
Complement System and the Kidney: Its Role in Renal Diseases, Kidney Transplantation and Renal Cell Carcinoma
 , , , , and
, , , , and 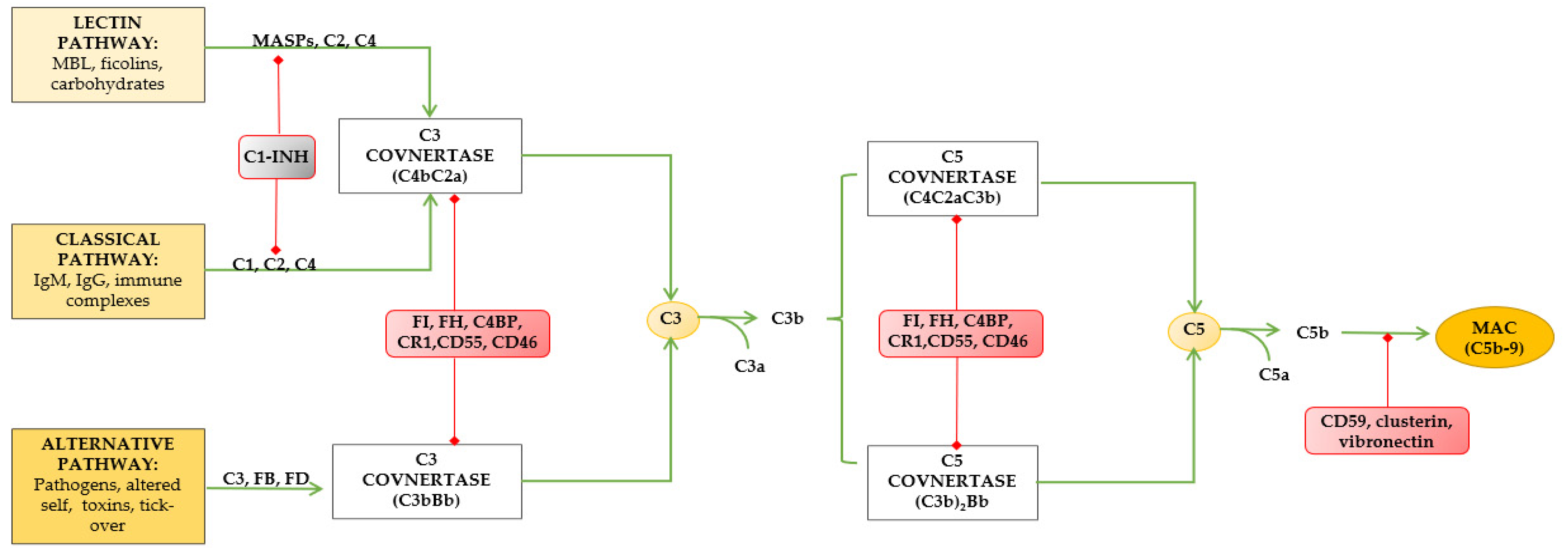{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Complement System (Components, Activation Cascades, Regulators, Local Activation)
3. Complosome: Definition and Roles
4. Complement and Kidney Diseases
5. Complement and Kidney Transplantation
6. Overview of Complement System in Cancer
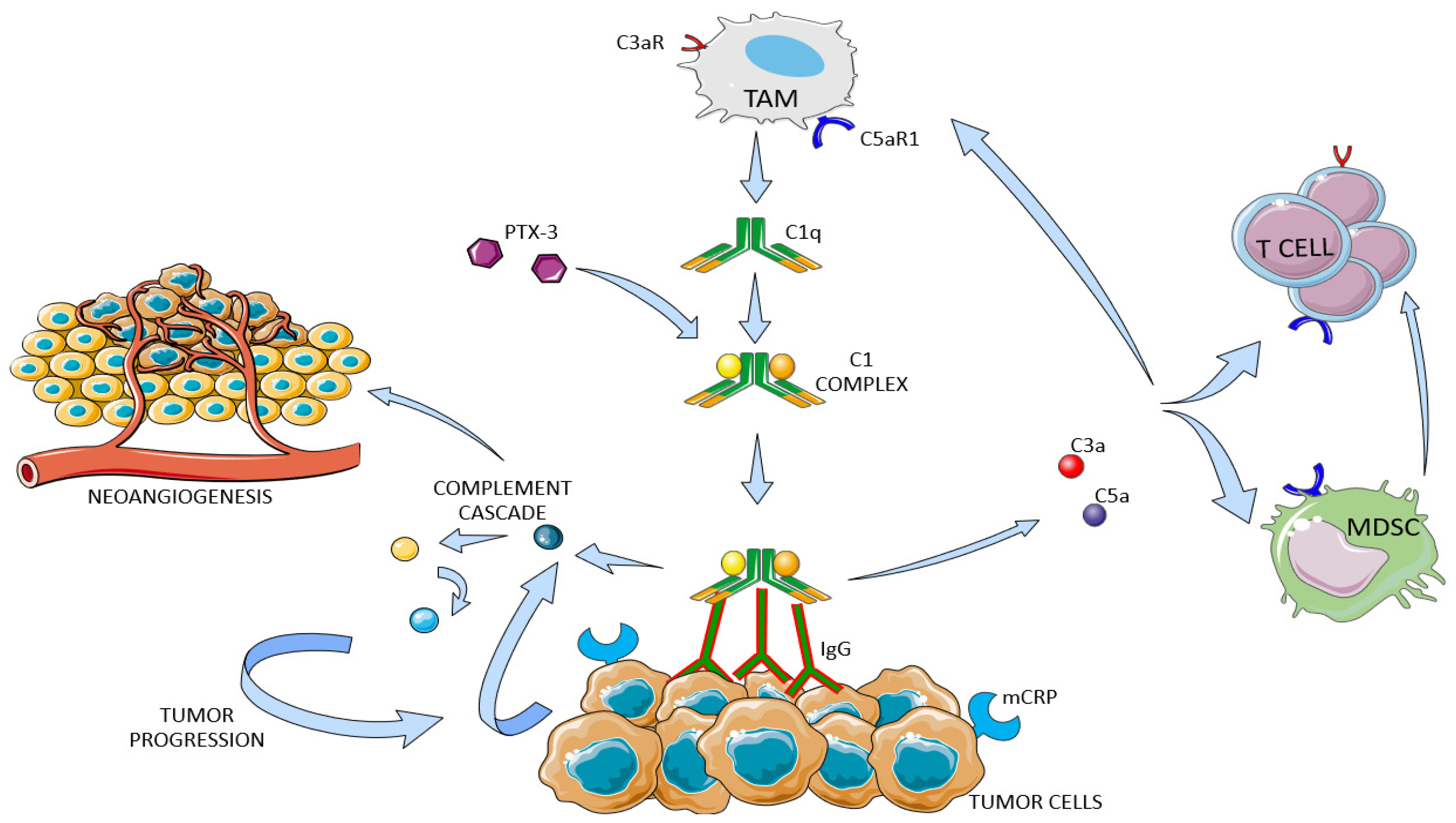
7. Complement System in Kidney Cancer
8. Clinical Applications
9. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Walport, M.J. Complement. First od two parts. N. Engl. J. Med. 2001, 344, 1058–1066. [Google Scholar] [CrossRef] [PubMed]
- Walport, M.J. Complement. Secondo of two parts. N. Engl. J. Med. 2001, 344, 1140–1144. [Google Scholar] [CrossRef] [PubMed]
- West, E.E.; Kolev, M.; Kemper, C. Complement and the Regulation of T Cell Responses. Annu. Rev. Immunol. 2018, 36, 309–338. [Google Scholar] [CrossRef] [PubMed]
- Killick, J.; Morisse, G.; Sieger, D.; Astier, A.L. Complement as a Regulator of Adaptive Immunity. Semin. Immunopathol. 2018, 40, 37–48. [Google Scholar] [CrossRef]
- Lubbers, R.; van Essen, M.F.; van Kooten, C.; Trouw, L.A. Production of Complement Components by Cells of the Immune System. Clin. Exp. Immunol. 2017, 188, 183–194. [Google Scholar] [CrossRef]
- Leonardi, L.; La Torre, F.; Soresina, A.; Federici, S.; Cancrini, C.; Castagnoli, R.; Cinicola, B.L.; Corrente, S.; Giardino, G.; Lougaris, V.; et al. Inherited Defects in the Complement System. Pediatr. Allergy Immunol. 2022, 33 (Suppl. 27), 73–76. [Google Scholar] [CrossRef]
- Defendi, F.; Thielens, N.M.; Clavarino, G.; Cesbron, J.-Y.; Dumestre-Pérard, C. The Immunopathology of Complement Proteins and Innate Immunity in Autoimmune Disease. Clin. Rev. Allergy Immunol. 2020, 58, 229–251. [Google Scholar] [CrossRef]
- Petr, V.; Thurman, J.M. The Role of Complement in Kidney Disease. Nat. Rev. Nephrol. 2023, 19, 771–787. [Google Scholar] [CrossRef]
- Freiwald, T.; Afzali, B. Renal Diseases and the Role of Complement: Linking Complement to Immune Effector Pathways and Therapeutics. Adv. Immunol. 2021, 152, 1–81. [Google Scholar] [CrossRef] [PubMed]
- Thurman, J.M. Complement and the Kidney: An Overview. Adv. Chronic Kidney Dis. 2020, 27, 86–94. [Google Scholar] [CrossRef]
- Revel, M.; Daugan, M.; Sautés-Fridman, C.; Fridman, W.; Roumenina, L. Complement System: Promoter or Suppressor of Cancer Progression? Antibodies 2020, 9, 57. [Google Scholar] [CrossRef] [PubMed]
- Afshar-Kharghan, V. The Role of the Complement System in Cancer. J. Clin. Investig. 2017, 127, 780–789. [Google Scholar] [CrossRef] [PubMed]
- Rutkowski, M.J.; Sughrue, M.E.; Kane, A.J.; Mills, S.A.; Parsa, A.T. Cancer and the Complement Cascade. Mol. Cancer Res. 2010, 8, 1453–1465. [Google Scholar] [CrossRef] [PubMed]
- Bukavina, L.; Bensalah, K.; Bray, F.; Carlo, M.; Challacombe, B.; Karam, J.A.; Kassouf, W.; Mitchell, T.; Montironi, R.; O’Brien, T.; et al. Epidemiology of Renal Cell Carcinoma: 2022 Update. Eur. Urol. 2022, 82, 529–542. [Google Scholar] [CrossRef] [PubMed]
- Battaglia, M.; Lucarelli, G. The Role of Renal Surgery in the Era of Targeted Therapy: The Urologist’s Perspective. Urologia 2015, 82, 137–138. [Google Scholar] [CrossRef]
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer Statistics, 2022. CA A Cancer J Clin. 2022, 72, 7–33. [Google Scholar] [CrossRef]
- Lucarelli, G.; Rutigliano, M.; Sanguedolce, F.; Galleggiante, V.; Giglio, A.; Cagiano, S.; Bufo, P.; Maiorano, E.; Ribatti, D.; Ranieri, E.; et al. Increased Expression of the Autocrine Motility Factor Is Associated With Poor Prognosis in Patients With Clear Cell-Renal Cell Carcinoma. Medicine 2015, 94, e2117. [Google Scholar] [CrossRef]
- Lucarelli, G.; Rutigliano, M.; Sallustio, F.; Ribatti, D.; Giglio, A.; Lepore Signorile, M.; Grossi, V.; Sanese, P.; Napoli, A.; Maiorano, E.; et al. Integrated Multi-Omics Characterization Reveals a Distinctive Metabolic Signature and the Role of NDUFA4L2 in Promoting Angiogenesis, Chemoresistance, and Mitochondrial Dysfunction in Clear Cell Renal Cell Carcinoma. Aging 2018, 10, 3957–3985. [Google Scholar] [CrossRef]
- Lucarelli, G.; Ferro, M.; Loizzo, D.; Bianchi, C.; Terracciano, D.; Cantiello, F.; Bell, L.N.; Battaglia, S.; Porta, C.; Gernone, A.; et al. Integration of Lipidomics and Transcriptomics Reveals Reprogramming of the Lipid Metabolism and Composition in Clear Cell Renal Cell Carcinoma. Metabolites 2020, 10, 509. [Google Scholar] [CrossRef]
- Lucarelli, G.; Galleggiante, V.; Rutigliano, M.; Sanguedolce, F.; Cagiano, S.; Bufo, P.; Lastilla, G.; Maiorano, E.; Ribatti, D.; Giglio, A.; et al. Metabolomic Profile of Glycolysis and the Pentose Phosphate Pathway Identifies the Central Role of Glucose-6-Phosphate Dehydrogenase in Clear Cell-Renal Cell Carcinoma. Oncotarget 2015, 6, 13371–13386. [Google Scholar] [CrossRef]
- Lucarelli, G.; Ferro, M.; Battaglia, M. Multi-Omics Approach Reveals the Secrets of Metabolism of Clear Cell-Renal Cell Carcinoma. Transl. Androl. Urol. 2016, 5, 801–803. [Google Scholar] [CrossRef] [PubMed]
- di Meo, N.A.; Lasorsa, F.; Rutigliano, M.; Loizzo, D.; Ferro, M.; Stella, A.; Bizzoca, C.; Vincenti, L.; Pandolfo, S.D.; Autorino, R.; et al. Renal Cell Carcinoma as a Metabolic Disease: An Update on Main Pathways, Potential Biomarkers, and Therapeutic Targets. Int. J. Mol. Sci. 2022, 23, 14360. [Google Scholar] [CrossRef]
- Ragone, R.; Sallustio, F.; Piccinonna, S.; Rutigliano, M.; Vanessa, G.; Palazzo, S.; Lucarelli, G.; Ditonno, P.; Battaglia, M.; Fanizzi, F.P.; et al. Renal Cell Carcinoma: A Study through NMR-Based Metabolomics Combined with Transcriptomics. Diseases 2016, 4, 7. [Google Scholar] [CrossRef] [PubMed]
- di Meo, N.A.; Lasorsa, F.; Rutigliano, M.; Milella, M.; Ferro, M.; Battaglia, M.; Ditonno, P.; Lucarelli, G. The Dark Side of Lipid Metabolism in Prostate and Renal Carcinoma: Novel Insights into Molecular Diagnostic and Biomarker Discovery. Expert. Rev. Mol. Diagn. 2023, 23, 297–313. [Google Scholar] [CrossRef] [PubMed]
