
Identification and Expressional Analysis of siRNAs Responsive to Fusarium graminearum Infection in Wheat
 and
and
Abstract
:1. Introduction
2. Results
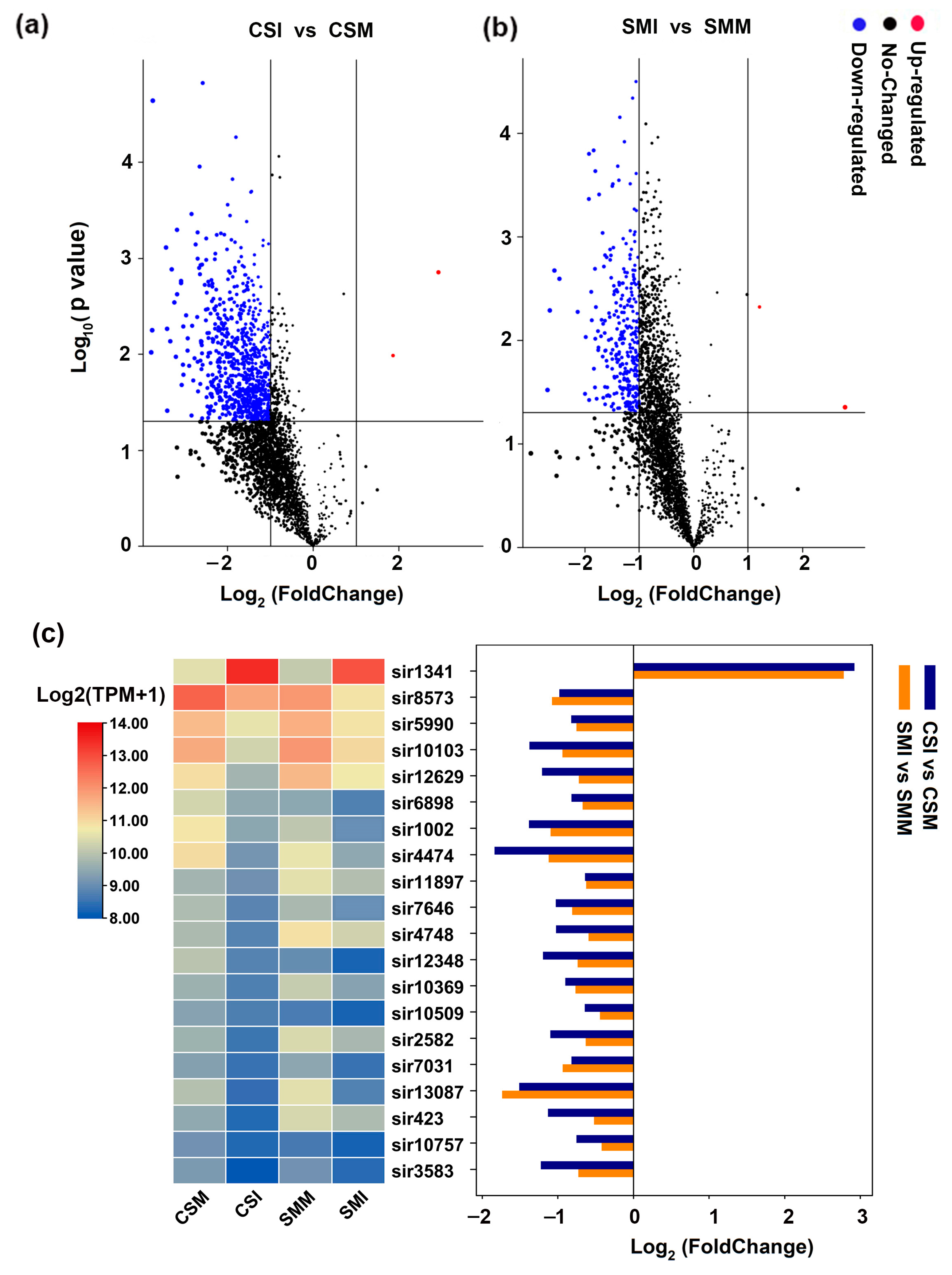
2.1. Identification of siRNAs in Wheat Spike via Small RNA Deep Sequencing
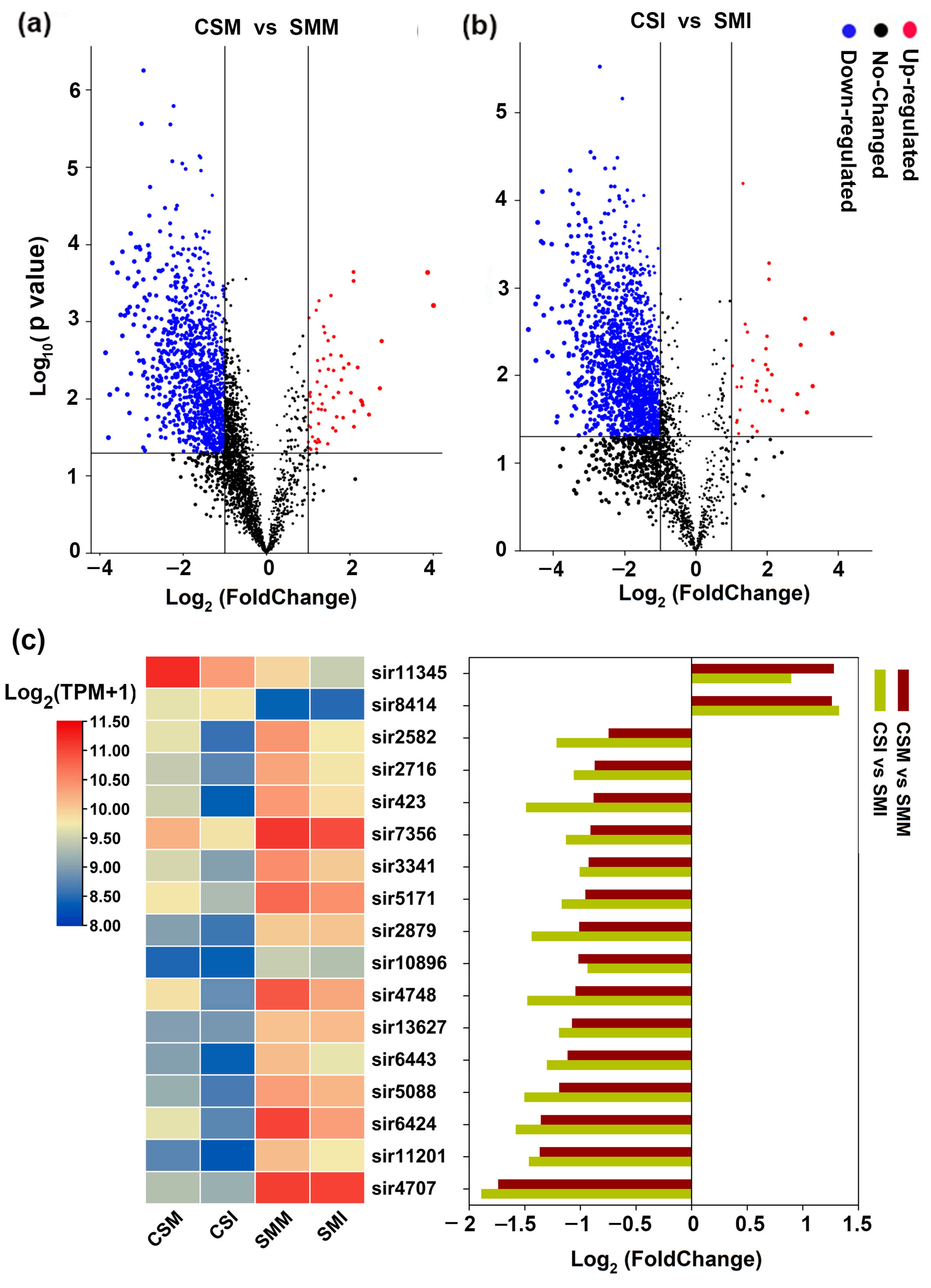
2.2. Most of siRNAs Were Down-Regulated after Infection with F. graminearum
2.3. More siRNAs Were Enriched in Sumai 3 Than in CS
2.4. The Expression of TaDCL3 Genes Were Repressed by F. graminearum
2.5. Disease Resistance-Related Genes Were Up-Regulated after F. graminearum Infection
3. Discussion
3.1. Inhibition of TaDCL3 by F. graminearum Is the Main Reason for Downregulation of siRNA
3.2. Inhibition of siRNA Released Resistance-Related Genes Thereby Improving the Wheat Resistance to FHB
3.3. Potential Application of siRNA into Breeding of FHB-Resistance Wheat
4. Materials and Methods
4.1. Plant Materials and F. graminearum Inoculation
4.2. RNA Extraction
4.3. Small RNA and mRNA Library Construction
4.4. Sequencing Data Analysis
4.5. qRT-PCR Validation of TaDCL3
4.6. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Alisaac, E.; Mahlein, A.-K. Fusarium head blight on wheat: Biology, modern detection and diagnosis and integrated disease management. Toxins 2023, 15, 192. [Google Scholar] [CrossRef]
- Chen, Y.; Kistler, H.C.; Ma, Z. Fusarium graminearum trichothecene mycotoxins: Biosynthesis, regulation, and management. Annu. Rev. Phytopathol. 2019, 57, 15–39. [Google Scholar] [CrossRef]
- McMullen, M.; Jones, R.; Gallenberg, D. Scab of wheat and barley: A re-emerging disease of devastating impact. Plant Dis. 1997, 81, 1340–1348. [Google Scholar] [CrossRef]
- Walter, S.; Nicholson, P.; Doohan, F.M. Action and reaction of host and pathogen during Fusarium head blight disease. New Phytol. 2010, 185, 54–66. [Google Scholar] [CrossRef]
