
Mechanisms of Cadmium Neurotoxicity


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
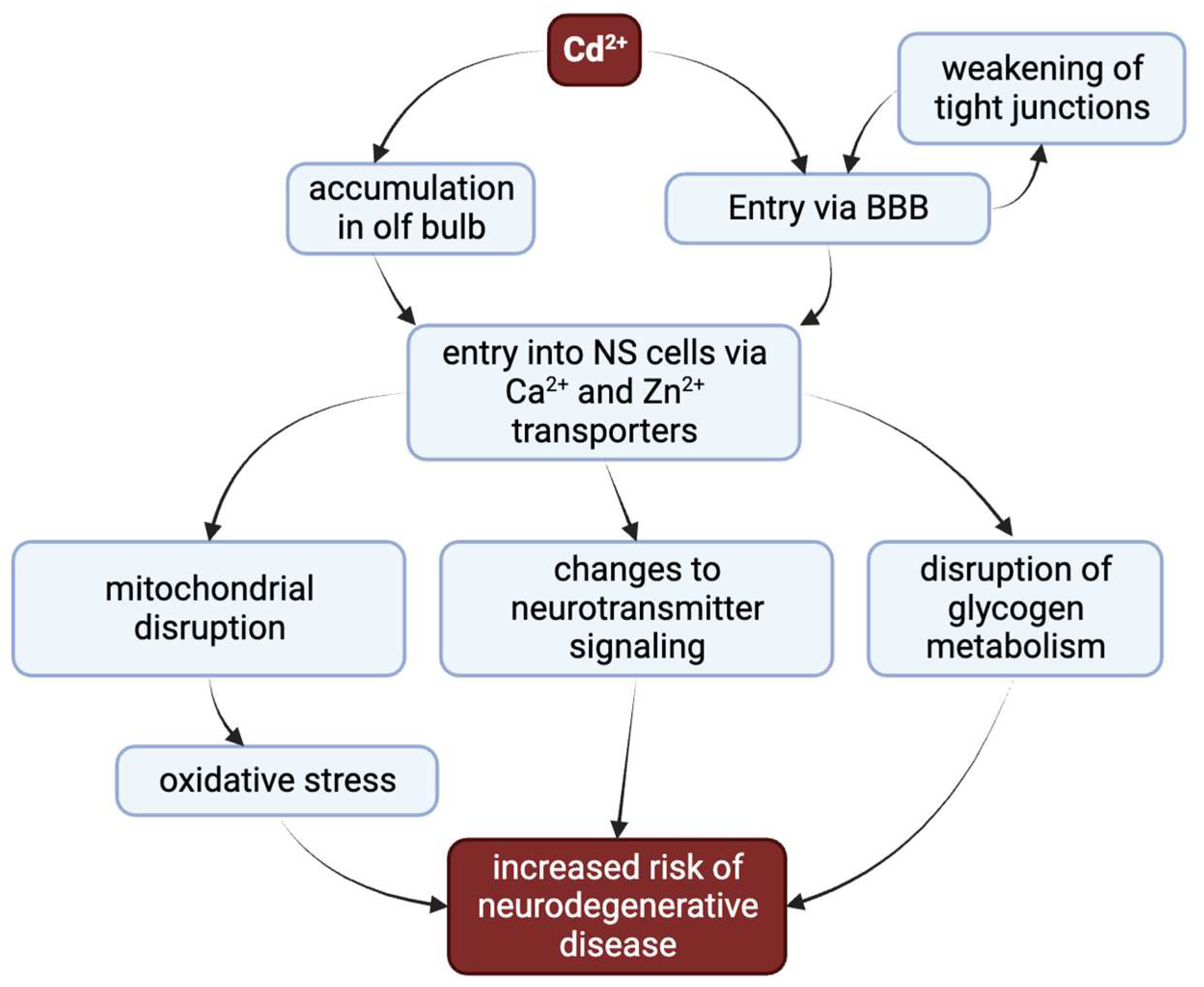
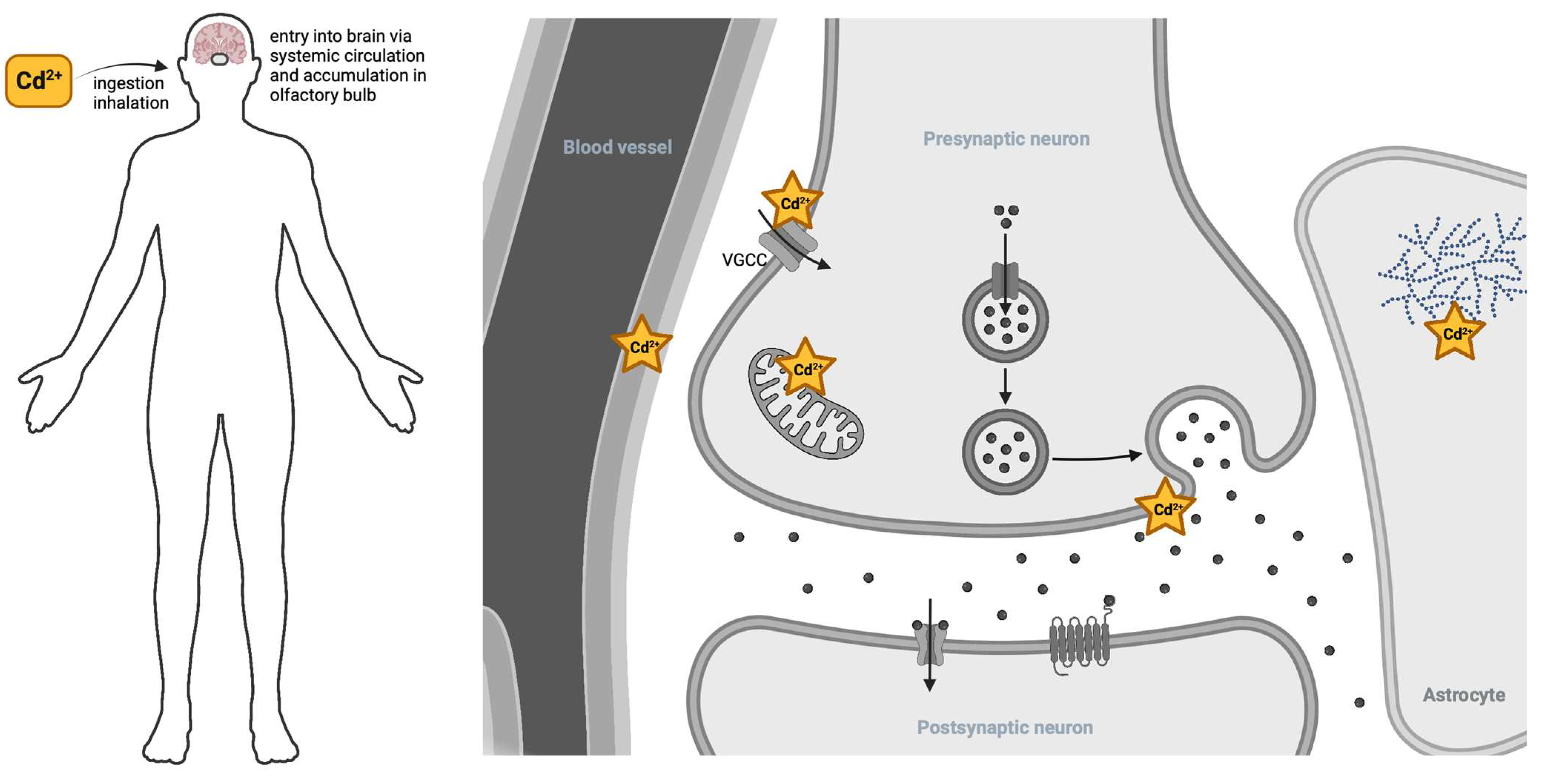
2. Cadmium Entry to the Nervous System
3. Cadmium Effects on Mitochondrial Respiration
3.1. Cadmium Interference with the Electron Transport Chain
3.2. Cadmium Opens the Permeability Transition Pore
3.3. Recent Focuses of Mitochondrial Apoptosis/Dysfunction: ER Stress and SIRT1
3.4. Cadmium-Induced Autophagy
4. The Role of Cadmium in Synaptic Transmission
4.1. Cadmium-Induced Asynchronous Neurotransmitter Release
4.2. Cadmium Disruption of Neurotransmission
5. Cadmium and Other Metals
5.1. Cadmium Disruption of Zinc Signaling and Homeostasis
5.2. Cadmium Contributions to Metal Imbalance in Alzheimer’s Disease
6. Cadmium and the Blood–Brain Barrier