- Bianchi, C.; Meregalli, C.; Bombelli, S.; Di Stefano, V.; Salerno, F.; Torsello, B.; De Marco, S.; Bovo, G.; Cifola, I.; Mangano, E.; et al. The Glucose and Lipid Metabolism Reprogramming Is Grade-Dependent in Clear Cell Renal Cell Carcinoma Primary Cultures and Is Targetable to Modulate Cell Viability and Proliferation. Oncotarget 2017, 8, 113502–113515. [Google Scholar] [CrossRef]
- Lucarelli, G.; Ferro, M.; Ditonno, P.; Battaglia, M. The Urea Cycle Enzymes Act as Metabolic Suppressors in Clear Cell Renal Cell Carcinoma. Transl. Cancer Res. 2018, 7, S766–S769. [Google Scholar] [CrossRef]
- Breda, A.; Lucarelli, G.; Rodriguez-Faba, O.; Guirado, L.; Facundo, C.; Bettocchi, C.; Gesualdo, L.; Castellano, G.; Grandaliano, G.; Battaglia, M.; et al. Clinical and Pathological Outcomes of Renal Cell Carcinoma (RCC) in Native Kidneys of Patients with End-Stage Renal Disease: A Long-Term Comparative Retrospective Study with RCC Diagnosed in the General Population. World J. Urol. 2015, 33, 1–7. [Google Scholar] [CrossRef]
- Vavallo, A.; Simone, S.; Lucarelli, G.; Rutigliano, M.; Galleggiante, V.; Grandaliano, G.; Gesualdo, L.; Campagna, M.; Cariello, M.; Ranieri, E.; et al. Pre-Existing Type 2 Diabetes Mellitus Is an Independent Risk Factor for Mortality and Progression in Patients with Renal Cell Carcinoma. Medicine 2014, 93, e183. [Google Scholar] [CrossRef]
- Bombelli, S.; Torsello, B.; De Marco, S.; Lucarelli, G.; Cifola, I.; Grasselli, C.; Strada, G.; Bovo, G.; Perego, R.A.; Bianchi, C. 36-kDa Annexin A3 Isoform Negatively Modulates Lipid Storage in Clear Cell Renal Cell Carcinoma Cells. Am. J. Pathol. 2020, 190, 2317–2326. [Google Scholar] [CrossRef]
- De Marco, S.; Torsello, B.; Minutiello, E.; Morabito, I.; Grasselli, C.; Bombelli, S.; Zucchini, N.; Lucarelli, G.; Strada, G.; Perego, R.A.; et al. The Cross-Talk between Abl2 Tyrosine Kinase and TGFβ1 Signalling Modulates the Invasion of Clear Cell Renal Cell Carcinoma Cells. FEBS Lett. 2022, 597, 1098–1113. [Google Scholar] [CrossRef]
- Gluba-Brzózka, A.; Rysz, J.; Ławiński, J.; Franczyk, B. Renal Cell Cancer and Obesity. Int. J. Mol. Sci. 2022, 23, 3404. [Google Scholar] [CrossRef] [PubMed]
- Saly, D.L.; Eswarappa, M.S.; Street, S.E.; Deshpande, P. Renal Cell Cancer and Chronic Kidney Disease. Adv. Chronic Kidney Dis. 2021, 28, 460–468.e1. [Google Scholar] [CrossRef] [PubMed]
- Heidegger, I.; Pircher, A.; Pichler, R. Targeting the Tumor Microenvironment in Renal Cell Cancer Biology and Therapy. Front. Oncol. 2019, 9, 490. [Google Scholar] [CrossRef] [PubMed]
- Vuong, L.; Kotecha, R.R.; Voss, M.H.; Hakimi, A.A. Tumor Microenvironment Dynamics in Clear-Cell Renal Cell Carcinoma. Cancer Discov. 2019, 9, 1349–1357. [Google Scholar] [CrossRef]
- Tamma, R.; Rutigliano, M.; Lucarelli, G.; Annese, T.; Ruggieri, S.; Cascardi, E.; Napoli, A.; Battaglia, M.; Ribatti, D. Microvascular Density, Macrophages, and Mast Cells in Human Clear Cell Renal Carcinoma with and without Bevacizumab Treatment. Urol. Oncol. 2019, 37, 355.e11–355.e19. [Google Scholar] [CrossRef]
- Cavaillon, J.-M.; Sansonetti, P.; Goldman, M. 100th Anniversary of Jules Bordet’s Nobel Prize: Tribute to a Founding Father of Immunology. Front. Immunol. 2019, 10, 2114. [Google Scholar] [CrossRef]
- Merle, N.S.; Church, S.E.; Fremeaux-Bacchi, V.; Roumenina, L.T. Complement System Part I—Molecular Mechanisms of Activation and Regulation. Front. Immunol. 2015, 6, 262. [Google Scholar] [CrossRef]
- Merle, N.S.; Noe, R.; Halbwachs-Mecarelli, L.; Fremeaux-Bacchi, V.; Roumenina, L.T. Complement System Part II: Role in Immunity. Front. Immunol. 2015, 6, 257. [Google Scholar] [CrossRef]
- Abbas, A.K.; Lichtman, A.H.; Pillai, S. Cellular and Molecular Immunology, 8th ed.; Elsevier Saunders: Philadelphia, PA, USA, 2015; ISBN 978-0-323-22275-4. [Google Scholar]
- Liszewski, M.K.; Atkinson, J.P. Complement Regulators in Human Disease: Lessons from Modern Genetics. J. Intern. Med. 2015, 277, 294–305. [Google Scholar] [CrossRef]
- Carroll, M.C.; Isenman, D.E. Regulation of Humoral Immunity by Complement. Immunity 2012, 37, 199–207. [Google Scholar] [CrossRef]
- Ricklin, D.; Hajishengallis, G.; Yang, K.; Lambris, J.D. Complement: A Key System for Immune Surveillance and Homeostasis. Nat. Immunol. 2010, 11, 785–797. [Google Scholar] [CrossRef]
- Strainic, M.G.; Liu, J.; Huang, D.; An, F.; Lalli, P.N.; Muqim, N.; Shapiro, V.S.; Dubyak, G.R.; Heeger, P.S.; Medof, M.E. Locally Produced Complement Fragments C5a and C3a Provide Both Costimulatory and Survival Signals to Naive CD4+ T Cells. Immunity 2008, 28, 425–435. [Google Scholar] [CrossRef] [PubMed]
- Lalli, P.N.; Strainic, M.G.; Yang, M.; Lin, F.; Medof, M.E.; Heeger, P.S. Locally Produced C5a Binds to T Cell-Expressed C5aR to Enhance Effector T-Cell Expansion by Limiting Antigen-Induced Apoptosis. Blood 2008, 112, 1759–1766. [Google Scholar] [CrossRef] [PubMed]
- Clarke, E.V.; Tenner, A.J. Complement Modulation of T Cell Immune Responses during Homeostasis and Disease. J. Leukoc. Biol. 2014, 96, 745–756. [Google Scholar] [CrossRef] [PubMed]
- West, E.E.; Kemper, C. Complosome—The Intracellular Complement System. Nat. Rev. Nephrol. 2023, 19, 426–439. [Google Scholar] [CrossRef] [PubMed]
- Singh, P.; Kemper, C. Complement, Complosome, and Complotype: A Perspective. Eur. J. Immunol. 2023, e2250042. [Google Scholar] [CrossRef]
- King, B.C.; Blom, A.M. Intracellular Complement: Evidence, Definitions, Controversies, and Solutions. Immunol. Rev. 2023, 313, 104–119. [Google Scholar] [CrossRef]
- Niyonzima, N.; Rahman, J.; Kunz, N.; West, E.E.; Freiwald, T.; Desai, J.V.; Merle, N.S.; Gidon, A.; Sporsheim, B.; Lionakis, M.S.; et al. Mitochondrial C5aR1 Activity in Macrophages Controls IL-1β Production Underlying Sterile Inflammation. Sci. Immunol. 2021, 6, eabf2489. [Google Scholar] [CrossRef]