- Vogelgsang, S.; Hecker, A.; Musa, T.; Dorn, B.; Forrer, H.R. On-farm experiments over 5 years in a grain maize/winter wheat rotation: Effect of maize residue treatments on Fusarium graminearum infection and deoxynivalenol contamination in wheat. Mycotoxin Res. 2011, 27, 81–96. [Google Scholar] [CrossRef]
- Bai, G.; Shaner, G. Management and resistance in wheat and barley to Fusarium head blight. Annu. Rev. Phytopathol. 2004, 42, 135–161. [Google Scholar] [CrossRef]
- Buerstmayr, H.; Ban, T.; Anderson, J.A. QTL mapping and marker assisted selection for Fusarium head blight resistance in wheat. Cereal Res. Commun. 2008, 36, 1–3. [Google Scholar] [CrossRef]
- Cai, J.; Wang, S.; Su, Z.; Li, T.; Zhang, X.; Bai, G. Meta-analysis of QTL for Fusarium head blight resistance in Chinese wheat landraces. Crop J. 2019, 7, 784–798. [Google Scholar] [CrossRef]
- Zheng, T.; Hua, C.; Li, L.; Sun, Z.; Yuan, M.; Bai, G.; Humphreys, G.; Li, T. Integration of meta-QTL discovery with omics: Towards a molecular breeding platform for improving wheat resistance to Fusarium head blight. Crop J. 2020, 9, 739–749. [Google Scholar] [CrossRef]
- Waldron, B.L.; Moreno-Sevilla, B.; Anderson, J.A.; Stack, R.W.; Frohberg, R.C. RFLP Mapping of QTL for Fusarium head blight resistance in wheat. Crop Sci. 1999, 39, 805–811. [Google Scholar] [CrossRef]
- Schweiger, W.; Steiner, B.; Vautrin, S.; Nussbaumer, T.; Siegwart, G.; Zamini, M.; Jungreithmeier, F.; Gratl, V.; Lemmens, M.; Mayer, K.F.; et al. Suppressed recombination and unique candidate genes in the divergent haplotype encoding Fhb1, a major Fusarium head blight resistance locus in wheat. Theor. Appl. Genet. 2016, 129, 1607–1623. [Google Scholar] [CrossRef]