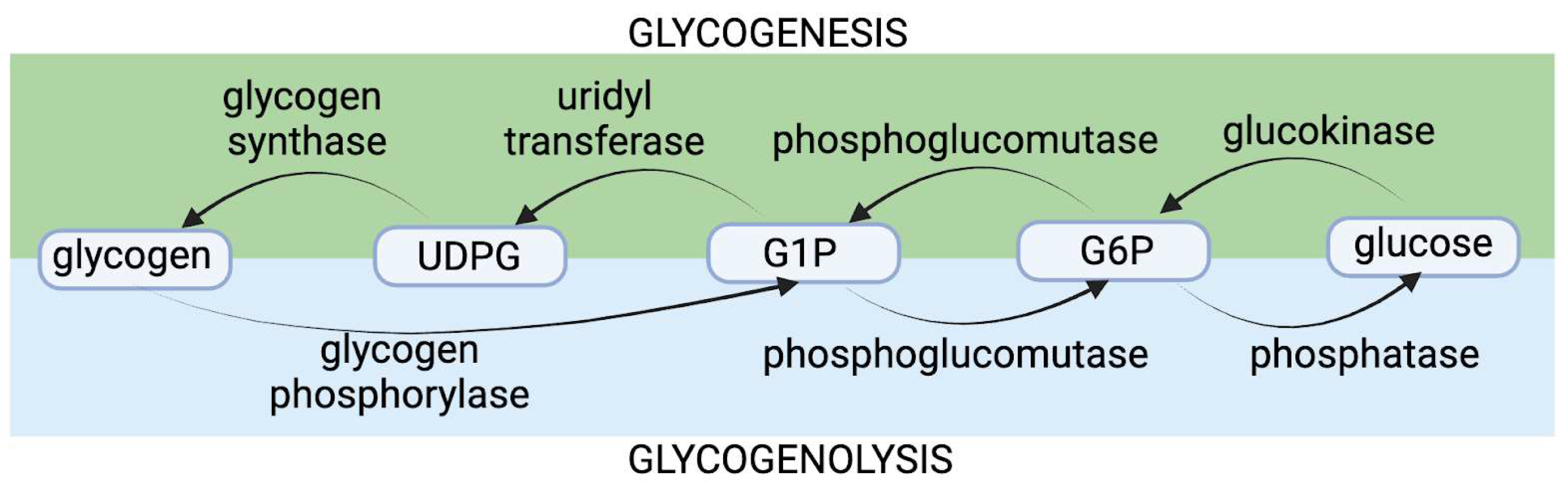
7. Cadmium’s Effects on Glycogen Metabolism
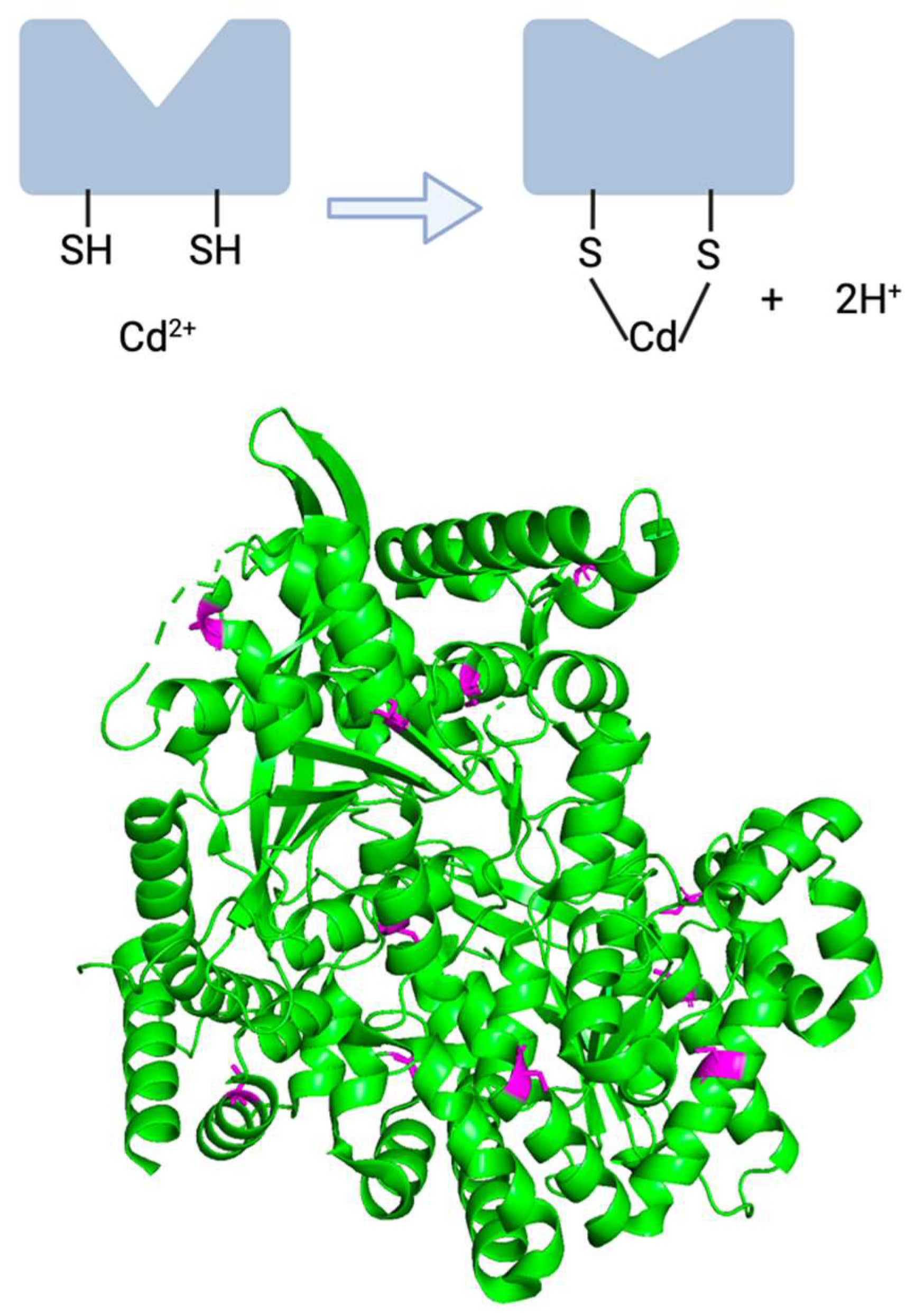
7.1. Cadmium and Glycogen Phosphorylase Impairment: The Original Hypothesis
7.2. Evidence Contradicting the Original Theory Regarding Cadmium’s Functional Impairment of bGP
7.3. Human Data Pointing towards Cadmium as a Glycogen-Disruptor
7.4. Next Steps for Resolving Cadmium’s Effects on Neuronal Glycogen
8. Possible Protective Measures against Cadmium Neurotoxicity
9. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Substance Priority List|ATSDR. Available online: https://www.atsdr.cdc.gov/spl/index.html (accessed on 10 October 2023).
- Public Health Statement Cadmium. Agency for Toxic Substances and Disease Registry. 2012. Available online: https://www.atsdr.cdc.gov/ToxProfiles/tp5-c1-b.pdf (accessed on 10 October 2023).
