
Mitochondrial GpC and CpG DNA Hypermethylation Cause Metabolic Stress-Induced Mitophagy and Cholestophagy

 , , , , , ,
, , , , , ,  , and
, and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. Overexpression of Mitochondrial Targeted DNMTs Promotes GpC and CpG mtDNA Hypermethylation
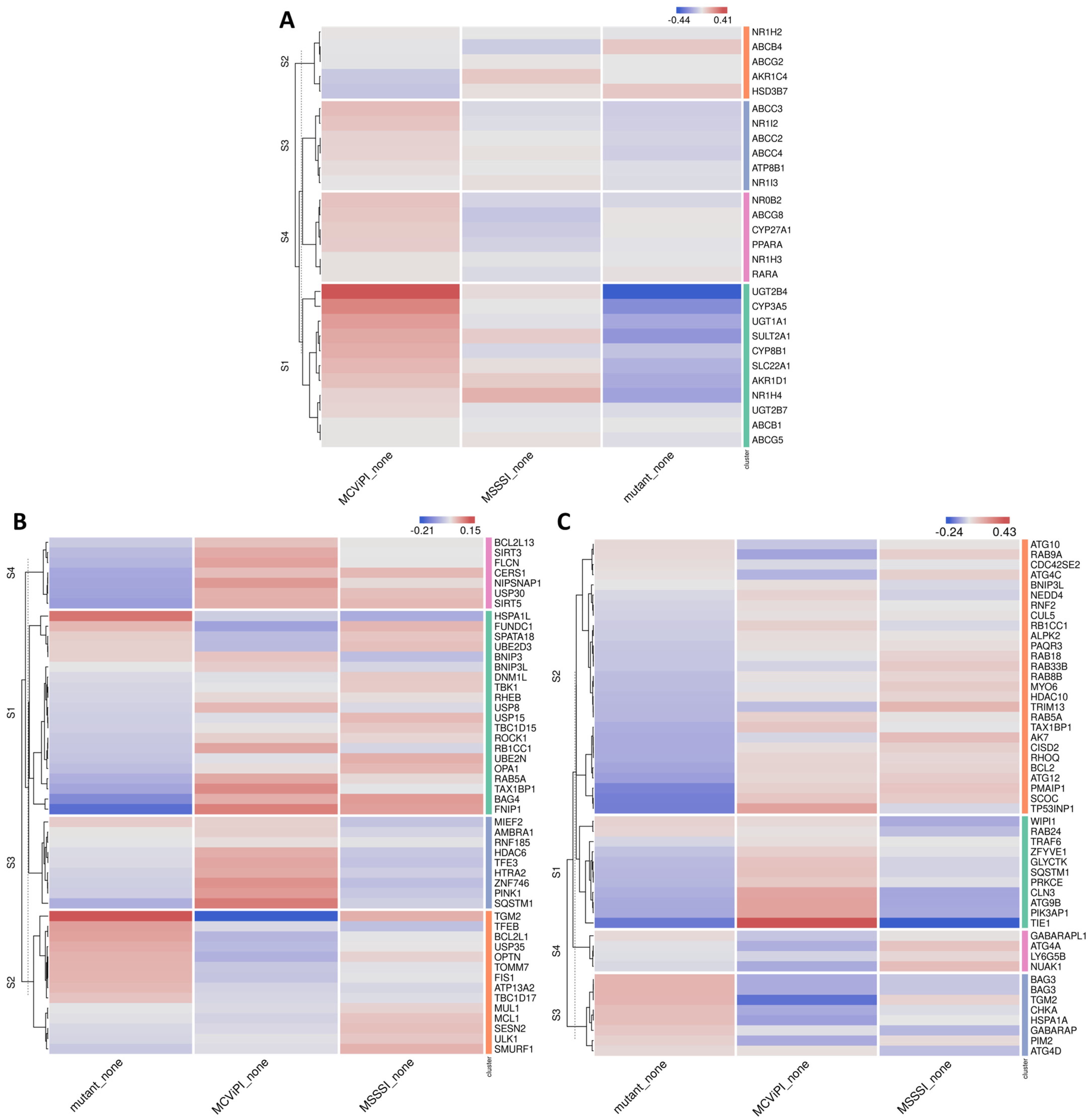
2.2. MtDNA GpC Hypermethylation Promotes Specific Changes in Bile Acid Metabolic Gene Expression
2.3. MtDNA GpC/CpG Hypermethylation Modulates Mito-Nuclear Epigenetic Crosstalk
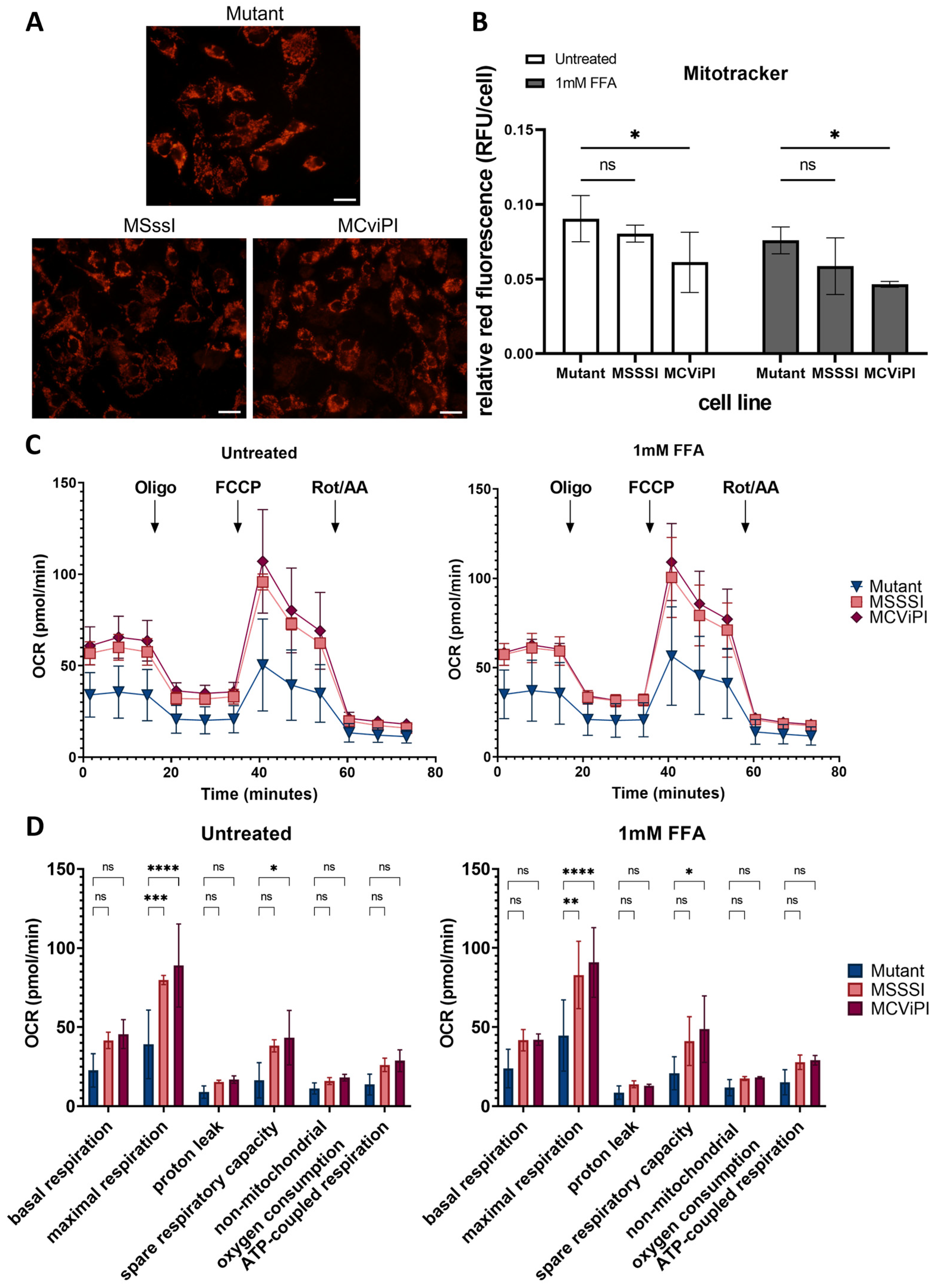
2.4. MtDNA GpC/CpG Hypermethylation Promotes Functional Mitochondrial Changes in Respiration and Morphological Features Associated with Mitophagy Stress Response
2.5. MtDNA GpC-CpG Hypermethylation Promotes Cholestasis-Associated Autophagy-Mitophagy Stress Response
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. FFA Medium
4.3. Nanopore Sequencing
4.4. RNA Extraction and RNA Sequencing
4.5. Quantitative Polymerase Chain Reaction (qPCR)
4.6. Methylation Analysis
4.7. Lipid Quantification with Adipored
4.8. Lipid Peroxidation
4.9. Mitotracker
4.10. Immunofluorescence Staining
4.11. Cell Death Assay—Ferroptosis Screening
4.12. Seahorse
4.13. Electron Microscopy
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Younossi, Z.M.; Koenig, A.B.; Abdelatif, D.; Fazel, Y.; Henry, L.; Wymer, M. Global epidemiology of nonalcoholic fatty liver disease-Meta-analytic assessment of prevalence, incidence, and outcomes. Hepatology 2016, 64, 73–84. [Google Scholar] [CrossRef] [PubMed]
- Loomba, R.; Sanyal, A.J. The global NAFLD epidemic. Nat. Rev. Gastroenterol. Hepatol. 2013, 10, 686–690. [Google Scholar] [CrossRef]
- Chalasani, N.; Younossi, Z.; LaVine, J.E.; Charlton, M.; Cusi, K.; Rinella, M.; Harrison, S.A.; Brunt, E.M.; Sanyal, A.J. The diagnosis and management of nonalcoholic fatty liver disease: Practice guidance from the American Association for the Study of Liver Diseases. Hepatology 2018, 67, 328–357. [Google Scholar] [CrossRef] [PubMed]
- Stepanova, M.; Younossi, Z.M. Independent Association Between Nonalcoholic Fatty Liver Disease and Cardiovascular Disease in the US Population. Clin. Gastroenterol. Hepatol. 2012, 10, 646–650. [Google Scholar] [CrossRef] [PubMed]
- Romero-Gómez, M.; Zelber-Sagi, S.; Trenell, M. Treatment of NAFLD with diet, physical activity and exercise. J. Hepatol. 2017, 67, 829–846. [Google Scholar] [CrossRef]
- Vilar-Gomez, E.; Martinez-Perez, Y.; Calzadilla-Bertot, L.; Torres-Gonzalez, A.; Gra-Oramas, B.; Gonzalez-Fabian, L.; Friedman, S.L.; Diago, M.; Romero-Gomez, M. Weight Loss Through Lifestyle Modification Significantly Reduces Features of Nonalcoholic Steatohepatitis. Gastroenterology 2015, 149, 367–378.e5. [Google Scholar] [CrossRef]