- Sorbara, M.T.; Foerster, E.G.; Tsalikis, J.; Abdel-Nour, M.; Mangiapane, J.; Sirluck-Schroeder, I.; Tattoli, I.; van Dalen, R.; Isenman, D.E.; Rohde, J.R.; et al. Complement C3 Drives Autophagy-Dependent Restriction of Cyto-Invasive Bacteria. Cell Host Microbe 2018, 23, 644–652.e5. [Google Scholar] [CrossRef]
- Kolev, M.; Dimeloe, S.; Le Friec, G.; Navarini, A.; Arbore, G.; Povoleri, G.A.; Fischer, M.; Belle, R.; Loeliger, J.; Develioglu, L.; et al. Complement Regulates Nutrient Influx and Metabolic Reprogramming during Th1 Cell Responses. Immunity 2015, 42, 1033–1047. [Google Scholar] [CrossRef]
- King, B.C.; Renström, E.; Blom, A.M. Intracellular Cytosolic Complement Component C3 Regulates Cytoprotective Autophagy in Pancreatic Beta Cells by Interaction with ATG16L1. Autophagy 2019, 15, 919–921. [Google Scholar] [CrossRef] [PubMed]
- Golec, E.; Ekström, A.; Noga, M.; Omar-Hmeadi, M.; Lund, P.-E.; Villoutreix, B.O.; Krus, U.; Wozniak, K.; Korsgren, O.; Renström, E.; et al. Alternative Splicing Encodes Functional Intracellular CD59 Isoforms That Mediate Insulin Secretion and Are Down-Regulated in Diabetic Islets. Proc. Natl. Acad. Sci. USA 2022, 119, e2120083119. [Google Scholar] [CrossRef] [PubMed]
- Kremlitzka, M.; Nowacka, A.A.; Mohlin, F.C.; Bompada, P.; De Marinis, Y.; Blom, A.M. Interaction of Serum-Derived and Internalized C3 With DNA in Human B Cells—A Potential Involvement in Regulation of Gene Transcription. Front. Immunol. 2019, 10, 493. [Google Scholar] [CrossRef]
- Elvington, M.; Liszewski, M.K.; Bertram, P.; Kulkarni, H.S.; Atkinson, J.P. A C3(H20) Recycling Pathway Is a Component of the Intracellular Complement System. J. Clin. Investig. 2017, 127, 970–981. [Google Scholar] [CrossRef] [PubMed]
- Hansen, C.B.; Willer, A.; Bayarri-Olmos, R.; Kemper, C.; Garred, P. Expression of Complement C3, C5, C3aR and C5aR1 Genes in Resting and Activated CD4+ T Cells. Immunobiology 2019, 224, 307–315. [Google Scholar] [CrossRef]
- Le Friec, G.; Sheppard, D.; Whiteman, P.; Karsten, C.M.; Shamoun, S.A.-T.; Laing, A.; Bugeon, L.; Dallman, M.J.; Melchionna, T.; Chillakuri, C.; et al. The CD46-Jagged1 Interaction Is Critical for Human TH1 Immunity. Nat. Immunol. 2012, 13, 1213–1221. [Google Scholar] [CrossRef]
- Brandstadter, J.D.; Maillard, I. Notch Signalling in T Cell Homeostasis and Differentiation. Open Biol. 2019, 9, 190187. [Google Scholar] [CrossRef]
- Hess, C.; Kemper, C. Complement-Mediated Regulation of Metabolism and Basic Cellular Processes. Immunity 2016, 45, 240–254. [Google Scholar] [CrossRef]
- Arbore, G.; West, E.E.; Spolski, R.; Robertson, A.A.B.; Klos, A.; Rheinheimer, C.; Dutow, P.; Woodruff, T.M.; Yu, Z.X.; O’Neill, L.A.; et al. T Helper 1 Immunity Requires Complement-Driven NLRP3 Inflammasome Activity in CD4+ T Cells. Science 2016, 352, aad1210. [Google Scholar] [CrossRef]
- Arbore, G.; West, E.E.; Rahman, J.; Le Friec, G.; Niyonzima, N.; Pirooznia, M.; Tunc, I.; Pavlidis, P.; Powell, N.; Li, Y.; et al. Complement Receptor CD46 Co-Stimulates Optimal Human CD8+ T Cell Effector Function via Fatty Acid Metabolism. Nat. Commun. 2018, 9, 4186. [Google Scholar] [CrossRef]
- Benoit, M.E.; Clarke, E.V.; Morgado, P.; Fraser, D.A.; Tenner, A.J. Complement Protein C1q Directs Macrophage Polarization and Limits Inflammasome Activity during the Uptake of Apoptotic Cells. J. Immunol. 2012, 188, 5682–5693. [Google Scholar] [CrossRef] [PubMed]
- Rahman, J.; Singh, P.; Merle, N.S.; Niyonzima, N.; Kemper, C. Complement’s Favourite Organelle-Mitochondria? Br. J. Pharmacol. 2021, 178, 2771–2785. [Google Scholar] [CrossRef] [PubMed]
- Viret, C.; Rozières, A.; Duclaux-Loras, R.; Boschetti, G.; Nancey, S.; Faure, M. Regulation of Anti-Microbial Autophagy by Factors of the Complement System. Microb. Cell 2020, 7, 93–105. [Google Scholar] [CrossRef]
- Li, Y.; Sha, Y.; Wang, H.; He, L.; Li, L.; Wen, S.; Sheng, L.; Hu, W.; Zhou, H. Intracellular C3 Prevents Hepatic Steatosis by Promoting Autophagy and Very-Low-Density Lipoprotein Secretion. FASEB J. 2021, 35, e22037. [Google Scholar] [CrossRef]
- Schäfer, N.; Rasras, A.; Ormenisan, D.M.; Amslinger, S.; Enzmann, V.; Jägle, H.; Pauly, D. Complement Factor H-Related 3 Enhanced Inflammation and Complement Activation in Human RPE Cells. Front. Immunol. 2021, 12, 769242. [Google Scholar] [CrossRef]
- Ishii, M.; Beeson, G.; Beeson, C.; Rohrer, B. Mitochondrial C3a Receptor Activation in Oxidatively Stressed Epithelial Cells Reduces Mitochondrial Respiration and Metabolism. Front. Immunol. 2021, 12, 628062. [Google Scholar] [CrossRef]
- Zeng, J.; Xu, H.; Huang, C.; Sun, Y.; Xiao, H.; Yu, G.; Zhou, H.; Zhang, Y.; Yao, W.; Xiao, W.; et al. CD46 Splice Variant Enhances Translation of Specific mRNAs Linked to an Aggressive Tumor Cell Phenotype in Bladder Cancer. Mol. Ther. Nucleic Acids 2021, 24, 140–153. [Google Scholar] [CrossRef] [PubMed]
- Xiao, F.; Guo, J.; Tomlinson, S.; Yuan, G.; He, S. The Role of the Complosome in Health and Disease. Front. Immunol. 2023, 14, 1146167. [Google Scholar] [CrossRef]
- Mahajan, S.; Jacob, A.; Kelkar, A.; Chang, A.; Mcskimming, D.; Neelamegham, S.; Quigg, R.J.; Alexander, J.J. Local Complement Factor H Protects Kidney Endothelial Cell Structure and Function. Kidney Int. 2021, 100, 824–836. [Google Scholar] [CrossRef]
- Portilla, D.; Xavier, S. Role of Intracellular Complement Activation in Kidney Fibrosis. Br. J. Pharmacol. 2021, 178, 2880–2891. [Google Scholar] [CrossRef]
- Daina, E.; Cortinovis, M.; Remuzzi, G. Kidney Diseases. Immunol. Rev. 2023, 313, 239–261. [Google Scholar] [CrossRef] [PubMed]
- Noris, M.; Mescia, F.; Remuzzi, G. STEC-HUS, Atypical HUS and TTP Are All Diseases of Complement Activation. Nat. Rev. Nephrol. 2012, 8, 622–633. [Google Scholar] [CrossRef] [PubMed]
- Fremeaux-Bacchi, V.; Fakhouri, F.; Garnier, A.; Bienaimé, F.; Dragon-Durey, M.-A.; Ngo, S.; Moulin, B.; Servais, A.; Provot, F.; Rostaing, L.; et al. Genetics and Outcome of Atypical Hemolytic Uremic Syndrome: A Nationwide French Series Comparing Children and Adults. Clin. J. Am. Soc. Nephrol. 2013, 8, 554–562. [Google Scholar] [CrossRef] [PubMed]