- Klaassen, C.; Watkins, J.B., III. Casarett and Doull’s Essentials of Toxicology, 1st ed.; McGraw-Hill Professional: New York, NY, USA, 2003. [Google Scholar]
- The Facts on Cadmium. Available online: https://sites.dartmouth.edu/toxmetal/more-metals/cadmium-an-illusive-presence/the-facts-on-cadmium/ (accessed on 1 October 2023).
- Mena, C.; Cabrera, C.; Lorenzo, M.L.; López, M.C. Cadmium levels in wine, beer and other alcoholic beverages: Possible sources of contamination. Sci. Total Environ. 1996, 181, 201. [Google Scholar] [CrossRef] [PubMed]
- Satarug, S.; Moore, M.R. Adverse Health Effects of Chronic Exposure to Low-Level Cadmium in Foodstuffs and Cigarette Smoke. Environ. Health Perspect. 2004, 112, 1099–1103. [Google Scholar] [CrossRef] [PubMed]
- Sunderman, F.W. Nasal toxicity, carcinogenicity, and olfactory uptake of metals. Ann. Clin. Lab. Sci. 2001, 31, 3–24. [Google Scholar]
- Suwazono, Y.; Kido, T.; Nakagawa, H.; Nishijo, M.; Honda, R.; Kobayashi, E.; Dochi, M.; Nogawa, K. Biological half-life of cadmium in the urine of inhabitants after cessation of cadmium exposure. Biomarkers 2009, 14, 77–81. [Google Scholar] [CrossRef]
- International Agency for Research on Cancer. Beryllium, Cadmium, Mercury, and Exposures in the Glass Manufacturing Industry; International Agency for Research on Cancer: Lyon, France, 1993; Volume 58, pp. 1–415. [Google Scholar]
- What Is the Biological Fate of Cadmium in the Body? Available online: https://www.atsdr.cdc.gov/csem/cadmium/Biological-Fate.html#:~:text=Cadmium%20is%20transported%20in%20the,the%20major%20mechanism%20of%20elimination (accessed on 12 October 2023).
- Vinceti, M.; Filippini, T.; Mandrioli, J.; Violi, F.; Bargellini, A.; Weuve, J.; Fini, N.; Grill, P.; Michalke, B. Lead, cadmium and mercury in cerebrospinal fluid and risk of amyotrophic lateral sclerosis: A case-control study. J. Trace Elem. Med. Biol. 2017, 43, 121–125. [Google Scholar] [CrossRef]
- Ma, Y.; Su, Q.; Yue, C.; Zou, H.; Zhu, J.; Zhao, H.; Song, R.; Liu, Z. The Effect of Oxidative Stress-Induced Autophagy by Cadmium Exposure in Kidney, Liver, and Bone Damage, and Neurotoxicity. Int. J. Mol. Sci. 2022, 23, 13491. [Google Scholar] [CrossRef]
- Viaene, M.K.; Masschelein, R.; Leenders, J.; De Groof, M.; Swerts, L.J.V.C.; Roels, H.A. Neurobehavioural effects of occupational exposure to cadmium: A cross sectional epidemiological study. Occup. Environ. Med. 2000, 57, 19–27. [Google Scholar] [CrossRef]
- Li, H.; Wang, Z.; Fu, Z.; Yan, M.; Wu, N.; Wu, H.; Yin, P. Associations between blood cadmium levels and cognitive function in a cross-sectional study of US adults aged 60 years or older. BMJ Open 2018, 8, e020533. [Google Scholar] [CrossRef]
- Ciesielski, T.; Weuve, J.; Bellinger, D.C.; Schwartz, J.; Lanphear, B.; Wright, R.O. Cadmium Exposure and Neurodevelopmental Outcomes in U.S. Children. Environ. Health Perspect. 2012, 120, 758–763. [Google Scholar] [CrossRef]
- Kippler, M.; Tofail, F.; Hamadani, J.D.; Gardner, R.M.; Grantham-McGregor, S.M.; Bottai, M.; Vahter, M. Early-life cadmium exposure and child development in 5-year-old girls and boys: A cohort study in rural bangladesh. Environ. Health Perspect. 2012, 120, 1462–1468. [Google Scholar] [CrossRef]
- Forcella, M.; Lau, P.; Oldani, M.; Melchioretto, P.; Bogni, A.; Gribaldo, L.; Fusi, P.; Urani, C. Neuronal specific and non-specific responses to cadmium possibly involved in neurodegeneration: A toxicogenomics study in a human neuronal cell model. Neurotoxicology 2020, 76, 162–173. [Google Scholar] [CrossRef] [PubMed]
- Raj, K.; Kaur, P.; Gupta, G.D.; Singh, S. Metals associated neurodegeneration in Parkinson’s disease: Insight to physiological, pathological mechanisms and management. Neurosci. Lett. 2021, 753, 135873. [Google Scholar] [CrossRef] [PubMed]
- Min, J.; Min, K. Blood cadmium levels and Alzheimer’s disease mortality risk in older US adults. Environ. Health 2016, 15, 69. [Google Scholar] [CrossRef]
- Branca, J.J.V.; Morucci, G.; Pacini, A. Cadmium-induced neurotoxicity: Still much ado. Neural Regen. Res. 2018, 13, 1879–1882. [Google Scholar] [PubMed]
- Witkowska, D.; Słowik, J.; Chilicka, K. Heavy Metals and Human Health: Possible Exposure Pathways and the Competition for Protein Binding Sites. Molecules 2021, 26, 6060. [Google Scholar] [CrossRef]
- Tjälve, H.; Henriksson, J.; Tallkvist, J.; Larsson, B.S.; Lindquist, N.G. Uptake of Manganese and Cadmium from the Nasal Mucosa into the Central Nervous System via Olfactory Pathways in Rats. Pharmacol. Toxicol. 1996, 79, 347–356. [Google Scholar] [CrossRef]
- Choong, G.; Liu, Y.; Templeton, D.M. Interplay of calcium and cadmium in mediating cadmium toxicity. Chem.-Biol. Interact. 2014, 211, 54–65. [Google Scholar] [CrossRef]
- Usai, C.; Barberis, A.; Moccagatta, L.; Marchetti, C. Pathways of Cadmium Influx in Mammalian Neurons. J. Neurochem. 1999, 72, 2154–2161. [Google Scholar] [CrossRef]
- Braga, M.F.M.; Rowan, E.G. Reversal by cysteine of the cadmium-induced block of skeletal neuromuscular transmission in vitro. Br. J. Pharmacol. 1992, 107, 95–100. [Google Scholar] [CrossRef]
- Minami, A.; Takeda, A.; Nishibaba, D.; Takefuta, S.; Oku, N. Cadmium toxicity in synaptic neurotransmission in the brain. Brain Res. 2001, 894, 336. [Google Scholar] [CrossRef]
- Thévenod, F.; Schulz, I. H+-dependent calcium uptake into an IP3-sensitive calcium pool from rat parotid gland. Am. J. Physiol.-Gastrointest. Liver Physiol. 1988, 255, 429–440. [Google Scholar] [CrossRef] [PubMed]
- Tsentsevitsky, A.; Petrov, A. Synaptic mechanisms of cadmium neurotoxicity. Neural Regen. Res. 2021, 16, 1762–1763. [Google Scholar] [PubMed]
- Mimouna, S.B.; Chemek, M.; Boughammoura, S.; Banni, M.; Messaoudi, I. Early-Life Exposure to Cadmium Triggers Distinct Zn-Dependent Protein Expression Patterns and Impairs Brain Development. Biol. Trace Elem. Res. 2018, 184, 409–421. [Google Scholar] [CrossRef]