- Juanola, O.; Martínez-López, S.; Francés, R.; Gómez-Hurtado, I. Non-Alcoholic Fatty Liver Disease: Metabolic, Genetic, Epigenetic and Environmental Risk Factors. Int. J. Environ. Res. Public Health 2021, 18, 5227. [Google Scholar] [CrossRef]
- Speliotes, E.K.; Butler, J.L.; Palmer, C.D.; Voight, B.F.; Hirschhorn, J.N. PNPLA3 variants specifically confer increased risk for histologic nonalcoholic fatty liver disease but not metabolic disease. Hepatology 2010, 52, 904–912. [Google Scholar] [CrossRef]
- Tiffon, C. The Impact of Nutrition and Environmental Epigenetics on Human Health and Disease. Int. J. Mol. Sci. 2018, 19, 3425. [Google Scholar] [CrossRef]
- Lai, Z.; Chen, J.; Ding, C.; Wong, K.; Chen, X.; Pu, L.; Huang, Q.; Chen, X.; Cheng, Z.; Liu, Y.; et al. Association of Hepatic Global DNA Methylation and Serum One-Carbon Metabolites with Histological Severity in Patients with NAFLD. Obesity 2020, 28, 197–205. [Google Scholar] [CrossRef]
- Vachher, M.; Bansal, S.; Kumar, B.; Yadav, S.; Burman, A. Deciphering the role of aberrant DNA methylation in NAFLD and NASH. Heliyon 2022, 8, e11119. [Google Scholar] [CrossRef]
- Sun, Q.-F.; Tang, L.-J.; Wang, M.-J.; Zhu, P.-W.; Li, Y.-Y.; Ma, H.-L.; Huang, O.-Y.; Hong, L.; Li, G.; Byrne, C.D.; et al. Potential Blood DNA Methylation Biomarker Genes for Diagnosis of Liver Fibrosis in Patients With Biopsy-Proven Non-alcoholic Fatty Liver Disease. Front. Med. 2022, 9, 864570. [Google Scholar] [CrossRef] [PubMed]
- Sokolowska, K.E.; Maciejewska-Markiewicz, D.; Bińkowski, J.; Palma, J.; Taryma-Leśniak, O.; Kozlowska-Petriczko, K.; Borowski, K.; Baśkiewicz-Hałasa, M.; Hawryłkowicz, V.; Załęcka, P.; et al. Identified in blood diet-related methylation changes stratify liver biopsies of NAFLD patients according to fibrosis grade. Clin. Epigenetics 2022, 14, 157. [Google Scholar] [CrossRef] [PubMed]
- Buzova, D.; Braghini, M.R.; Bianco, S.D.; Re, O.L.; Raffaele, M.; Frohlich, J.; Kisheva, A.; Crudele, A.; Mosca, A.; Sartorelli, M.R.; et al. Profiling of cell-free DNA methylation and histone signatures in pediatric NAFLD: A pilot study. Hepatol. Commun. 2022, 6, 3311–3323. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.; Nano, J.; Ding, J.; Zheng, Y.; Hennein, R.; Liu, C.; Speliotes, E.K.; Huan, T.; Song, C.; Mendelson, M.M.; et al. A Peripheral Blood DNA Methylation Signature of Hepatic Fat Reveals a Potential Causal Pathway for Nonalcoholic Fatty Liver Disease. Diabetes 2019, 68, 1073–1083. [Google Scholar] [CrossRef]
- Hardy, T.; Zeybel, M.; Day, C.P.; Dipper, C.; Masson, S.; McPherson, S.; Henderson, E.; Tiniakos, D.; White, S.; French, J.; et al. Plasma DNA methylation: A potential biomarker for stratification of liver fibrosis in non-alcoholic fatty liver disease. Gut 2017, 66, 1321–1328. [Google Scholar] [CrossRef]