- Michael, M.; Bagga, A.; Sartain, S.E.; Smith, R.J.H. Haemolytic Uraemic Syndrome. Lancet 2022, 400, 1722–1740. [Google Scholar] [CrossRef]
- Zuber, J.; Le Quintrec, M.; Morris, H.; Frémeaux-Bacchi, V.; Loirat, C.; Legendre, C. Targeted Strategies in the Prevention and Management of Atypical HUS Recurrence after Kidney Transplantation. Transplant. Rev. 2013, 27, 117–125. [Google Scholar] [CrossRef]
- Sellier-Leclerc, A.-L.; Fremeaux-Bacchi, V.; Dragon-Durey, M.-A.; Macher, M.-A.; Niaudet, P.; Guest, G.; Boudailliez, B.; Bouissou, F.C.; Deschenes, G.; Gie, S.; et al. Differential Impact of Complement Mutations on Clinical Characteristics in Atypical Hemolytic Uremic Syndrome. J. Am. Soc. Nephrol. 2007, 18, 2392–2400. [Google Scholar] [CrossRef]
- Pérez-Caballero, D.; González-Rubio, C.; Gallardo, M.E.; Vera, M.; López-Trascasa, M.; Rodríguez De Córdoba, S.; Sánchez-Corral, P. Clustering of Missense Mutations in the C-Terminal Region of Factor H in Atypical Hemolytic Uremic Syndrome. Am. J. Human. Genet. 2001, 68, 478–484. [Google Scholar] [CrossRef]
- Fremeaux-Bacchi, V. Complement Factor I: A Susceptibility Gene for Atypical Haemolytic Uraemic Syndrome. J. Med. Genet. 2004, 41, e84. [Google Scholar] [CrossRef]
- Noris, M.; Brioschi, S.; Caprioli, J.; Todeschini, M.; Bresin, E.; Porrati, F.; Gamba, S.; Remuzzi, G. Familial Haemolytic Uraemic Syndrome and an MCP Mutation. Lancet 2003, 362, 1542–1547. [Google Scholar] [CrossRef]
- Delvaeye, M.; Noris, M.; De Vriese, A.; Esmon, C.T.; Esmon, N.L.; Ferrell, G.; Del-Favero, J.; Plaisance, S.; Claes, B.; Lambrechts, D.; et al. Thrombomodulin Mutations in Atypical Hemolytic–Uremic Syndrome. N. Engl. J. Med. 2009, 361, 345–357. [Google Scholar] [CrossRef]
- Zipfel, P.F.; Wiech, T.; Stea, E.D.; Skerka, C. CFHR Gene Variations Provide Insights in the Pathogenesis of the Kidney Diseases Atypical Hemolytic Uremic Syndrome and C3 Glomerulopathy. J. Am. Soc. Nephrol. 2020, 31, 241–256. [Google Scholar] [CrossRef] [PubMed]
- Iatropoulos, P.; Noris, M.; Mele, C.; Piras, R.; Valoti, E.; Bresin, E.; Curreri, M.; Mondo, E.; Zito, A.; Gamba, S.; et al. Complement Gene Variants Determine the Risk of Immunoglobulin-Associated MPGN and C3 Glomerulopathy and Predict Long-Term Renal Outcome. Mol. Immunol. 2016, 71, 131–142. [Google Scholar] [CrossRef] [PubMed]
- Nicolas, C.; Vuiblet, V.; Baudouin, V.; Macher, M.-A.; Vrillon, I.; Biebuyck-Gouge, N.; Dehennault, M.; Gié, S.; Morin, D.; Nivet, H.; et al. C3 Nephritic Factor Associated with C3 Glomerulopathy in Children. Pediatr. Nephrol. 2014, 29, 85–94. [Google Scholar] [CrossRef] [PubMed]
- Ronco, P.; Plaisier, E.; Debiec, H. Advances in Membranous Nephropathy. JCM 2021, 10, 607. [Google Scholar] [CrossRef] [PubMed]
- Ma, H.; Sandor, D.G.; Beck, L.H. The Role of Complement in Membranous Nephropathy. Semin. Nephrol. 2013, 33, 531–542. [Google Scholar] [CrossRef]
- Hoxha, E.; Reinhard, L.; Stahl, R.A.K. Membranous Nephropathy: New Pathogenic Mechanisms and Their Clinical Implications. Nat. Rev. Nephrol. 2022, 18, 466–478. [Google Scholar] [CrossRef]
- Hilhorst, M.; Van Paassen, P.; Van Rie, H.; Bijnens, N.; Heerings-Rewinkel, P.; Van Breda Vriesman, P.; Cohen Tervaert, J.W.; Limburg Renal Registry. Complement in ANCA-Associated Glomerulonephritis. Nephrol. Dial. Transplant. 2017, 32, 1302–1313. [Google Scholar] [CrossRef]
- Endre, Z.H.; Pussell, B.A.; Charlesworth, J.A.; Coovadia, H.M.; Seedat, Y.K. C3 Metabolism in Acute Glomerulonephritis: Implications for Sites of Complement Activation. Kidney Int. 1984, 25, 937–941. [Google Scholar] [CrossRef]
- Kang, Y.; Xu, B.; Shi, S.; Zhou, X.; Chen, P.; Liu, L.; Li, Y.; Leng, Y.; Lv, J.; Zhu, L.; et al. Mesangial C3 Deposition, Complement-Associated Variant, and Disease Progression in IgA Nephropathy. Clin. J. Am. Soc. Nephrol. 2023; ahead of print. [Google Scholar] [CrossRef]
- Medjeral-Thomas, N.R.; Cook, H.T.; Pickering, M.C. Complement Activation in IgA Nephropathy. Semin. Immunopathol. 2021, 43, 679–690. [Google Scholar] [CrossRef]
- Chiu, Y.-L.; Lin, W.-C.; Shu, K.-H.; Fang, Y.-W.; Chang, F.-C.; Chou, Y.-H.; Wu, C.-F.; Chiang, W.-C.; Lin, S.-L.; Chen, Y.-M.; et al. Alternative Complement Pathway Is Activated and Associated with Galactose-Deficient IgA1 Antibody in IgA Nephropathy Patients. Front. Immunol. 2021, 12, 638309. [Google Scholar] [CrossRef] [PubMed]
- Song, D.; Guo, W.; Wang, F.; Li, Y.; Song, Y.; Yu, F.; Zhao, M. Complement Alternative Pathway’s Activation in Patients With Lupus Nephritis. Am. J. Med. Sci. 2017, 353, 247–257. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.; Kim, T.; Kim, M.; Lee, H.Y.; Kim, Y.; Kang, M.S.; Kim, J. Activation of the Alternative Complement Pathway Predicts Renal Outcome in Patients with Lupus Nephritis. Lupus 2020, 29, 862–871. [Google Scholar] [CrossRef]
- Wang, F.; Yu, F.; Tan, Y.; Song, D.; Zhao, M. Serum Complement Factor H Is Associated with Clinical and Pathological Activities of Patients with Lupus Nephritis. Rheumatology 2012, 51, 2269–2277. [Google Scholar] [CrossRef] [PubMed]
- Trambas, I.A.; Coughlan, M.T.; Tan, S.M. Therapeutic Potential of Targeting Complement C5a Receptors in Diabetic Kidney Disease. Int. J. Mol. Sci. 2023, 24, 8758. [Google Scholar] [CrossRef]
- Kronbichler, A.; Bajema, I.; Geetha, D.; Säemann, M. Novel Aspects in the Pathophysiology and Diagnosis of Glomerular Diseases. Ann. Rheum. Dis. 2023, 82, 585–593. [Google Scholar] [CrossRef]
- Caillard, P.; Vigneau, C.; Halimi, J.-M.; Hazzan, M.; Thervet, E.; Heitz, M.; Juillard, L.; Audard, V.; Rabant, M.; Hertig, A.; et al. Prognostic Value of Complement Serum C3 Level and Glomerular C3 Deposits in Anti-Glomerular Basement Membrane Disease. Front. Immunol. 2023, 14, 1190394. [Google Scholar] [CrossRef]
- Ma, R.; Cui, Z.; Hu, S.-Y.; Jia, X.-Y.; Yang, R.; Zheng, X.; Ao, J.; Liu, G.; Liao, Y.-H.; Zhao, M.-H. The Alternative Pathway of Complement Activation May Be Involved in the Renal Damage of Human Anti-Glomerular Basement Membrane Disease. PLoS ONE 2014, 9, e91250. [Google Scholar] [CrossRef]