- McAllister, B.B.; Dyck, R.H. Zinc transporter 3 (ZnT3) and vesicular zinc in central nervous system function. Neurosci. Biobehav. Rev. 2017, 80, 329–350. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Xiao, G.; Liu, L.; Lang, M. Zinc transporters in Alzheimer’s disease. Mol. Brain 2019, 12, 106. [Google Scholar] [CrossRef]
- Ben Mimouna, S.; Le Charpentier, T.; Lebon, S.; Van Steenwinckel, J.; Messaoudi, I.I.; Gressens, P. Involvement of the synapse-specific zinc transporter ZnT3 in cadmium-induced hippocampal neurotoxicity. J. Cell. Physiol. 2019, 234, 15872. [Google Scholar] [CrossRef]
- Rolfe, D.F.; Brown, G.C. Cellular energy utilization and molecular origin of standard metabolic rate in mammals. Physiol. Rev. 1997, 77, 731–758. [Google Scholar] [CrossRef]
- Brand, M.D.; Orr, A.L.; Perevoshchikova, I.V.; Quinlan, C.L. The role of mitochondrial function and cellular bioenergetics in ageing and disease. Br. J. Dermatol. 2013, 169 (Suppl. S2), 1–8. [Google Scholar] [CrossRef]
- Zorova, L.D.; Popkov, V.A.; Plotnikov, E.Y.; Silachev, D.N.; Pevzner, I.B.; Jankauskas, S.S.; Babenko, V.A.; Zorov, S.D.; Balakireva, A.V.; Juhaszova, M.; et al. Mitochondrial membrane potential. Anal. Biochem. 2018, 552, 50–59. [Google Scholar] [CrossRef]
- Belyaeva, E.; Glazunov, V.; Korotkov, S. Cd2-promoted mitochondrial permeability transition: A comparison with other heavy metals. Acta Biochim. Pol. 2004, 51, 545–551. [Google Scholar] [CrossRef]
- Branca, J.J.V.; Pacini, A.; Gulisano, M.; Taddei, N.; Fiorillo, C.; Becatti, M. Cadmium-Induced Cytotoxicity: Effects on Mitochondrial Electron Transport Chain. Front. Cell Dev. Biol. 2020, 8, 604377. [Google Scholar] [CrossRef] [PubMed]
- Xu, S.; Pi, H.; Chen, Y.; Zhang, N.; Guo, P.; Lu, Y.; He, M.; Xie, J.; Zhong, M.; Zhang, Y.; et al. Cadmium induced Drp1-dependent mitochondrial fragmentation by disturbing calcium homeostasis in its hepatotoxicity. Cell Death Dis. 2013, 4, e540. [Google Scholar] [CrossRef] [PubMed]
- Weidemann, M.J.; Erdelt, H.; Klingenberg, M. Adenine Nucleotide Translocation of Mitochondria. Eur. J. Biochem. 1970, 16, 313–335. [Google Scholar] [CrossRef] [PubMed]
- Appleby, R.D.; Porteous, W.K.; Hughes, G.; James, A.M.; Shannon, D.; Wei, Y.; Murphy, M.P. Quantitation and origin of the mitochondrial membrane potential in human cells lacking mitochondrial DNA. Eur. J. Biochem. 1999, 262, 108–116. [Google Scholar] [CrossRef]
- Li, M.; Xia, T.; Jiang, C.; Li, L.; Fu, J.; Zhou, Z. Cadmium directly induced the opening of membrane permeability pore of mitochondria which possibly involved in cadmium-triggered apoptosis. Toxicology 2003, 194, 19–33. [Google Scholar] [CrossRef]
- Bround, M.J.; Bers, D.M.; Molkentin, J.D. A 20/20 view of ANT function in mitochondrial biology and necrotic cell death. J. Mol. Cell Cardiol. 2020, 144, A3–A13. [Google Scholar] [CrossRef]
- Loeffler, M.; Kroemer, G. The mitochondrion in cell death control: Certainties and incognita. Exp. Cell Res. 2000, 256, 19–26. [Google Scholar] [CrossRef]
- Morishima, N.; Nakanishi, K.; Takenouchi, H.; Shibata, T.; Yasuhiko, Y. An endoplasmic reticulum stress-specific caspase cascade in apoptosis: Cytochrome c-independent activation of caspase-9 by caspase-12. J. Biol. Chem. 2002, 277, 34287–34294. [Google Scholar] [CrossRef]
- Kim, S.; Cheon, H.; Kim, S.; Juhnn, Y.; Kim, Y. Cadmium induces neuronal cell death through reactive oxygen species activated by GADD153. BMC Cell Biol. 2013, 14, 4. [Google Scholar] [CrossRef]
- Ji, Y.; Wang, H.; Zhao, X.; Wang, Q.; Zhang, C.; Zhang, Y.; Zhao, M.; Chen, Y.; Meng, X.; Xu, D. Crosstalk between endoplasmic reticulum stress and mitochondrial pathway mediates cadmium-induced germ cell apoptosis in testes. Toxicol. Sci. 2011, 124, 446–459. [Google Scholar] [CrossRef]
- Nakagawa, T.; Zhu, H.; Morishima, N.; Li, E.; Xu, J.; Yankner, B.A.; Yuan, J. Caspase-12 mediates endoplasmic-reticulum-specific apoptosis and cytotoxicity by amyloid-β. Nature 2000, 403, 98–103. [Google Scholar] [CrossRef] [PubMed]
- Shati, A.A. Resveratrol protects against cadmium chloride-induced hippocampal neurotoxicity by inhibiting ER stress and GAAD 153 and activating sirtuin 1/AMPK/Akt. Environ. Toxicol. 2019, 34, 1340–1353. [Google Scholar] [CrossRef] [PubMed]
- Rapizzi, E.; Pinton, P.; Szabadkai, G.; Wieckowski, M.R.; Vandecasteele, G.; Baird, G.; Tuft, R.A.; Fogarty, K.E.; Rizzuto, R. Recombinant expression of the voltage-dependent anion channel enhances the transfer of Ca2+ microdomains to mitochondria. J. Cell Biol. 2002, 159, 613–624. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Zhu, Q.; Cao, B.; Cai, Y.; Wen, S.; Bian, J.; Zou, H.; Song, R.; Gu, J.; Liu, X. Ca2+ transfer via the ER-mitochondria tethering complex in neuronal cells contribute to cadmium-induced autophagy. Cell Biol. Toxicol. 2021, 38, 469–485. [Google Scholar] [CrossRef]
- Ghavami, S.; Shojaei, S.; Yeganeh, B.; Ande, S.R.; Jangamreddy, J.R.; Mehrpour, M.; Christoffersson, J.; Chaabane, W.; Moghadam, A.R.; Kashani, H.H.; et al. Autophagy and apoptosis dysfunction in neurodegenerative disorders. Prog. Neurobiol. 2014, 112, 24–49. [Google Scholar] [CrossRef]
- Chatterjee, S.; Sarkar, S.; Bhattacharya, S. Toxic metals and autophagy. Chem. Res. Toxicol. 2014, 27, 1887–1900. [Google Scholar] [CrossRef]
- Wang, T.; Wang, Q.; Song, R.; Zhang, Y.; Zhang, K.; Yuan, Y.; Bian, J.; Liu, X.; Gu, J.; Liu, Z. Autophagy plays a cytoprotective role during cadmium-induced oxidative damage in primary neuronal cultures. Biol. Trace Elem. Res. 2015, 168, 481–489. [Google Scholar] [CrossRef]
- Chen, Y.; McMillan-Ward, E.; Kong, J.; Israels, S.J.; Gibson, S.B. Oxidative stress induces autophagic cell death independent of apoptosis in transformed and cancer cells. Cell Death Differ. 2008, 15, 171–182. [Google Scholar] [CrossRef]
- Zhang, H.; Dong, X.; Zhao, R.; Zhang, R.; Xu, C.; Wang, X.; Liu, C.; Hu, X.; Huang, S.; Chen, L. Cadmium results in accumulation of autophagosomes-dependent apoptosis through activating Akt-impaired autophagic flux in neuronal cells. Cell Signal 2019, 55, 26–39. [Google Scholar] [CrossRef]