- Loomba, R.; Gindin, Y.; Jiang, Z.; Lawitz, E.; Caldwell, S.; Djedjos, C.S.; Xu, R.; Chung, C.; Myers, R.P.; Subramanian, G.M.; et al. DNA methylation signatures reflect aging in patients with nonalcoholic steatohepatitis. JCI Insight 2018, 3, e96685. [Google Scholar] [CrossRef]
- Hao, Z.; Wu, T.; Cui, X.; Zhu, P.; Tan, C.; Dou, X.; Hsu, K.-W.; Lin, Y.-T.; Peng, P.-H.; Zhang, L.-S.; et al. N6-Deoxyadenosine Methylation in Mammalian Mitochondrial DNA. Mol. Cell 2020, 78, 382–395.e8. [Google Scholar] [CrossRef]
- Stoccoro, A.; Coppedè, F. Mitochondrial DNA Methylation and Human Diseases. Int. J. Mol. Sci. 2021, 22, 4594. [Google Scholar] [CrossRef]
- Rebelo, A.P.; Williams, S.L.; Moraes, C.T. In vivo methylation of mtDNA reveals the dynamics of protein–mtDNA interactions. Nucleic Acids Res. 2009, 37, 6701–6715. [Google Scholar] [CrossRef]
- Mposhi, A.; Cortés-Mancera, F.; Heegsma, J.; de Meijer, V.E.; van de Sluis, B.; Sydor, S.; Bechmann, L.P.; Theys, C.; de Rijk, P.; De Pooter, T.; et al. Mitochondrial DNA methylation in metabolic associated fatty liver disease. Front. Nutr. 2023, 10, 964337. [Google Scholar] [CrossRef]
- Adeva-Andany, M.M.; Carneiro-Freire, N.; Seco-Filgueira, M.; Fernández-Fernández, C.; Mouriño-Bayolo, D. Mitochondrial β-oxidation of saturated fatty acids in humans. Mitochondrion 2019, 46, 73–90. [Google Scholar] [CrossRef] [PubMed]
- Koliaki, C.; Szendroedi, J.; Kaul, K.; Jelenik, T.; Nowotny, P.; Jankowiak, F.; Herder, C.; Carstensen, M.; Krausch, M.; Knoefel, W.T.; et al. Adaptation of Hepatic Mitochondrial Function in Humans with Non-Alcoholic Fatty Liver Is Lost in Steatohepatitis. Cell Metab. 2015, 21, 739–746. [Google Scholar] [CrossRef] [PubMed]
- Nassir, F. NAFLD: Mechanisms, Treatments, and Biomarkers. Biomolecules 2022, 12, 824. [Google Scholar] [CrossRef] [PubMed]
- Shock, L.S.; Thakkar, P.V.; Peterson, E.J.; Moran, R.G.; Taylor, S.M. DNA methyltransferase 1, cytosine methylation, and cytosine hydroxymethylation in mammalian mitochondria. Proc. Natl. Acad. Sci. USA 2011, 108, 3630–3635. [Google Scholar] [CrossRef]
- Pirola, C.J.; Gianotti, T.F.; Burgueño, A.L.; Rey-Funes, M.; Loidl, C.F.; Mallardi, P.; Martino, J.S.; Castaño, G.O.; Sookoian, S. Epigenetic modification of liver mitochondrial DNA is associated with histological severity of nonalcoholic fatty liver disease. Gut 2013, 62, 1356–1363. [Google Scholar] [CrossRef]
- van der Wijst, M.G.P.; van Tilburg, A.Y.; Ruiters, M.H.J.; Rots, M.G. Experimental mitochondria-targeted DNA methylation identifies GpC methylation, not CpG methylation, as potential regulator of mitochondrial gene expression. Sci. Rep. 2017, 7, 177. [Google Scholar] [CrossRef]
- Chiang, J.Y.L. Bile Acid Regulation of Gene Expression: Roles of Nuclear Hormone Receptors. Endocr. Rev. 2002, 23, 443–463. [Google Scholar] [CrossRef]