- Lucarelli, G.; Bettocchi, C.; Battaglia, M.; Impedovo, S.V.; Vavallo, A.; Grandaliano, G.; Castellano, G.; Schena, F.P.; Selvaggi, F.P.; Ditonno, P. Extended Criteria Donor Kidney Transplantation: Comparative Outcome Analysis between Single versus Double Kidney Transplantation at 5 Years. Transplant. Proc. 2010, 42, 1104–1107. [Google Scholar] [CrossRef]
- Ditonno, P.; Lucarelli, G.; Impedovo, S.V.; Spilotros, M.; Grandaliano, G.; Selvaggi, F.P.; Bettocchi, C.; Battaglia, M. Obesity in Kidney Transplantation Affects Renal Function but Not Graft and Patient Survival. Transplant. Proc. 2011, 43, 367–372. [Google Scholar] [CrossRef]
- Losappio, V.; Stallone, G.; Infante, B.; Schena, A.; Rossini, M.; Maiorano, A.; Fiorentino, M.; Ditonno, P.; Lucarelli, G.; Battaglia, M.; et al. A Single-Center Cohort Study to Define the Role of Pretransplant Biopsy Score in the Long-Term Outcome of Kidney Transplantation. Transplantation 2014, 97, 934–939. [Google Scholar] [CrossRef] [PubMed]
- Salvadori, M.; Rosso, G.; Bertoni, E. Update on Ischemia-Reperfusion Injury in Kidney Transplantation: Pathogenesis and Treatment. World J. Transplant. 2015, 5, 52–67. [Google Scholar] [CrossRef] [PubMed]
- Loizzo, D.; Di Meo, N.A.; Peluso, M.R.; Rutigliano, M.; Matera, M.; Miacola, C.; Palella, G.; Tedeschi, M.; Spilotros, M.; Ferro, M.; et al. Novel Insights into the Molecular Mechanisms of Ischemia/Reperfusion Injury in Kidney Transplantation. Transplantology 2021, 2, 191–207. [Google Scholar] [CrossRef]
- Nauser, C.L.; Farrar, C.A.; Sacks, S.H. Complement Recognition Pathways in Renal Transplantation. J. Am. Soc. Nephrol. 2017, 28, 2571–2578. [Google Scholar] [CrossRef] [PubMed]
- Qi, R.; Qin, W. Role of Complement System in Kidney Transplantation: Stepping From Animal Models to Clinical Application. Front. Immunol. 2022, 13, 811696. [Google Scholar] [CrossRef]
- Gibson, B.; Connelly, C.; Moldakhmetova, S.; Sheerin, N.S. Complement Activation and Kidney Transplantation; a Complex Relationship. Immunobiology 2023, 228, 152396. [Google Scholar] [CrossRef]
- Santarsiero, D.; Aiello, S. The Complement System in Kidney Transplantation. Cells 2023, 12, 791. [Google Scholar] [CrossRef]
- Zheng, X.; Feng, B.; Chen, G.; Zhang, X.; Li, M.; Sun, H.; Liu, W.; Vladau, C.; Liu, R.; Jevnikar, A.M.; et al. Preventing Renal IschemiaReperfusion Injury Using Small Interfering RNA by Targeting Complement 3 Gene. Am. J. Transplant. 2006, 6, 2099–2108. [Google Scholar] [CrossRef]
- Farrar, C.A.; Tran, D.; Li, K.; Wu, W.; Peng, Q.; Schwaeble, W.; Zhou, W.; Sacks, S.H. Collectin-11 Detects Stress-Induced L-Fucose Pattern to Trigger Renal Epithelial Injury. J. Clin. Investig. 2016, 126, 1911–1925. [Google Scholar] [CrossRef]
- Asgari, E.; Farrar, C.A.; Lynch, N.; Ali, Y.M.; Roscher, S.; Stover, C.; Zhou, W.; Schwaeble, W.J.; Sacks, S.H. Mannan-Binding Lectin-Associated Serine Protease 2 Is Critical for the Development of Renal Ischemia Reperfusion Injury and Mediates Tissue Injury in the Absence of Complement C4. FASEB J. 2014, 28, 3996–4003. [Google Scholar] [CrossRef]
- Yamada, K.; Miwa, T.; Liu, J.; Nangaku, M.; Song, W.-C. Critical Protection from Renal Ischemia Reperfusion Injury by CD55 and CD59. J. Immunol. 2004, 172, 3869–3875. [Google Scholar] [CrossRef] [PubMed]
- Bongoni, A.K.; Lu, B.; Salvaris, E.J.; Roberts, V.; Fang, D.; McRae, J.L.; Fisicaro, N.; Dwyer, K.M.; Cowan, P.J. Overexpression of Human CD55 and CD59 or Treatment with Human CD55 Protects against Renal Ischemia-Reperfusion Injury in Mice. J. Immunol. 2017, 198, 4837–4845. [Google Scholar] [CrossRef] [PubMed]
- Castellano, G.; Melchiorre, R.; Loverre, A.; Ditonno, P.; Montinaro, V.; Rossini, M.; Divella, C.; Battaglia, M.; Lucarelli, G.; Annunziata, G.; et al. Therapeutic Targeting of Classical and Lectin Pathways of Complement Protects from Ischemia-Reperfusion-Induced Renal Damage. Am. J. Pathol. 2010, 176, 1648–1659. [Google Scholar] [CrossRef] [PubMed]
- Ferrer, I.R.; Hester, J.; Bushell, A.; Wood, K.J. Induction of Transplantation Tolerance through Regulatory Cells: From Mice to Men. Immunol. Rev. 2014, 258, 102–116. [Google Scholar] [CrossRef]
- Peng, Q.; Wu, W.; Wu, K.-Y.; Cao, B.; Qiang, C.; Li, K.; Sacks, S.H.; Zhou, W. The C5a/C5aR1 Axis Promotes Progression of Renal Tubulointerstitial Fibrosis in a Mouse Model of Renal Ischemia/Reperfusion Injury. Kidney Int. 2019, 96, 117–128. [Google Scholar] [CrossRef] [PubMed]
- Castellano, G.; Franzin, R.; Stasi, A.; Divella, C.; Sallustio, F.; Pontrelli, P.; Lucarelli, G.; Battaglia, M.; Staffieri, F.; Crovace, A.; et al. Complement Activation During Ischemia/Reperfusion Injury Induces Pericyte-to-Myofibroblast Transdifferentiation Regulating Peritubular Capillary Lumen Reduction Through pERK Signaling. Front. Immunol. 2018, 9, 1002. [Google Scholar] [CrossRef]
- Divella, C.; Stasi, A.; Franzin, R.; Rossini, M.; Pontrelli, P.; Sallustio, F.; Netti, G.S.; Ranieri, E.; Lacitignola, L.; Staffieri, F.; et al. Pentraxin-3-Mediated Complement Activation in a Swine Model of Renal Ischemia/Reperfusion Injury. Aging 2021, 13, 10920–10933. [Google Scholar] [CrossRef]
- Curci, C.; Castellano, G.; Stasi, A.; Divella, C.; Loverre, A.; Gigante, M.; Simone, S.; Cariello, M.; Montinaro, V.; Lucarelli, G.; et al. Endothelial-to-Mesenchymal Transition and Renal Fibrosis in Ischaemia/Reperfusion Injury Are Mediated by Complement Anaphylatoxins and Akt Pathway. Nephrol. Dial. Transplant. 2014, 29, 799–808. [Google Scholar] [CrossRef]
- Castellano, G.; Franzin, R.; Sallustio, F.; Stasi, A.; Banelli, B.; Romani, M.; De Palma, G.; Lucarelli, G.; Divella, C.; Battaglia, M.; et al. Complement Component C5a Induces Aberrant Epigenetic Modifications in Renal Tubular Epithelial Cells Accelerating Senescence by Wnt4/Βcatenin Signaling after Ischemia/Reperfusion Injury. Aging 2019, 11, 4382–4406. [Google Scholar] [CrossRef]
- Pesce, F.; Stea, E.D.; Divella, C.; Accetturo, M.; Laghetti, P.; Gallo, P.; Rossini, M.; Cianciotta, F.; Crispino, L.; Granata, A.; et al. DelCFHR3-1 Influences Graft Survival in Transplant Patients with IgA Nephropathy via Complement-Mediated Cellular Senescence. Am. J. Transplant. 2021, 21, 838–845. [Google Scholar] [CrossRef]