- Zhang, L.; Xia, Q.; Zhou, Y.; Li, J. Endoplasmic reticulum stress and autophagy contribute to cadmium-induced cytotoxicity in retinal pigment epithelial cells. Toxicol. Lett. 2019, 311, 105–113. [Google Scholar] [CrossRef]
- Xu, C.; Liu, C.; Liu, L.; Zhang, R.; Zhang, H.; Chen, S.; Luo, Y.; Chen, L.; Huang, S. Rapamycin prevents cadmium-induced neuronal cell death via targeting both mTORC1 and mTORC2 pathways. Neuropharmacology 2015, 97, 35–45. [Google Scholar] [CrossRef] [PubMed]
- Xu, C.; Chen, S.; Xu, M.; Chen, X.; Wang, X.; Zhang, H.; Dong, X.; Zhang, R.; Chen, X.; Gao, W.; et al. Cadmium Impairs Autophagy Leading to Apoptosis by Ca2+-Dependent Activation of JNK Signaling Pathway in Neuronal Cells. Neurochem. Res. 2021, 46, 2033–2045. [Google Scholar] [CrossRef] [PubMed]
- Hu, T.; Wei, X.; Zhang, X.; Cheng, F.; Shuai, X.; Zhang, L.; Kang, L. Protective effect of Potentilla anserine polysaccharide (PAP) on hydrogen peroxide induced apoptosis in murine splenic lymphocytes. Carbohydr. Polym. 2010, 79, 356–361. [Google Scholar] [CrossRef]
- QIN, X.; LI, L.; Qi, L.V.; YU, B.; YANG, S.; Tao, H.E.; ZHANG, Y. Neuroprotection of n-butanol extract from roots of Potentilla anserina on hypoxic injury in primary hippocampal neurons. Chin. Herb. Med. 2012, 4, 195–200. [Google Scholar]
- Shuai, X.; Hu, T.; Zhang, X.; Cheng, F.; Chen, J. Inhibitory action of Potentilla anserine polysaccharide fraction on H2O2-induced apoptosis of murine splenic lymphocytes. Yao Xue Xue Bao = Acta Pharm. Sin. 2009, 44, 987–993. [Google Scholar]
- Shen, R.; Liu, D.; Hou, C.; Liu, D.; Zhao, L.; Cheng, J.; Wang, D.; Bai, D. Protective effect of Potentilla anserina polysaccharide on cadmium-induced nephrotoxicity in vitro and in vivo. Food Funct. 2017, 8, 3636–3646. [Google Scholar] [CrossRef]
- Cheng, J.; Liu, D.; Zhao, L.; Zhao, Q.; Zhang, X.; Wang, B.; Bai, D. Potentilla anserine L. polysaccharide inhibits cadmium-induced neurotoxicity by attenuating autophagy. Neurochem. Int. 2021, 147, 105045. [Google Scholar] [CrossRef]
- Ide, M.; Sonoda, N.; Inoue, T.; Kimura, S.; Minami, Y.; Makimura, H.; Hayashida, E.; Hyodo, F.; Yamato, M.; Takayanagi, R. The dipeptidyl peptidase-4 inhibitor, linagliptin, improves cognitive impairment in streptozotocin-induced diabetic mice by inhibiting oxidative stress and microglial activation. PLoS ONE 2020, 15, e0228750. [Google Scholar] [CrossRef]
- Siddiqui, N.; Ali, J.; Parvez, S.; Zameer, S.; Najmi, A.K.; Akhtar, M. Linagliptin, a DPP-4 inhibitor, ameliorates Aβ (1− 42) peptides induced neurodegeneration and brain insulin resistance (BIR) via insulin receptor substrate-1 (IRS-1) in rat model of Alzheimer’s disease. Neuropharmacology 2021, 195, 108662. [Google Scholar] [CrossRef]
- Arab, H.H.; Eid, A.H.; Alsufyani, S.E.; Ashour, A.M.; El-Sheikh, A.A.K.; Darwish, H.W.; Georgy, G.S. Neuroprotective Impact of Linagliptin against Cadmium-Induced Cognitive Impairment and Neuropathological Aberrations: Targeting SIRT1/Nrf2 Axis, Apoptosis, and Autophagy. Pharmaceuticals 2023, 16, 1065. [Google Scholar] [CrossRef]
- Pagani, M.R.; Reisin, R.C.; Uchitel, O.D. Calcium Signaling Pathways Mediating Synaptic Potentiation Triggered by Amyotrophic Lateral Sclerosis IgG in Motor Nerve Terminals. J. Neurosci. 2006, 26, 2661–2672. [Google Scholar] [CrossRef] [PubMed]
- Adeoye, T.; Shah, S.I.; Demuro, A.; Rabson, D.A.; Ullah, G. Upregulated Ca2+ Release from the Endoplasmic Reticulum Leads to Impaired Presynaptic Function in Familial Alzheimer’s Disease. Cells 2022, 11, 2167. [Google Scholar] [CrossRef] [PubMed]
- Tsentsevitsky, A.N.; Zakyrjanova, G.F.; Petrov, A.M. Cadmium desynchronizes neurotransmitter release in the neuromuscular junction: Key role of ROS. Free Radic. Biol. Med. 2020, 155, 19–28. [Google Scholar] [CrossRef] [PubMed]
- Kaeser, P.S.; Regehr, W.G. Molecular Mechanisms for Synchronous, Asynchronous, and Spontaneous Neurotransmitter Release. Annu. Rev. Physiol. 2014, 76, 333. [Google Scholar] [CrossRef]
- Moriyama, Y.; Maeda, M.; Futai, M. The role of V-ATPase in neuronal and endocrine systems. J. Exp. Biol. 1992, 172, 171–178. [Google Scholar] [CrossRef]
- Kosmidis, E.; Shuttle, C.G.; Preobraschenski, J.; Ganzella, M.; Johnson, P.J.; Veshaguri, S.; Holmkvist, J.; Møller, M.P.; Marantos, O.; Marcoline, F.; et al. Regulation of the mammalian-brain V-ATPase through ultraslow mode-switching. Nature 2022, 611, 827. [Google Scholar] [CrossRef]
- Borisova, T.; Krisanova, N.; Sivko, R.; Kasatkina, L.; Borysov, A.; Griffin, S.; Wireman, M. Presynaptic malfunction: The neurotoxic effects of cadmium and lead on the proton gradient of synaptic vesicles and glutamate transport. Neurochem. Int. 2011, 59, 272–279. [Google Scholar] [CrossRef]
- Moyano, P.; de Frias, M.; Lobo, M.; Anadon, M.J.; Sola, E.; Pelayo, A.; Díaz, M.J.; Frejo, M.T.; Del Pino, J. Cadmium induced ROS alters M1 and M3 receptors, leading to SN56 cholinergic neuronal loss, through AChE variants disruption. Toxicology 2018, 394, 54–62. [Google Scholar] [CrossRef]
- Lee, S.R. Critical Role of Zinc as Either an Antioxidant or a Prooxidant in Cellular Systems. Oxidative Med. Cell. Longev. 2018, 2018, 9156285-11. [Google Scholar] [CrossRef]
- Garfinkel, D. Is aging inevitable?: The intracellular zinc deficiency hypothesis of aging. Med. Hypotheses 1986, 19, 117–137. [Google Scholar] [CrossRef]
- Hong Bin, Q.; Garfinkel, D. The cadmium toxicity hypothesis of aging: A possible explanation for the zinc deficiency hypothesis of aging. Med. Hypotheses 1994, 42, 380–384. [Google Scholar]
- Paoletti, P.; Vergnano, A.M.; Barbour, B.; Casado, M. Zinc at glutamatergic synapses. Neuroscience 2009, 158, 126–136. [Google Scholar] [CrossRef] [PubMed]
- Beyer, N.; Coulson, D.T.R.; Heggarty, S.; Ravid, R.; Brent Irvine, G.; Hellemans, J.; Johnston, J.A. ZnT3 mRNA levels are reduced in Alzheimer’s disease post-mortem brain|Molecular Neurodegeneration|Full Text. Mol. Neurodegener. 2009, 4, 53. [Google Scholar] [CrossRef] [PubMed]