- Ratman, D.; Mylka, V.; Bougarne, N.; Pawlak, M.; Caron, S.; Hennuyer, N.; Paumelle, R.; De Cauwer, L.; Thommis, J.; Rider, M.H.; et al. Chromatin recruitment of activated AMPK drives fasting response genes co-controlled by GR and PPARα. Nucleic Acids Res. 2016, 44, 10539–10553. [Google Scholar] [CrossRef]
- Iacobazzi, V.; Castegna, A.; Infantino, V.; Andria, G. Mitochondrial DNA methylation as a next-generation biomarker and diagnostic tool. Mol. Genet. Metab. 2013, 110, 25–34. [Google Scholar] [CrossRef]
- Naviaux, R.K. Mitochondrial control of epigenetics. Cancer Biol. Ther. 2008, 7, 1191–1193. [Google Scholar] [CrossRef]
- Parmentier, C.; Couttet, P.; Wolf, A.; Zaccharias, T.; Heyd, B.; Bachellier, P.; Uteng, M.; Richert, L. Evaluation of transcriptomic signature as a valuable tool to study drug-induced cholestasis in primary human hepatocytes. Arch. Toxicol. 2017, 91, 2879–2893. [Google Scholar] [CrossRef] [PubMed]
- Bordi, M.; De Cegli, R.; Testa, B.; Nixon, R.A.; Ballabio, A.; Cecconi, F. A gene toolbox for monitoring autophagy transcription. Cell Death Dis. 2021, 12, 1044. [Google Scholar] [CrossRef] [PubMed]
- Schyman, P.; Xu, Z.; Desai, V.; Wallqvist, A. TOXPANEL: A Gene-Set Analysis Tool to Assess Liver and Kidney Injuries. Front. Pharmacol. 2021, 12, 601511. [Google Scholar] [CrossRef]
- Panzitt, K.; Fickert, P.; Wagner, M. Regulation of autophagy by bile acids and in cholestasis-CholestoPHAGY or CholeSTOPagy. Biochim. Biophys. Acta (BBA)-Mol. Basis Dis. 2021, 1867, 166017. [Google Scholar] [CrossRef]
- Pinto, C.; Ninfole, E.; Benedetti, A.; Marzioni, M.; Maroni, L. Involvement of Autophagy in Ageing and Chronic Cholestatic Diseases. Cells 2021, 10, 2772. [Google Scholar] [CrossRef] [PubMed]
- Lüth, T.; Wasner, K.; Klein, C.; Schaake, S.; Tse, R.; Pereira, S.L.; Laß, J.; Sinkkonen, L.; Grünewald, A.; Trinh, J. Nanopore Single-Molecule Sequencing for Mitochondrial DNA Methylation Analysis: Investigating Parkin-Associated Parkinsonism as a Proof of Concept. Front. Aging Neurosci. 2021, 13, 713084. [Google Scholar] [CrossRef] [PubMed]
- Goldsmith, C.; Rodríguez-Aguilera, J.R.; El-Rifai, I.; Jarretier-Yuste, A.; Hervieu, V.; Raineteau, O.; Saintigny, P.; de Sánchez, V.C.; Dante, R.; Ichim, G.; et al. Low biological fluctuation of mitochondrial CpG and non-CpG methylation at the single-molecule level. Sci. Rep. 2021, 11, 8032. [Google Scholar] [CrossRef]
- Bellizzi, D.; D’Aquila, P.; Scafone, T.; Giordano, M.; Riso, V.; Riccio, A.; Passarino, G. The Control Region of Mitochondrial DNA Shows an Unusual CpG and Non-CpG Methylation Pattern. DNA Res. 2013, 20, 537–547. [Google Scholar] [CrossRef]