- Li, T.; Zhang, H.; Huang, Y.; Su, Z.; Deng, Y.; Liu, H.; Mai, C.; Yu, G.; Li, H.; Yu, L.; et al. Effects of the Fhb1 gene on Fusarium head blight resistance and agronomic traits of winter wheat. Crop J. 2019, 7, 799–808. [Google Scholar] [CrossRef]
- Ma, X.; Liu, C.; Cao, X. Plant transfer RNA-derived fragments: Biogenesis and functions. J. Integr. Plant Biol. 2021, 63, 1399–1409. [Google Scholar] [CrossRef]
- Borges, F.; Martienssen, R.A. The expanding world of small RNAs in plants. Nat. Rev. Mol. Cell Bio. 2015, 16, 727–741. [Google Scholar] [CrossRef]
- Tang, G. siRNA and miRNA: An insight into RISCs. Trends Biochem. Sci. 2005, 30, 106–114. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Xue, Y.; Zhang, L.; Zhong, Z.; Feng, S.; Wang, C.; Xiao, L.; Yang, Z.; Harris, C.J.; Wu, Z.; et al. Mechanism of siRNA production by a plant Dicer-RNA complex in dicing-competent conformation. Science 2021, 374, 1152–1157. [Google Scholar] [CrossRef] [PubMed]
- Dalmay, T.; Hamilton, A.; Rudd, S.; Angell, S.; Baulcombe, D.C. An RNA-Dependent RNA polymerase gene in Arabidopsis is required for posttranscriptional gene silencing mediated by a transgene but not by a virus. Cell 2000, 101, 543–553. [Google Scholar] [CrossRef]
- Zhan, J.; Meyers, B. Plant small RNAs: Their biogenesis, regulatory roles, and functions. Annu. Rev. Plant Biol. 2023, 74, 21–51. [Google Scholar] [CrossRef]
- Song, X.; Li, Y.; Cao, X.; Qi, Y. MicroRNAs and their regulatory roles in plant-environment interactions. Annu. Rev. Plant Biol. 2019, 70, 489–525. [Google Scholar] [CrossRef]
- Wu, H.; Li, B.; Iwakawa, H.O.; Pan, Y.; Tang, X.; Ling-Hu, Q.; Liu, Y.; Sheng, S.; Feng, L.; Zhang, H.; et al. Plant 22-nt siRNAs mediate translational repression and stress adaptation. Nature 2020, 581, 89–93. [Google Scholar] [CrossRef]