- Adlard, P.A.; Parncutt, J.M.; Finkelstein, D.I.; Bush, A.I. Cognitive loss in zinc transporter-3 knock-out mice: A phenocopy for the synaptic and memory deficits of Alzheimer’s disease? J. Neurosci. 2010, 30, 1631–1636. [Google Scholar] [CrossRef] [PubMed]
- Hajji, N.; Calvert, C.; Ritchie, C.W.; Sastre, M. The Role of Metals in Alzheimer’s Disease. Mech. Met. Involv. Neurodegener. Dis. 2013, 80. [Google Scholar] [CrossRef]
- Cuajungco, M.P.; Lees, G.J. Zinc and Alzheimer’s disease: Is there a direct link? Brain Res. Rev. 1997, 23, 219. [Google Scholar] [CrossRef]
- Atrián-Blasco, E.; Conte-Daban, A.; Hureau, C. Mutual interference of Cu and Zn ions in Alzheimer’s disease: Perspectives at the molecular level. Dalton Trans. 2017, 46, 1275–12759. [Google Scholar] [CrossRef]
- Yuan, Y.; Niu, F.; Liu, Y.; Lu, N. Zinc and its effects on oxidative stress in Alzheimer’s disease. Neurol. Sci. 2014, 35, 923–928. [Google Scholar] [CrossRef]
- Lavanya, R.D.; Reddy, B.S.; Abdul Sattar, S.; Rao, A.D.P. Trace element imbalances in blood serum of Alzheimer’s disease patients. Spectrosc. Lett. 2021, 54, 458–471. [Google Scholar] [CrossRef]
- Li, K.; Li, A.; Mei, Y.; Zhao, J.; Zhou, Q.; Li, Y.; Yang, M.; Xu, Q. Trace elements and Alzheimer dementia in population-based studies: A bibliometric and meta-analysis. Environ. Pollut. 2023, 318, 120782. [Google Scholar] [CrossRef]
- Xu, L.; Zhang, W.; Liu, X.; Zhang, C.; Wang, P.; Zhao, X. Circulatory Levels of Toxic Metals (Aluminum, Cadmium, Mercury, Lead) in Patients with Alzheimer’s Disease: A Quantitative Meta-Analysis and Systematic Review. J. Alzheimer’s Dis. 2018, 62, 361–372. [Google Scholar] [CrossRef] [PubMed]
- Jin, W. Regulation of BDNF-TrkB Signaling and Potential Therapeutic Strategies for Parkinson’s Disease. J. Clin. Med. 2020, 9, 257. [Google Scholar] [CrossRef] [PubMed]
- Syme, C.D.; Nadal, R.C.; Rigby, S.E.J.; John, H. Viles Copper Binding to the Amyloid-beta (abeta) Peptide Associated with Alzheimer’s Disease. J. Biol. Chem. 2004, 279, 18169. [Google Scholar] [CrossRef] [PubMed]
- Boopathi, S.; Kolandaivel, P. Fe2+ binding on amyloid [beta]-peptide promotes aggregation. Proteins Struct. Funct. Bioinform. 2016, 84, 1257. [Google Scholar] [CrossRef]
- Ward, R.J.; Zucca, F.A.; Duyn, J.H.; Crichton, R.R.; Zecca, L. The role of iron in brain ageing and neurodegenerative disorders. Lancet Neurol. 1045, 13, 1045. [Google Scholar] [CrossRef]
- Bauer, H.; Krizbai, I.A.; Bauer, H.; Traweger, A. “You Shall Not Pass”–tight junctions of the blood brain barrier. Front. Neurosci. 2014, 8, 392. [Google Scholar] [CrossRef]
- Luissint, A.; Artus, C.; Glacial, F.; Ganeshamoorthy, K.; Couraud, P. Tight junctions at the blood brain barrier: Physiological architecture and disease-associated dysregulation|Fluids and Barriers of the CNS|Full Text. Fluids Barriers CNS 2012, 9, 23. [Google Scholar] [CrossRef]
- Wang, L.; Xiong, X.; Zhang, L.; Shen, J. Neurovascular Unit: A critical role in ischemic stroke. CNS Neurosci. Ther. 2021, 27, 7–16. [Google Scholar] [CrossRef]
- Mendez-Armenta, M.; Rios, C. Cadmium neurotoxicity. Environ. Toxicol. Pharmacol. 2007, 23, 350–358. [Google Scholar] [CrossRef]
- Rai, A.; Maurya, S.K.; Khare, P.; Srivastava, A.; Bandyopadhyay, S. Characterization of Developmental Neurotoxicity of As, Cd, and Pb Mixture: Synergistic Action of Metal Mixture in Glial and Neuronal Functions. Toxicol. Sci. 2010, 118, 586–601. [Google Scholar] [CrossRef]
- Carrino, D.; Branca, J.J.V.; Becatti, M.; Paternostro, F.; Morucci, G.; Gulisano, M.; Di Cesare Mannelli, L.; Pacini, A. Alcohol-Induced Blood-Brain Barrier Impairment: An In Vitro Study. Int. J. Environ. Res. Public Health 2021, 18, 2683. [Google Scholar] [CrossRef] [PubMed]
- Pal, R.; Nath, R.; Dipgill, K. Influence of ethanol on cadmium accumulation and its impact on lipid peroxidation and membrane bound functional enzymes (Na+, K+-ATPASE and acetylcholinesterase) in various regions of adult rat brain. Neurochem. Int. 1993, 23, 451. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; Xu, Z.; Wen, L.; Lei, D.; Li, S.; Wang, J.; Huang, J.; Wang, N.; Durkan, C.; Liao, X.; et al. Cadmium-induced dysfunction of the blood-brain barrier depends on ROS-mediated inhibition of PTPase activity in zebrafish. J. Hazard. Mater. 2021, 412, 125198. [Google Scholar] [CrossRef] [PubMed]
- Branca, J.J.V.; Maresca, M.; Morucci, G.; Mello, T.; Becatti, M.; Pazzagli, L.; Colzi, I.; Gonnelli, C.; Carrino, D.; Paternostro, F.; et al. Effects of Cadmium on ZO-1 Tight Junction Integrity of the Blood Brain Barrier. Int. J. Mol. Sci. 2019, 20, 6010. [Google Scholar] [CrossRef] [PubMed]
- Lohmann, C.; Krischke, M.; Wegener, J.; Galla, H. Tyrosine phosphatase inhibition induces loss of blood–brain barrier integrity by matrix metalloproteinase-dependent and -independent pathways. Brain Res. 2004, 995, 184–196. [Google Scholar] [CrossRef]
- Kim, S.; Cheon, H.; Kim, S.; Kim, Y. GSK-3β-mediated regulation of cadmium-induced cell death and survival. Cell Mol. Biol. Lett. 2018, 23, 9. [Google Scholar] [CrossRef]
- Maier, J.; Locatelli, L.; Fedele, G.; Cazzaniga, A.; Mazur, A. Magnesium and the Brain: A Focus on Neuroinflammation and Neurodegeneration. Int. J. Mol. Sci. 2022, 24, 223. [Google Scholar] [CrossRef]
- Streit, W.J.; Mrak, R.E.; Griffin, W.S.T. Microglia and neuroinflammation: A pathological perspective|Journal of Neuroinflammation|Full Text. J. Neuroinflamm. 2004, 1, 14. [Google Scholar] [CrossRef]
- DiSabato, D.; Quan, N.; Godbout, J.P. Neuroinflammation: The Devil Is in the Details. J. Neurochem. 2016, 139, 136–153. [Google Scholar] [CrossRef]
- Gilhus, N.E.; Deuschl, G. Neuroinflammation—A common thread in neurological disorders. Nat. Rev. Neurol. 2019, 15, 429–430. [Google Scholar] [CrossRef]