- Dou, X.; Boyd-Kirkup, J.D.; McDermott, J.; Zhang, X.; Li, F.; Rong, B.; Zhang, R.; Miao, B.; Chen, P.; Cheng, H.; et al. The strand-biased mitochondrial DNA methylome and its regulation by DNMT3A. Genome Res. 2019, 29, 1622–1634. [Google Scholar] [CrossRef]
- Boughanem, H.; Böttcher, Y.; Tomé-Carneiro, J.; Hazas, M.L.d.L.; Dávalos, A.; Cayir, A.; Macias-González, M. The emergent role of mitochondrial RNA modifications in metabolic alterations. WIREs RNA 2023, 14, e1753. [Google Scholar] [CrossRef] [PubMed]
- Delaunay, S.; Pascual, G.; Feng, B.; Klann, K.; Behm, M.; Hotz-Wagenblatt, A.; Richter, K.; Zaoui, K.; Herpel, E.; Münch, C.; et al. Mitochondrial RNA modifications shape metabolic plasticity in metastasis. Nature 2022, 607, 593–603. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Patterson, A.D.; Krausz, K.W.; Tanaka, N.; Gonzalez, F.J. Metabolomics reveals an essential role for peroxisome proliferator-activated receptor α in bile acid homeostasis. J. Lipid Res. 2012, 53, 1625–1635. [Google Scholar] [CrossRef] [PubMed]
- Schiöth, H.B.; Boström, A.; Murphy, S.K.; Erhart, W.; Hampe, J.; Moylan, C.; Mwinyi, J. A targeted analysis reveals relevant shifts in the methylation and transcription of genes responsible for bile acid homeostasis and drug metabolism in non-alcoholic fatty liver disease. BMC Genom. 2016, 17, 462. [Google Scholar] [CrossRef]
- Wiese, M.; Bannister, A.J. Two genomes, one cell: Mitochondrial-nuclear coordination via epigenetic pathways. Mol. Metab. 2020, 38, 100942. [Google Scholar] [CrossRef]
- Pennisi, G.; Pipitone, R.M.; Cabibi, D.; Enea, M.; Romero-Gomez, M.; Viganò, M.; Bugianesi, E.; Wong, V.W.; Fracanzani, A.L.; Sebastiani, G.; et al. A cholestatic pattern predicts major liver-related outcomes in patients with non-alcoholic fatty liver disease. Liver Int. 2022, 42, 1037–1048. [Google Scholar] [CrossRef]
- Gottlieb, A.; Canbay, A. Why Bile Acids Are So Important in Non-Alcoholic Fatty Liver Disease (NAFLD) Progression. Cells 2019, 8, 1358. [Google Scholar] [CrossRef]
- Ferslew, B.C.; Xie, G.; Johnston, C.K.; Su, M.; Stewart, P.W.; Jia, W.; Brouwer, K.L.; Barritt, A.S. Altered Bile Acid Metabolome in Patients with Nonalcoholic Steatohepatitis. Dig. Dis. Sci. 2015, 60, 3318–3328. [Google Scholar] [CrossRef]
- Van Brantegem, P.; Chatterjee, S.; De Bruyn, T.; Annaert, P.; Deferm, N. Drug-induced cholestasis assay in primary hepatocytes. MethodsX 2020, 7, 101080. [Google Scholar] [CrossRef]
- Xu, X.; Poulsen, K.L.; Wu, L.; Liu, S.; Miyata, T.; Song, Q.; Wei, Q.; Zhao, C.; Lin, C.; Yang, J. Targeted therapeutics and novel signaling pathways in non-alcohol-associated fatty liver/steatohepatitis (NAFL/NASH). Signal Transduct. Target. Ther. 2022, 7, 287. [Google Scholar] [CrossRef]
- Jiao, T.; Ma, Y.; Guo, X.; Ye, Y.; Xie, C. Bile acid and receptors: Biology and drug discovery for nonalcoholic fatty liver disease. Acta Pharmacol. Sin. 2022, 43, 1103–1119. [Google Scholar] [CrossRef] [PubMed]
