan, A.; Cohen, R.D.; Dal Poggetto, G.; Huang, Z.; Yang, H.; Martin, G.E.; Sherer, E.C.; Reibarkh, M.; Wang, X. Unequivocal Identification of Two-Bond Heteronuclear Correlations in Natural Products at Nanomole Scale by i-HMBC. Nat. Commun. 2023, 14, 1–11. [Google Scholar] [CrossRef]
- Bigler, P.; Furrer, J. D-HMBC versus LR-HSQMBC: Which Experiment Provides Theoretically and Experimentally the Best Results? Magn. Reson. Chem. 2019, 57, 129–143. [Google Scholar] [CrossRef]
- Schleucher, J.; Schwendinger, M.; Sattler, M.; Schmidt, P.; Schedletzky, O.; Glaser, S.J.; Sorensen, O.W.; Griesinger, C. A General Enhancement Scheme in Heteronuclear Multidimensional NMR Employing Pulsed Field Gradients. J. Biomol. NMR 1994, 4, 301–306. [Google Scholar] [CrossRef]
- Cicero, D.O.; Barbato, G.; Bazzo, R. Sensitivity Enhancement of a Two-Dimensional Experiment for the Measurement of Heteronuclear Long Range Coupling Constants, by a New Scheme of Coherence. J. Magn. Reson. 2001, 148, 209–213. [Google Scholar] [CrossRef] [PubMed]
- Kiryutin, A.S.; Zhukov, I.V.; Ferrage, F.; Bodenhausen, G.; Yurkovskaya, A.V.; Ivanov, K.L. Sequential Assignment of NMR Spectra of Peptides at Natural Isotopic Abundance with Zero- and Ultra-Low-Field Total Correlation Spectroscopy (ZULF-TOCSY). Phys. Chem. Chem. Phys. 2021, 23, 9715–9720. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.; Chen, J.; Zeng, Q.; Luo, Y.; Chen, Z.; Lin, Y. Simultaneous Acquirement of Pure Shift 2D Homonuclear Correlation Spectra. J. Magn. Reson. 2022, 339, 107229. [Google Scholar] [CrossRef]
- Epasto, L.M.; Honegger, P.; Che, K.; Kozak, F.; Jörg, F.; Schröder, C.; Kurzbach, D. Nuclear Overhauser Spectroscopy in Hyperpolarized Water—Chemical vs. Magnetic Exchange. Chem. Commun. 2022, 58, 11661–11664. [Google Scholar] [CrossRef] [PubMed]
- Stott, K.; Keeler, J.; Hwang, T.-L.L.T.-L.; Shaka, A.J.J.; Stonehouse, J.; Keeler, J.; Hwang, T.-L.L.T.-L.; Shaka, A.J.J.; Stonehouse, J. Excitation Sculpting in High-Resolution Nuclear Magnetic Resonance Spectroscopy: Application to Selective NOE Experiments. J. Am. Chem. Soc. 1995, 117, 4199–4200. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian G16, Revis. A.03; Gaussian 09 Revis. C.01; Gaussian Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Armstrong, A.; Boto, R.A.; Dingwall, P.; Contreras-García, J.; Harvey, M.J.; Mason, N.J.; Rzepa, H.S. The Houk–List Transition States for Organocatalytic Mechanisms Revisited. Chem. Sci. 2014, 5, 2057–2071. [Google Scholar] [CrossRef]
- Wright, S.E.; Cole, J.C.; Cruz-Cabeza, A.J. Conformational Change in Molecular Crystals: Impact of Solvate Formation and Importance of Conformational Free Energies. Cryst. Growth Des. 2021, 21, 6924–6936. [Google Scholar] [CrossRef]
- Belov, K.V.; Batista de Carvalho, L.A.E.; Dyshin, A.A.; Efimov, S.V.; Khodov, I.A. The Role of Hidden Conformers in Determination of Conformational Preferences of Mefenamic Acid by NOESY Spectroscopy. Pharmaceutics 2022, 14, 2276. [Google Scholar] [CrossRef]
- Cutini, M.; Maschio, L.; Ugliengo, P. Exfoliation Energy of Layered Materials by DFT-D: Beware of Dispersion! J. Chem. Theory Comput. 2020, 16, 5244–5252. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.; Song, S.; Sim, E.; Burke, K. Halogen and Chalcogen Binding Dominated by Density-Driven Errors. J. Phys. Chem. Lett. 2019, 10, 295–301. [Google Scholar] [CrossRef] [PubMed]
- O’Reilly, R.J.; Balanay, M.P. A Quantum Chemical Study of the Effect of Substituents in Governing the Strength of the S–F Bonds of Sulfenyl-Type Fluorides toward Homolytic Dissociation and Fluorine Atom Transfer. Chem. Data Collect. 2019, 20, 100186. [Google Scholar] [CrossRef]










Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Belov, K.; Brel, V.; Sobornova, V.; Fedorova, I.; Khodov, I. Conformational Analysis of 1,5-Diaryl-3-Oxo-1,4-Pentadiene Derivatives: A Nuclear Overhauser Effect Spectroscopy Investigation. Int. J. Mol. Sci. 2023, 24, 16707. https://doi.org/10.3390/ijms242316707
Belov K, Brel V, Sobornova V, Fedorova I, Khodov I. Conformational Analysis of 1,5-Diaryl-3-Oxo-1,4-Pentadiene Derivatives: A Nuclear Overhauser Effect Spectroscopy Investigation. International Journal of Molecular Sciences. 2023; 24(23):16707. https://doi.org/10.3390/ijms242316707
Chicago/Turabian StyleBelov, Konstantin, Valery Brel, Valentina Sobornova, Irina Fedorova, and Ilya Khodov. 2023. "Conformational Analysis of 1,5-Diaryl-3-Oxo-1,4-Pentadiene Derivatives: A Nuclear Overhauser Effect Spectroscopy Investigation" International Journal of Molecular Sciences 24, no. 23: 16707. https://doi.org/10.3390/ijms242316707