- Wells, E.; Hacohen, Y.; Waldman, A.; Tillema, J.M.; Soldatos, A.; Ances, B.; Benseler, S.; Bielekova, B.; Dale, R.C.; Dalmau, J.; et al. Neuroimmune disorders of the central nervous system in children in the molecular era. Nat. Rev. Neurol. 2018, 14, 433–445. [Google Scholar] [CrossRef] [PubMed]
- Martínez-Hernández, M.I.; Acosta-Saavedra, L.C.; Hernández-Kelly, L.C.; Loaeza-Loaeza, J.; Ortega, A. Microglial Activation in Metal Neurotoxicity: Impact in Neurodegenerative Diseases. Biomed. Res. Int. 2023, 2023, 7389508. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Yang, S.; Qian, S.Y.; Hong, J.; Kadiiska, M.B.; Tennant, R.W.; Waalkes, M.P.; Liu, J. Cadmium-Induced Toxicity in Rat Primary Mid-brain Neuroglia Cultures: Role of Oxidative Stress from Microglia. Toxicol. Sci. 2007, 98, 488–494. [Google Scholar] [CrossRef]
- Khan, A.; Ikram, M.; Muhammad, T.; Park, J.; Kim, M.O. Caffeine Modulates Cadmium-Induced Oxidative Stress, Neuroinflammation, and Cognitive Impairments by Regulating Nrf-2/HO-1 In Vivo and In Vitro. J. Clin. Med. 2019, 8, 680. [Google Scholar] [CrossRef]
- Górska, A.; Markiewicz-Gospodarek, A.; Markiewicz, R.; Chilimoniuk, Z.; Borowski, B.; Trubalski, M.; Czarnek, K. Distribution of Iron, Copper, Zinc and Cadmium in Glia, Their Influence on Glial Cells and Relationship with Neurodegenerative Diseases. Brain Sci. 2023, 13, 911. [Google Scholar] [CrossRef] [PubMed]
- Raichle, M.E.; Posner, J.B.; Plum, F. Cerebral Blood Flow During. Arch. Neurol. 1970, 23, 394–403. [Google Scholar] [CrossRef]
- Mathieu, C.; de la Sierra-Gallay, I.L.; Duval, R.; Xu, X.; Cocaign, A.; Léger, T.; Woffendin, G.; Camadro, J.; Etchebest, C.; Haouz, A.; et al. Insights into Brain Glycogen Metabolism: The Structure of Human Brain Glycogen Phosphorylase. J. Biol. Chem. 2016, 291, 18072–18083. [Google Scholar] [CrossRef]
- Wang, Y.; Ma, K.; Wang, P.; Baba, O.; Zhang, H.; Parent, J.M.; Zheng, P.; Liu, Y.; Minassian, B.A.; Liu, Y. Laforin Prevents Stress-Induced Polyglucosan Body Formation and Lafora Disease Progression in Neurons. Mol. Neurobiol. 2013, 48, 49–61. [Google Scholar] [CrossRef]
- Saez, I.; Duran, J.; Sinadinos, C.; Beltran, A.; Yanes, O.; María, F.T.; Carlos, M.-P.; Marco, M.; Joan, J.G. Neurons Have an Active Glycogen Metabolism that Contributes to Tolerance to Hypoxia. J. Cereb. Blood Flow Metab. 2014, 34, 945–955. [Google Scholar] [CrossRef]
- Tischner, K. Cadmium. In Experimental and Clinical Neurotoxicology, 1st ed.; Oxford Univeristy Press: Oxford, UK, 1980; pp. 345–355. [Google Scholar]
- Mathieu, C.; Dupret, J.; Rodrigues-Lima, F. The Structure and the Regulation of Glycogen Phosphorylases in Brain. In Brain Glycogen Metabolism; DiNuzzo, M., Schousboe, A., Eds.; Springer International Publishing: Cham, Switzerland, 2019; pp. 125–145. [Google Scholar]
- Rai, A.; Singh, P.K.; Singh, V.; Kumar, V.; Mishra, R.; Thakur, A.K.; Mahadevan, A.; Shankar, S.K.; Jana, N.R.; Ganesh, S. Glycogen synthase protects neurons from cytotoxicity of mutant huntingtin by enhancing the autophagy flux. Cell Death Dis. 2018, 9, 201. [Google Scholar] [CrossRef]
- Smith, E.E.; Taylor, P.M.; Whelan, W.J. Enzymic Processes in Glycogen Metabolism. In Carbohydrate Metabolism and Its Disorders; Dickens, F., Whelan, W.J., Randale, P.J., Eds.; Academic Press: London, UK; New York, NY, USA, 1968; Volume I, pp. 89–133. [Google Scholar]
- Lew, C.R.; Guin, S.; Theodorescu, D. Targeting glycogen metabolism in bladder cancer. Nat. Rev. Urol. 2015, 12, 383–391. [Google Scholar] [CrossRef] [PubMed]
- Kielan, Z.; Ziółkowska, B.; Falkus, B.; Jethon, Z. Effect of cadmium intoxication on glucose utilization in energy metabolism of muscles. Acta Physiol. Pol. 1989, 40, 535–543. [Google Scholar] [PubMed]
- Hazelhoff Roelfzema, W.; Hacker, H.J.; Van Noorden, C.J.F. Effects of Cadmium Exposure on Glycogen Phosphorylase Activity in Rat Placenta as Demonstrated by Histochemical Means. Histochemistry 1989, 91, 305–308. [Google Scholar] [CrossRef]
- Miseta, A.; Csutora, P. Relationship Between the Occurrence of Cysteine in Proteins and the Complexity of Organisms. Mol. Biol. Evol. 2000, 17, 1232–1239. [Google Scholar] [CrossRef] [PubMed]
- Smith, C.; Dicaire, M.; Brais, B.; La Piana, R. Neurological Involvement in Glycogen Storage Disease Type IXa due to PHKA2 Mutation. Can. J. Neurol. Sci. 2020, 47, 400–403. [Google Scholar] [CrossRef]
- Muzetti, J.H.; do Valle, D.A.; Santos, M.L.S.F.; Telles, B.A.; Cordeiro, M.L. Neurological Characteristics of Pediatric Glycogen Storage Disease. Front. Endocrinol. 2021, 12, 685272. [Google Scholar] [CrossRef]
- Canibano-Fraile, R.; Harlaar, L.; dos Santos, C.A.; Hoogeveen-Westerveld, M.; Demmers, J.A.A.; Snijders, T.; Lijnzaad, P.; Verdijk, R.M.; van der Beek Nadine, A.M.E.; van Doorn, P.A.; et al. Lysosomal glycogen accumulation in Pompe disease results in disturbed cytoplasmic glycogen metabolism. J. Inherit. Metab. Dis. 2023, 46, 101–115. [Google Scholar] [CrossRef] [PubMed]
- Taylor, K.M.; Meyers, E.; Phipps, M.; Kishnani, P.S.; Cheng, S.H.; Scheule, R.K.; Moreland, R.J. Dysregulation of Multiple Facets of Glycogen Metabolism in a Murine Model of Pompe Disease. PLoS ONE 2013, 8, e56181. [Google Scholar] [CrossRef] [PubMed]
- Khanh, D.N.N.; Vy, N.T.T.; Phuong, T.H.; Nhi, P.T.; Thang, N.Q.; Sy, D.T.; Phuong, N.T.K. Effects of Cadmium and Lead on Muscle and Liver Glycogen Levels of Climbing Perch (Anabas testudineus). Bull. Environ. Contam. Toxicol. 2022, 108, 854–860. [Google Scholar] [CrossRef]
- Dwivedi, K. International Journal of Advanced Research in Biological Sciences Effect of Cadmium on Liver Glycogen Reserve and its Size in Albino Rats. Int. J. Adv. Res. Biol. Sci. 2021, 8, 150–154. [Google Scholar]
- Buha, A.; Đukić-Ćosić, D.; Ćurčić, M.; Bulat, Z.; Antonijević, B.; Moulis, J.; Goumenou, M.; Wallace, D. Emerging Links between Cadmium Exposure and Insulin Resistance: Human, Animal, and Cell Study Data. Toxics 2020, 8, 63. [Google Scholar] [CrossRef] [PubMed]