- Abrigo, J.; Olguín, H.; Gutierrez, D.; Tacchi, F.; Arrese, M.; Cabrera, D.; Valero-Breton, M.; Elorza, A.A.; Simon, F.; Cabello-Verrugio, C. Bile Acids Induce Alterations in Mitochondrial Function in Skeletal Muscle Fibers. Antioxidants 2022, 11, 1706. [Google Scholar] [CrossRef] [PubMed]
- Abrigo, J.; Olguín, H.; Tacchi, F.; Orozco-Aguilar, J.; Valero-Breton, M.; Soto, J.; Castro-Sepúlveda, M.; Elorza, A.A.; Simon, F.; Cabello-Verrugio, C. Cholic and deoxycholic acids induce mitochondrial dysfunction, impaired biogenesis and autophagic flux in skeletal muscle cells. Biol. Res. 2023, 56, 30. [Google Scholar] [CrossRef]
- Rolo, A.P. Bile Acids Affect Liver Mitochondrial Bioenergetics: Possible Relevance for Cholestasis Therapy. Toxicol. Sci. 2000, 57, 177–185. [Google Scholar] [CrossRef]
- Legaki, A.-I.; Moustakas, I.I.; Sikorska, M.; Papadopoulos, G.; Velliou, R.-I.; Chatzigeorgiou, A. Hepatocyte Mitochondrial Dynamics and Bioenergetics in Obesity-Related Non-Alcoholic Fatty Liver Disease. Curr. Obes. Rep. 2022, 11, 126–143. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Ding, W.-X.; Li, T. Cholesterol and bile acid-mediated regulation of autophagy in fatty liver diseases and atherosclerosis. Biochim. Biophys. Acta (BBA)-Mol. Cell Biol. Lipids 2018, 1863, 726–733. [Google Scholar] [CrossRef] [PubMed]
- Ma, K.; Chen, G.; Li, W.; Kepp, O.; Zhu, Y.; Chen, Q. Mitophagy, Mitochondrial Homeostasis, and Cell Fate. Front. Cell Dev. Biol. 2020, 8, 467. [Google Scholar] [CrossRef]
- Ma, X.; McKeen, T.; Zhang, J.; Ding, W.-X. Role and Mechanisms of Mitophagy in Liver Diseases. Cells 2020, 9, 837. [Google Scholar] [CrossRef]
- Moore, M.P.; Cunningham, R.P.; Meers, G.M.; Johnson, S.A.; Wheeler, A.A.; Ganga, R.R.; Spencer, N.M.; Pitt, J.B.; Diaz-Arias, A.; Swi, A.I.A.; et al. Compromised hepatic mitochondrial fatty acid oxidation and reduced markers of mitochondrial turnover in human NAFLD. Hepatology 2022, 76, 1452–1465. [Google Scholar] [CrossRef]
- Zhou, T.; Chang, L.; Luo, Y.; Zhou, Y.; Zhang, J. Mst1 inhibition attenuates non-alcoholic fatty liver disease via reversing Parkin-related mitophagy. Redox Biol. 2019, 21, 101120. [Google Scholar] [CrossRef]
- Lin, D.; He, H.; Ji, H.; Willis, J.; Willard, L.; Jiang, Y.; Medeiros, D.M.; Wark, L.; Han, J.; Liu, Y.; et al. Wolfberries potentiate mitophagy and enhance mitochondrial biogenesis leading to prevention of hepatic steatosis in obese mice: The role of AMP-activated protein kinase α2 subunit. Mol. Nutr. Food Res. 2014, 58, 1005–1015. [Google Scholar] [CrossRef] [PubMed]
- Choi, S.; Dong, B.; Lin, C.J.; Heo, M.J.; Kim, K.H.; Sun, Z.; Wagner, M.; Putluri, N.; Suh, J.M.; Wang, M.C.; et al. Methyl-Sensing Nuclear Receptor Liver Receptor Homolog-1 Regulates Mitochondrial Function in Mouse Hepatocytes. Hepatology 2020, 71, 1055–1069. [Google Scholar] [CrossRef] [PubMed]