- Castellano, G.; Intini, A.; Stasi, A.; Divella, C.; Gigante, M.; Pontrelli, P.; Franzin, R.; Accetturo, M.; Zito, A.; Fiorentino, M.; et al. Complement Modulation of Anti-Aging Factor Klotho in Ischemia/Reperfusion Injury and Delayed Graft Function. Am. J. Transplant. 2016, 16, 325–333. [Google Scholar] [CrossRef]
- Błogowski, W.; Dołęgowska, B.; Sałata, D.; Budkowska, M.; Domański, L.; Starzyńska, T. Clinical Analysis of Perioperative Complement Activity during Ischemia/Reperfusion Injury Following Renal Transplantation. Clin. J. Am. Soc. Nephrol. 2012, 7, 1843–1851. [Google Scholar] [CrossRef] [PubMed]
- Arias-Cabrales, C.E.; Riera, M.; Pérez-Sáez, M.J.; Gimeno, J.; Benito, D.; Redondo, D.; Burballa, C.; Crespo, M.; Pascual, J.; Rodríguez, E. Activation of Final Complement Components after Kidney Transplantation as a Marker of Delayed Graft Function Severity. Clin. Kidney J. 2021, 14, 1190–1196. [Google Scholar] [CrossRef]
- De Vries, D.K.; Van Der Pol, P.; Van Anken, G.E.; Van Gijlswijk, D.J.; Damman, J.; Lindeman, J.H.; Reinders, M.E.J.; Schaapherder, A.F.; Kooten, C.V. Acute But Transient Release of Terminal Complement Complex After Reperfusion in Clinical Kidney Transplantation. Transplantation 2013, 95, 816–820. [Google Scholar] [CrossRef]
- Teh, B.K.; Yeo, J.G.; Chern, L.M.; Lu, J. C1q Regulation of Dendritic Cell Development from Monocytes with Distinct Cytokine Production and T Cell Stimulation. Mol. Immunol. 2011, 48, 1128–1138. [Google Scholar] [CrossRef] [PubMed]
- Frémeaux-Bacchi, V.; Legendre, C.M. The Emerging Role of Complement Inhibitors in Transplantation. Kidney Int. 2015, 88, 967–973. [Google Scholar] [CrossRef] [PubMed]
- Muñoz-Herrera, C.M.; Gutiérrez-Bautista, J.F.; López-Nevot, M.Á. Complement Binding Anti-HLA Antibodies and the Survival of Kidney Transplantation. J. Clin. Med. 2023, 12, 2335. [Google Scholar] [CrossRef]
- Stites, E.; Le Quintrec, M.; Thurman, J.M. The Complement System and Antibody-Mediated Transplant Rejection. J. Immunol. 2015, 195, 5525–5531. [Google Scholar] [CrossRef]
- Roufosse, C.; Simmonds, N.; Clahsen-van Groningen, M.; Haas, M.; Henriksen, K.J.; Horsfield, C.; Loupy, A.; Mengel, M.; Perkowska-Ptasińska, A.; Rabant, M.; et al. A 2018 Reference Guide to the Banff Classification of Renal Allograft Pathology. Transplantation 2018, 102, 1795–1814. [Google Scholar] [CrossRef]
- Lasorsa, F.; Rutigliano, M.; Milella, M.; Ferro, M.; Pandolfo, S.D.; Crocetto, F.; Tataru, O.S.; Autorino, R.; Battaglia, M.; Ditonno, P.; et al. Cellular and Molecular Players in the Tumor Microenvironment of Renal Cell Carcinoma. J. Clin. Med. 2023, 12, 3888. [Google Scholar] [CrossRef]
- Roumenina, L.T.; Daugan, M.V.; Petitprez, F.; Sautès-Fridman, C.; Fridman, W.H. Context-Dependent Roles of Complement in Cancer. Nat. Rev. Cancer 2019, 19, 698–715. [Google Scholar] [CrossRef] [PubMed]
- Mamidi, S.; Höne, S.; Kirschfink, M. The Complement System in Cancer: Ambivalence between Tumour Destruction and Promotion. Immunobiology 2017, 222, 45–54. [Google Scholar] [CrossRef] [PubMed]
- Ajona, D.; Ortiz-Espinosa, S.; Pio, R. Complement Anaphylatoxins C3a and C5a: Emerging Roles in Cancer Progression and Treatment. Semin. Cell Dev. Biol. 2019, 85, 153–163. [Google Scholar] [CrossRef] [PubMed]
- Kolev, M.; Das, M.; Gerber, M.; Baver, S.; Deschatelets, P.; Markiewski, M.M. Inside-Out of Complement in Cancer. Front. Immunol. 2022, 13, 931273. [Google Scholar] [CrossRef]
- Tirosh, I.; Izar, B.; Prakadan, S.M.; Wadsworth, M.H.; Treacy, D.; Trombetta, J.J.; Rotem, A.; Rodman, C.; Lian, C.; Murphy, G.; et al. Dissecting the Multicellular Ecosystem of Metastatic Melanoma by Single-Cell RNA-Seq. Science 2016, 352, 189–196. [Google Scholar] [CrossRef]
- Shu, C.; Zha, H.; Long, H.; Wang, X.; Yang, F.; Gao, J.; Hu, C.; Zhou, L.; Guo, B.; Zhu, B. C3a-C3aR Signaling Promotes Breast Cancer Lung Metastasis via Modulating Carcinoma Associated Fibroblasts. J. Exp. Clin. Cancer Res. 2020, 39, 11. [Google Scholar] [CrossRef]
- Medler, T.R.; Murugan, D.; Horton, W.; Kumar, S.; Cotechini, T.; Forsyth, A.M.; Leyshock, P.; Leitenberger, J.J.; Kulesz-Martin, M.; Margolin, A.A.; et al. Complement C5a Fosters Squamous Carcinogenesis and Limits T Cell Response to Chemotherapy. Cancer Cell 2018, 34, 561–578.e6. [Google Scholar] [CrossRef]
- Yasmin, H.; Bulla, R.; Madan, T.; Kishore, U. Complement and Cancer Immunity. In Handbook of Cancer and Immunology; Rezaei, N., Ed.; Springer International Publishing: Cham, Switzerland, 2022; pp. 1–19. ISBN 978-3-030-80962-1. [Google Scholar]
- Rozenberg, P.; Ziporen, L.; Gancz, D.; Saar-Ray, M.; Fishelson, Z. Cooperation between Hsp90 and Mortalin/GRP75 in Resistance to Cell Death Induced by Complement C5b-9. Cell Death Dis. 2018, 9, 150. [Google Scholar] [CrossRef]
- Zhang, R.; Liu, Q.; Li, T.; Liao, Q.; Zhao, Y. Role of the Complement System in the Tumor Microenvironment. Cancer Cell Int. 2019, 19, 300. [Google Scholar] [CrossRef]
- Zha, H.; Wang, X.; Zhu, Y.; Chen, D.; Han, X.; Yang, F.; Gao, J.; Hu, C.; Shu, C.; Feng, Y.; et al. Intracellular Activation of Complement C3 Leads to PD-L1 Antibody Treatment Resistance by Modulating Tumor-Associated Macrophages. Cancer Immunol. Res. 2019, 7, 193–207. [Google Scholar] [CrossRef]
- Markiewski, M.M.; DeAngelis, R.A.; Benencia, F.; Ricklin-Lichtsteiner, S.K.; Koutoulaki, A.; Gerard, C.; Coukos, G.; Lambris, J.D. Modulation of the Antitumor Immune Response by Complement. Nat. Immunol. 2008, 9, 1225–1235. [Google Scholar] [CrossRef]
- Kochanek, D.M.; Ghouse, S.M.; Karbowniczek, M.M.; Markiewski, M.M. Complementing Cancer Metastasis. Front. Immunol. 2018, 9, 1629. [Google Scholar] [CrossRef] [PubMed]
- Singel, K.L.; Emmons, T.R.; Khan, A.N.H.; Mayor, P.C.; Shen, S.; Wong, J.T.; Morrell, K.; Eng, K.H.; Mark, J.; Bankert, R.B.; et al. Mature Neutrophils Suppress T Cell Immunity in Ovarian Cancer Microenvironment. JCI Insight 2019, 4, e122311. [Google Scholar] [CrossRef]