- Fang, X.; Yu, S.X.; Lu, Y.; Bast, R.C.; Woodgett, J.R.; Mills, G.B. Phosphorylation and inactivation of glycogen synthase kinase 3 by protein kinase A. Proc. Natl. Acad. Sci. USA 2000, 97, 11960–11965. [Google Scholar] [CrossRef] [PubMed]
- Shati, A.A.; Alfaifi, M.Y. Trans-resveratrol Inhibits Tau Phosphorylation in the Brains of Control and Cadmium Chloride-Treated Rats by Activating PP2A and PI3K/Akt Induced-Inhibition of GSK3β. Neurochem. Res. 2019, 44, 357–373. [Google Scholar] [CrossRef] [PubMed]
- De Zwaan, A.; Zandee, D.I. The utilization of glycogen and accumulation of some intermediates during anaerobiosis in Mytilus edulis L. Comp. Biochem. Physiol. Part B Comp. Biochem. 1972, 43, 47–54. [Google Scholar] [CrossRef]
- Lee, B.; Kim, Y. Association of Blood Cadmium Level with Metabolic Syndrome After Adjustment for Confounding by Serum Ferritin and Other Factors: 2008–2012 Korean National Health and Nutrition Examination Survey. Biol. Trace Elem. Res. 2016, 171, 6–16. [Google Scholar] [CrossRef]
- Filippini, T.; Wise, L.A.; Vinceti, M. Cadmium exposure and risk of diabetes and prediabetes: A systematic review and dose-response meta-analysis. Environ. Int. 2022, 158, 106920. [Google Scholar] [CrossRef]
- Gong, P.; Chang, X.; Chen, X.; Bai, X.; Wen, H.; Pi, S.; Yang, W.; Wang, L.; Chen, F. Metabolomics study of cadmium-induced diabetic nephropathy and protective effect of caffeic acid phenethyl ester using UPLC-Q-TOF-MS combined with pattern recognition. Environ. Toxicol. Pharmacol. 2017, 54, 80–92. [Google Scholar] [CrossRef]
- Bak, L.K.; Walls, A.B.; Schousboe, A.; Waagepetersen, H.S. Astrocytic glycogen metabolism in the healthy and diseased brain. J. Biol. Chem. 2018, 293, 7108–7116. [Google Scholar] [CrossRef]
- Islam, F.; Shohag, S.; Akhter, S.; Islam, M.R.; Sultana, S.; Mitra, S.; Chandran, D.; Khandaker, M.U.; Ashraf, G.M.; Idris, A.M.; et al. Exposure of metal toxicity in Alzheimer’s disease: An extensive review. Front. Pharmacol. 2022, 13, 903099. [Google Scholar] [CrossRef]
- Duran, J.; Gruart, A.; García-Rocha, M.; Delgado-García, J.M.; Guinovart, J.J. Glycogen accumulation underlies neurodegeneration and autophagy impairment in Lafora disease. Hum. Mol. Genet. 2014, 23, 3147–3156. [Google Scholar] [CrossRef]
- Huat, T.J.; Camats-Perna, J.; Newcombe, E.A.; Valmas, N.; Kitazawa, M.; Medeiros, R. Metal Toxicity Links to Alzheimer’s Disease and Neuroinflammation. J. Mol. Biol. 2019, 431, 1843–1868. [Google Scholar] [CrossRef] [PubMed]
- Lavoie, S.; Allaman, I.; Petit, J.; Do, K.Q.; Magistretti, P.J. Altered Glycogen Metabolism in Cultured Astrocytes from Mice with Chronic Glutathione Deficit; Relevance for Neuroenergetics in Schizophrenia. PLoS ONE 2011, 6, e22875. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Zhu, H.; Wang, K.; Yang, Z.; Liu, Z. Protective effect of quercetin on rat testes against cadmium toxicity by alleviating oxidative stress and autophagy. Environ. Sci. Pollut. Res. 2020, 27, 25278–25286. [Google Scholar] [CrossRef] [PubMed]
- Alshammari, G.M.; Al-Qahtani, W.H.; Alshuniaber, M.A.; Yagoub, A.E.A.; Al-Khalifah, A.S.; Al-Harbi, L.N.; Alhussain, M.H.; AlSedairy, S.A.; Yahya, M.A. Quercetin improves the impairment in memory function and attenuates hippocampal damage in cadmium chloride-intoxicated male rats by suppressing acetylcholinesterase and concomitant activation of SIRT1 signaling. J. Funct. Foods 2021, 86, 104675. [Google Scholar] [CrossRef]
- Kini, R.D.; Arunkumar, N.; Gokul, M. Potential Protective Role of Beta Carotene on Cadmium Induced Brain and Kidney Damage. Indian J. Public Health Res. Dev. 2019, 10, 532–535. [Google Scholar] [CrossRef]
- Wiberg, E.; Wiberg, N. Inorganic Chemistry; Academic Press: Cambridge, MA, USA, 2001. [Google Scholar]
- Cranton, E.M.; Zheng, X.L.; Smith, I.M. Urinary trace and toxic elements and minerals in untimed urine specimens relative to urine creatinine. J. Adv. Med. 1989, 1, 331–397. [Google Scholar]
- Waters, R.S.; Bryden, N.A.; Patterson, K.Y.; Veillon, C.; Anderson, R.A. EDTA chelation effects on urinary losses of cadmium, calcium, chromium, cobalt, copper, lead, magnesium, and zinc. Biol. Trace Elem. Res. 2001, 83, 207–221. [Google Scholar] [CrossRef]
- Fulgenzi, A.; Vietti, D.; Ferrero, M.E. EDTA Chelation Therapy in the Treatment of Neurodegenerative Diseases: An Update. Biomedicines 2020, 8, 269. [Google Scholar] [CrossRef]
- Mostafa, D.G.; Khaleel, E.F.; Badi, R.M.; Abdel-Aleem, G.; Abdeen, H.M. Rutin hydrate inhibits apoptosis in the brains of cadmium chloride-treated rats via preserving the mitochondrial integrity and inhibiting endoplasmic reticulum stress. Neurol. Res. 2019, 41, 594–608. [Google Scholar] [CrossRef]
- Bjørklund, G.; Crisponi, G.; Nurchi, V.M.; Cappai, R.; Buha Djordjevic, A.; Aaseth, J. A Review on Coordination Properties of Thiol-Containing Chelating Agents Towards Mercury, Cadmium, and Lead. Molecules 2019, 24, 3247. [Google Scholar] [CrossRef]
- Rahimzadeh, M.R.; Rahimzadeh, M.R.; Kazemi, S.; Moghadamnia, A.A. Review Article Cadmium toxicity and treatment: An update. Casp. J. Intern. Med. 2017, 8, 135. [Google Scholar]
- Harris, C.J.; Voss, K.; Murchison, C.; Ralle, M.; Frahler, K.; Carter, R.; Rhoads, A.; Lind, B.; Robinson, E.; Quinn, J.F. Oral Zinc Reduces Amyloid Burden in Tg2576 Mice. J. Alzheimer’s Dis. 2014, 41, 179–192. [Google Scholar] [CrossRef] [PubMed]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Arruebarrena, M.A.; Hawe, C.T.; Lee, Y.M.; Branco, R.C. Mechanisms of Cadmium Neurotoxicity. Int. J. Mol. Sci. 2023, 24, 16558. https://doi.org/10.3390/ijms242316558
Arruebarrena MA, Hawe CT, Lee YM, Branco RC. Mechanisms of Cadmium Neurotoxicity. International Journal of Molecular Sciences. 2023; 24(23):16558. https://doi.org/10.3390/ijms242316558
Chicago/Turabian StyleArruebarrena, Madelyn A., Calvin T. Hawe, Young Min Lee, and Rachel C. Branco. 2023. "Mechanisms of Cadmium Neurotoxicity" International Journal of Molecular Sciences 24, no. 23: 16558. https://doi.org/10.3390/ijms242316558