- Pérez-Carreras, M.; Del Hoyo, P.; Martín, M.A.; Rubio, J.C.; Martín, A.; Castellano, G.; Colina, F.; Arenas, J.; Solis-Herruzo, J.A. Defective hepatic mitochondrial respiratory chain in patients with nonalcoholic steatohepatitis. Hepatology 2003, 38, 999–1007. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Chakraborty, B.; Safi, R.; Kazmin, D.; Chang, C.; McDonnell, D.P. Dysregulated cholesterol homeostasis results in resistance to ferroptosis increasing tumorigenicity and metastasis in cancer. Nat. Commun. 2021, 12, 5103. [Google Scholar] [CrossRef]
- Reumers, J.; De Rijk, P.; Zhao, H.; Liekens, A.; Smeets, D.; Cleary, J.; Van Loo, P.; Bossche, M.V.D.; Catthoor, K.; Sabbe, B.; et al. Optimized filtering reduces the error rate in detecting genomic variants by short-read sequencing. Nat. Biotechnol. 2012, 30, 61–68. [Google Scholar] [CrossRef]
- Schneider, V.A.; Graves-Lindsay, T.; Howe, K.; Bouk, N.; Chen, H.-C.; Kitts, P.A.; Murphy, T.D.; Pruitt, K.D.; Thibaud-Nissen, F.; Albracht, D.; et al. Evaluation of GRCh38 and de novo haploid genome assemblies demonstrates the enduring quality of the reference assembly. Genome Res. 2017, 27, 849–864. [Google Scholar] [CrossRef]
- Li, H. Minimap2: Pairwise alignment for nucleotide sequences. Bioinformatics 2018, 34, 3094–3100. [Google Scholar] [CrossRef]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef]
- Sedlazeck, F.J.; Rescheneder, P.; Smolka, M.; Fang, H.; Nattestad, M.; von Haeseler, A.; Schatz, M.C. Accurate detection of complex structural variations using single-molecule sequencing. Nat. Methods 2018, 15, 461–468. [Google Scholar] [CrossRef]
- Jiang, T.; Liu, Y.; Jiang, Y.; Li, J.; Gao, Y.; Cui, Z.; Liu, Y.; Liu, B.; Wang, Y. Long-read-based human genomic structural variation detection with cuteSV. Genome Biol. 2020, 21, 189. [Google Scholar] [CrossRef]
- Shao, H.; Ganesamoorthy, D.; Duarte, T.; Cao, M.D.; Hoggart, C.J.; Coin, L.J.M. npInv: Accurate detection and genotyping of inversions using long read sub-alignment. BMC Bioinform. 2018, 19, 261. [Google Scholar] [CrossRef] [PubMed]
- Edge, P.; Bansal, V. Longshot enables accurate variant calling in diploid genomes from single-molecule long read sequencing. Nat. Commun. 2019, 10, 4660. [Google Scholar] [CrossRef] [PubMed]
- Loman, N.J.; Quick, J.; Simpson, J.T. A complete bacterial genome assembled de novo using only nanopore sequencing data. Nat. Methods 2015, 12, 733–735. [Google Scholar] [CrossRef] [PubMed]
- Andrews, S. FastQC A Quality Control Tool for High Throughput Sequence Data [Online]. 2010. Available online: http://www.bioinformatics.babraham.ac.uk/projects/fastqc/ (accessed on 23 August 2023).