- Guglietta, S.; Chiavelli, A.; Zagato, E.; Krieg, C.; Gandini, S.; Ravenda, P.S.; Bazolli, B.; Lu, B.; Penna, G.; Rescigno, M. Coagulation Induced by C3aR-Dependent NETosis Drives Protumorigenic Neutrophils during Small Intestinal Tumorigenesis. Nat. Commun. 2016, 7, 11037. [Google Scholar] [CrossRef] [PubMed]
- Cho, M.S.; Vasquez, H.G.; Rupaimoole, R.; Pradeep, S.; Wu, S.; Zand, B.; Han, H.-D.; Rodriguez-Aguayo, C.; Bottsford-Miller, J.; Huang, J.; et al. Autocrine Effects of Tumor-Derived Complement. Cell Rep. 2014, 6, 1085–1095. [Google Scholar] [CrossRef] [PubMed]
- Cho, M.S.; Rupaimoole, R.; Choi, H.-J.; Noh, K.; Chen, J.; Hu, Q.; Sood, A.K.; Afshar-Kharghan, V. Complement Component 3 Is Regulated by TWIST1 and Mediates Epithelial-Mesenchymal Transition. J. Immunol. 2016, 196, 1412–1418. [Google Scholar] [CrossRef]
- Hu, W.-H.; Hu, Z.; Shen, X.; Dong, L.-Y.; Zhou, W.-Z.; Yu, X.-X. C5a Receptor Enhances Hepatocellular Carcinoma Cell Invasiveness via Activating ERK1/2-Mediated Epithelial-Mesenchymal Transition. Exp. Mol. Pathol. 2016, 100, 101–108. [Google Scholar] [CrossRef]
- Gu, J.; Ding, J.; Lu, C.; Lin, Z.; Chu, Y.; Zhao, G.; Guo, J.; Ge, D. Overexpression of CD88 Predicts Poor Prognosis in Non-Small-Cell Lung Cancer. Lung Cancer 2013, 81, 259–265. [Google Scholar] [CrossRef]
- Nunez-Cruz, S.; Gimotty, P.A.; Guerra, M.W.; Connolly, D.C.; Wu, Y.-Q.; DeAngelis, R.A.; Lambris, J.D.; Coukos, G.; Scholler, N. Genetic and Pharmacologic Inhibition of Complement Impairs Endothelial Cell Function and Ablates Ovarian Cancer Neovascularization. Neoplasia 2012, 14, 994–1004. [Google Scholar] [CrossRef]
- Lasorsa, F.; Rutigliano, M.; Milella, M.; Ferro, M.; Pandolfo, S.D.; Crocetto, F.; Autorino, R.; Battaglia, M.; Ditonno, P.; Lucarelli, G. Cancer Stem Cells in Renal Cell Carcinoma: Origins and Biomarkers. Int. J. Mol. Sci. 2023, 24, 13179. [Google Scholar] [CrossRef]
- Chen, J.; Ding, P.; Li, L.; Gu, H.; Zhang, X.; Zhang, L.; Wang, N.; Gan, L.; Wang, Q.; Zhang, W.; et al. CD59 Regulation by SOX2 Is Required for Epithelial Cancer Stem Cells to Evade Complement Surveillance. Stem Cell Rep. 2017, 8, 140–151. [Google Scholar] [CrossRef] [PubMed]
- de Weers, M.; Tai, Y.-T.; van der Veer, M.S.; Bakker, J.M.; Vink, T.; Jacobs, D.C.H.; Oomen, L.A.; Peipp, M.; Valerius, T.; Slootstra, J.W.; et al. Daratumumab, a Novel Therapeutic Human CD38 Monoclonal Antibody, Induces Killing of Multiple Myeloma and Other Hematological Tumors. J. Immunol. 2011, 186, 1840–1848. [Google Scholar] [CrossRef] [PubMed]
- El-Sahwi, K.; Bellone, S.; Cocco, E.; Cargnelutti, M.; Casagrande, F.; Bellone, M.; Abu-Khalaf, M.; Buza, N.; Tavassoli, F.A.; Hui, P.; et al. In Vitro Activity of Pertuzumab in Combination with Trastuzumab in Uterine Serous Papillary Adenocarcinoma. Br. J. Cancer 2010, 102, 134–143. [Google Scholar] [CrossRef] [PubMed]
- Macor, P.; Secco, E.; Mezzaroba, N.; Zorzet, S.; Durigutto, P.; Gaiotto, T.; De Maso, L.; Biffi, S.; Garrovo, C.; Capolla, S.; et al. Bispecific Antibodies Targeting Tumor-Associated Antigens and Neutralizing Complement Regulators Increase the Efficacy of Antibody-Based Immunotherapy in Mice. Leukemia 2015, 29, 406–414. [Google Scholar] [CrossRef] [PubMed]
- Roumenina, L.T.; Daugan, M.V.; Noé, R.; Petitprez, F.; Vano, Y.A.; Sanchez-Salas, R.; Becht, E.; Meilleroux, J.; Clec’h, B.L.; Giraldo, N.A.; et al. Tumor Cells Hijack Macrophage-Produced Complement C1q to Promote Tumor Growth. Cancer Immunol. Res. 2019, 7, 1091–1105. [Google Scholar] [CrossRef]
- Maeda, Y.; Kawano, Y.; Wada, Y.; Yatsuda, J.; Motoshima, T.; Murakami, Y.; Kikuchi, K.; Imamura, T.; Eto, M. C5aR Is Frequently Expressed in Metastatic Renal Cell Carcinoma and Plays a Crucial Role in Cell Invasion via the ERK and PI3 Kinase Pathways. Oncol. Rep. 2015, 33, 1844–1850. [Google Scholar] [CrossRef]
- Daugan, M.V.; Revel, M.; Thouenon, R.; Dragon-Durey, M.-A.; Robe-Rybkine, T.; Torset, C.; Merle, N.S.; Noé, R.; Verkarre, V.; Oudard, S.M.; et al. Intracellular Factor H Drives Tumor Progression Independently of the Complement Cascade. Cancer Immunol. Res. 2021, 9, 909–925. [Google Scholar] [CrossRef]
- Dong, Y.; Ma, W.-M.; Yang, W.; Hao, L.; Zhang, S.-Q.; Fang, K.; Hu, C.-H.; Zhang, Q.-J.; Shi, Z.-D.; Zhang, W.; et al. Identification of C3 and FN1 as Potential Biomarkers Associated with Progression and Prognosis for Clear Cell Renal Cell Carcinoma. BMC Cancer 2021, 21, 1135. [Google Scholar] [CrossRef]
- Reese, B.; Silwal, A.; Daugherity, E.; Daugherity, M.; Arabi, M.; Daly, P.; Paterson, Y.; Woolford, L.; Christie, A.; Elias, R.; et al. Complement as Prognostic Biomarker and Potential Therapeutic Target in Renal Cell Carcinoma. J. Immunol. 2020, 205, 3218–3229. [Google Scholar] [CrossRef]
- Daugan, M.V.; Revel, M.; Russick, J.; Dragon-Durey, M.-A.; Gaboriaud, C.; Robe-Rybkine, T.; Poillerat, V.; Grunenwald, A.; Lacroix, G.; Bougouin, A.; et al. Complement C1s and C4d as Prognostic Biomarkers in Renal Cancer: Emergence of Noncanonical Functions of C1s. Cancer Immunol. Res. 2021, 9, 891–908. [Google Scholar] [CrossRef]
- Wang, J.; Zhang, S.; Wang, Y.; Zhu, Y.; Xu, X.; Guo, J. Alternative Complement Pathway Signature Determines Immunosuppression and Resistance to Immunotherapy Plus Tyrosine Kinase Inhibitor Combinations in Renal Cell Carcinoma. Urol. Oncol. Semin. Orig. Investig. 2023, 41, 51.e13–51.e23. [Google Scholar] [CrossRef] [PubMed]
- Garlanda, C.; Bottazzi, B.; Magrini, E.; Inforzato, A.; Mantovani, A. PTX3, a Humoral Pattern Recognition Molecule, in Innate Immunity, Tissue Repair, and Cancer. Physiol. Rev. 2018, 98, 623–639. [Google Scholar] [CrossRef] [PubMed]
- Giacomini, A.; Ghedini, G.C.; Presta, M.; Ronca, R. Long Pentraxin 3: A Novel Multifaceted Player in Cancer. Biochim. Biophys. Acta (BBA)—Rev. Cancer 2018, 1869, 53–63. [Google Scholar] [CrossRef] [PubMed]