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef]
- Szklarczyk, D.; Gable, A.L.; Lyon, D.; Junge, A.; Wyder, S.; Huerta-Cepas, J.; Simonovic, M.; Doncheva, N.T.; Morris, J.H.; Bork, P.; et al. STRING v11: Protein-protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res. 2019, 47, D607–D613. [Google Scholar] [CrossRef]
- Aryee, M.J.; Jaffe, A.E.; Corrada-Bravo, H.; Ladd-Acosta, C.; Feinberg, A.P.; Hansen, K.D.; Irizarry, R.A. Minfi: A flexible and comprehensive Bioconductor package for the analysis of Infinium DNA methylation microarrays. Bioinformatics 2014, 30, 1363–1369. [Google Scholar] [CrossRef]
- Tian, Y.; Morris, T.J.; Webster, A.P.; Yang, Z.; Beck, S.; Feber, A.; Teschendorff, A.E. ChAMP: Updated methylation analysis pipeline for Illumina BeadChips. Bioinformatics 2017, 33, 3982–3984. [Google Scholar] [CrossRef]
- Teschendorff, A.E.; Marabita, F.; Lechner, M.; Bartlett, T.; Tegner, J.; Gomez-Cabrero, D.; Beck, S. A beta-mixture quantile normalization method for correcting probe design bias in Illumina Infinium 450 k DNA methylation data. Bioinformatics 2013, 29, 189–196. [Google Scholar] [CrossRef]
- Zhou, Y.; Zhou, B.; Pache, L.; Chang, M.; Khodabakhshi, A.H.; Tanaseichuk, O.; Benner, C.; Chanda, S.K. Metascape provides a biologist-oriented resource for the analysis of systems-level datasets. Nat. Commun. 2019, 10, 1523. [Google Scholar] [CrossRef]
- Schindelin, J.; Arganda-Carreras, I.; Frise, E.; Kaynig, V.; Longair, M.; Pietzsch, T.; Preibisch, S.; Rueden, C.; Saalfeld, S.; Schmid, B.; et al. Fiji: An open-source platform for biological-image analysis. Nat. Methods 2012, 9, 676–682. [Google Scholar] [CrossRef] [PubMed]
- Lam, J.; Katti, P.; Biete, M.; Mungai, M.; AshShareef, S.; Neikirk, K.; Lopez, E.G.; Vue, Z.; Christensen, T.A.; Beasley, H.K.; et al. A Universal Approach to Analyzing Transmission Electron Microscopy with ImageJ. Cells 2021, 10, 2177. [Google Scholar] [CrossRef] [PubMed]









Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Theys, C.; Ibrahim, J.; Mateiu, L.; Mposhi, A.; García-Pupo, L.; De Pooter, T.; De Rijk, P.; Strazisar, M.; İnce, İ.A.; Vintea, I.; et al. Mitochondrial GpC and CpG DNA Hypermethylation Cause Metabolic Stress-Induced Mitophagy and Cholestophagy. Int. J. Mol. Sci. 2023, 24, 16412. https://doi.org/10.3390/ijms242216412
Theys C, Ibrahim J, Mateiu L, Mposhi A, García-Pupo L, De Pooter T, De Rijk P, Strazisar M, İnce İA, Vintea I, et al. Mitochondrial GpC and CpG DNA Hypermethylation Cause Metabolic Stress-Induced Mitophagy and Cholestophagy. International Journal of Molecular Sciences. 2023; 24(22):16412. https://doi.org/10.3390/ijms242216412
Chicago/Turabian StyleTheys, Claudia, Joe Ibrahim, Ligia Mateiu, Archibold Mposhi, Laura García-Pupo, Tim De Pooter, Peter De Rijk, Mojca Strazisar, İkbal Agah İnce, Iuliana Vintea, and et al. 2023. "Mitochondrial GpC and CpG DNA Hypermethylation Cause Metabolic Stress-Induced Mitophagy and Cholestophagy" International Journal of Molecular Sciences 24, no. 22: 16412. https://doi.org/10.3390/ijms242216412