- Bonavita, E.; Gentile, S.; Rubino, M.; Maina, V.; Papait, R.; Kunderfranco, P.; Greco, C.; Feruglio, F.; Molgora, M.; Laface, I.; et al. PTX3 Is an Extrinsic Oncosuppressor Regulating Complement-Dependent Inflammation in Cancer. Cell 2015, 160, 700–714. [Google Scholar] [CrossRef] [PubMed]
- Netti, G.S.; Lucarelli, G.; Spadaccino, F.; Castellano, G.; Gigante, M.; Divella, C.; Rocchetti, M.T.; Rascio, F.; Mancini, V.; Stallone, G.; et al. PTX3 Modulates the Immunoflogosis in Tumor Microenvironment and Is a Prognostic Factor for Patients with Clear Cell Renal Cell Carcinoma. Aging 2020, 12, 7585–7602. [Google Scholar] [CrossRef] [PubMed]
- Lucarelli, G.; Rutigliano, M.; Loizzo, D.; di Meo, N.A.; Lasorsa, F.; Mastropasqua, M.; Maiorano, E.; Bizzoca, C.; Vincenti, L.; Battaglia, M.; et al. MUC1 Tissue Expression and Its Soluble Form CA15-3 Identify a Clear Cell Renal Cell Carcinoma with Distinct Metabolic Profile and Poor Clinical Outcome. Int. J. Mol. Sci. 2022, 23, 13968. [Google Scholar] [CrossRef]
- Lucarelli, G.; Ditonno, P.; Bettocchi, C.; Vavallo, A.; Rutigliano, M.; Galleggiante, V.; Larocca, A.M.V.; Castellano, G.; Gesualdo, L.; Grandaliano, G.; et al. Diagnostic and Prognostic Role of Preoperative Circulating CA 15-3, CA 125, and Beta-2 Microglobulin in Renal Cell Carcinoma. Dis. Markers 2014, 2014, 689795. [Google Scholar] [CrossRef]
- Lucarelli, G.; Netti, G.S.; Rutigliano, M.; Lasorsa, F.; Loizzo, D.; Milella, M.; Schirinzi, A.; Fontana, A.; Di Serio, F.; Tamma, R.; et al. MUC1 Expression Affects the Immunoflogosis in Renal Cell Carcinoma Microenvironment through Complement System Activation and Immune Infiltrate Modulation. Int. J. Mol. Sci. 2023, 24, 4814. [Google Scholar] [CrossRef]
- Lasorsa, F.; di Meo, N.A.; Rutigliano, M.; Milella, M.; Ferro, M.; Pandolfo, S.D.; Crocetto, F.; Tataru, O.S.; Autorino, R.; Battaglia, M.; et al. Immune Checkpoint Inhibitors in Renal Cell Carcinoma: Molecular Basis and Rationale for Their Use in Clinical Practice. Biomedicines 2023, 11, 1071. [Google Scholar] [CrossRef]
- Greenbaum, L.A.; Fila, M.; Ardissino, G.; Al-Akash, S.I.; Evans, J.; Henning, P.; Lieberman, K.V.; Maringhini, S.; Pape, L.; Rees, L.; et al. Eculizumab Is a Safe and Effective Treatment in Pediatric Patients with Atypical Hemolytic Uremic Syndrome. Kidney Int. 2016, 89, 701–711. [Google Scholar] [CrossRef]
- Vonbrunn, E.; Büttner-Herold, M.; Amann, K.; Daniel, C. Complement Inhibition in Kidney Transplantation: Where Are We Now? BioDrugs 2023, 37, 5–19. [Google Scholar] [CrossRef] [PubMed]
- Schröppel, B.; Akalin, E.; Baweja, M.; Bloom, R.D.; Florman, S.; Goldstein, M.; Haydel, B.; Hricik, D.E.; Kulkarni, S.; Levine, M.; et al. Peritransplant Eculizumab Does Not Prevent Delayed Graft Function in Deceased Donor Kidney Transplant Recipients: Results of Two Randomized Controlled Pilot Trials. Am. J. Transplant. 2020, 20, 564–572. [Google Scholar] [CrossRef]
- Kaabak, M.; Babenko, N.; Shapiro, R.; Zokoyev, A.; Dymova, O.; Kim, E. A Prospective Randomized, Controlled Trial of Eculizumab to Prevent Ischemia-Reperfusion Injury in Pediatric Kidney Transplantation. Pediatr. Transplant. 2018, 22, e13129. [Google Scholar] [CrossRef] [PubMed]
- Jordan, S.C.; Choi, J.; Aubert, O.; Haas, M.; Loupy, A.; Huang, E.; Peng, A.; Kim, I.; Louie, S.; Ammerman, N.; et al. A Phase I/II, Double-Blind, Placebo-Controlled Study Assessing Safety and Efficacy of C1 Esterase Inhibitor for Prevention of Delayed Graft Function in Deceased Donor Kidney Transplant Recipients. Am. J. Transplant. 2018, 18, 2955–2964. [Google Scholar] [CrossRef] [PubMed]
- Huang, E.; Vo, A.; Choi, J.; Ammerman, N.; Lim, K.; Sethi, S.; Kim, I.; Kumar, S.; Najjar, R.; Peng, A.; et al. Three-Year Outcomes of a Randomized, Double-Blind, Placebo-Controlled Study Assessing Safety and Efficacy of C1 Esterase Inhibitor for Prevention of Delayed Graft Function in Deceased Donor Kidney Transplant Recipients. Clin. J. Am. Soc. Nephrol. 2020, 15, 109–116. [Google Scholar] [CrossRef] [PubMed]
- Stegall, M.D.; Diwan, T.; Raghavaiah, S.; Cornell, L.D.; Burns, J.; Dean, P.G.; Cosio, F.G.; Gandhi, M.J.; Kremers, W.; Gloor, J.M. Terminal Complement Inhibition Decreases Antibody-Mediated Rejection in Sensitized Renal Transplant Recipients. Am. J. Transplant. 2011, 11, 2405–2413. [Google Scholar] [CrossRef]
- Vo, A.A.; Zeevi, A.; Choi, J.; Cisneros, K.; Toyoda, M.; Kahwaji, J.; Peng, A.; Villicana, R.; Puliyanda, D.; Reinsmoen, N.; et al. A Phase I/II Placebo-Controlled Trial of C1-Inhibitor for Prevention of Antibody-Mediated Rejection in HLA Sensitized Patients. Transplantation 2015, 99, 299–308. [Google Scholar] [CrossRef]
- Wooden, B.; Tarragon, B.; Navarro-Torres, M.; Bomback, A.S. Complement Inhibitors for Kidney Disease. Nephrol. Dial. Transplant. 2023, 38, gfad079. [Google Scholar] [CrossRef]
- Andrighetto, S.; Leventhal, J.; Zaza, G.; Cravedi, P. Complement and Complement Targeting Therapies in Glomerular Diseases. Int. J. Mol. Sci. 2019, 20, 6336. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lasorsa, F.; Rutigliano, M.; Milella, M.; Ferro, M.; Pandolfo, S.D.; Crocetto, F.; Simone, S.; Gesualdo, L.; Battaglia, M.; Ditonno, P.; et al. Complement System and the Kidney: Its Role in Renal Diseases, Kidney Transplantation and Renal Cell Carcinoma. Int. J. Mol. Sci. 2023, 24, 16515. https://doi.org/10.3390/ijms242216515
Lasorsa F, Rutigliano M, Milella M, Ferro M, Pandolfo SD, Crocetto F, Simone S, Gesualdo L, Battaglia M, Ditonno P, et al. Complement System and the Kidney: Its Role in Renal Diseases, Kidney Transplantation and Renal Cell Carcinoma. International Journal of Molecular Sciences. 2023; 24(22):16515. https://doi.org/10.3390/ijms242216515
Chicago/Turabian StyleLasorsa, Francesco, Monica Rutigliano, Martina Milella, Matteo Ferro, Savio Domenico Pandolfo, Felice Crocetto, Simona Simone, Loreto Gesualdo, Michele Battaglia, Pasquale Ditonno, and et al. 2023. "Complement System and the Kidney: Its Role in Renal Diseases, Kidney Transplantation and Renal Cell Carcinoma" International Journal of Molecular Sciences 24, no. 22: 16515. https://doi.org/10.3390/ijms242216515