- Henderson, I.R.; Zhang, X.; Lu, C.; Johnson, L.; Meyers, B.C.; Green, P.J.; Jacobsen, S.E. Dissecting Arabidopsis thaliana DICER function in small RNA processing, gene silencing and DNA methylation patterning. Nat. Genet. 2006, 38, 721–725. [Google Scholar] [CrossRef]
- Xie, Z.; Allen, E.; Wilken, A.; Carrington, J.C. DICER-LIKE 4 functions in trans-acting small interfering RNA biogenesis and vegetative phase change in Arabidopsis thaliana. Proc. Natl. Acad. Sci. USA 2005, 102, 12984–12989. [Google Scholar] [CrossRef]
- Katiyar-Agarwal, S.; Morgan, R.; Dahlbeck, D.; Borsani, O.; Villegas, A., Jr.; Zhu, J.K.; Staskawicz, B.J.; Jin, H. A pathogen-inducible endogenous siRNA in plant immunity. Proc. Natl. Acad. Sci. USA 2006, 103, 18002–18007. [Google Scholar] [CrossRef] [PubMed]
- Colombo, M.; Raposo, G.; Thery, C. Biogenesis, secretion, and intercellular interactions of exosomes and other extracellular vesicles. Annu. Rev. Cell Dev. Biol. 2014, 30, 255–289. [Google Scholar] [CrossRef] [PubMed]
- Hou, Y.; Zhai, Y.; Feng, L.; Karimi, H.Z.; Rutter, B.D.; Zeng, L.; Choi, D.S.; Zhang, B.; Gu, W.; Chen, X.; et al. A phytophthora effector suppresses Trans-Kingdom RNAi to promote disease susceptibility. Cell Host Microbe 2019, 25, 153–165, e5. [Google Scholar] [CrossRef]
- Kamthan, A.; Chaudhuri, A.; Kamthan, M.; Datta, A. Small RNAs in plants: Recent development and application for crop improvement. Front. Plant Sci. 2015, 6, 208. [Google Scholar] [CrossRef]
- Sun, Z.; Hu, Y.; Zhou, Y.; Jiang, N.; Hu, S.; Li, L.; Li, T. tRNA-derived fragments from wheat are potentially involved in susceptibility to Fusarium head blight. BMC Plant Biol. 2022, 22, 3. [Google Scholar] [CrossRef] [PubMed]
- Szittya, G.; Silhavy, D.; Molnar, A.; Havelda, Z.; Lovas, A.; Lakatos, L.; Banfalvi, Z.; Burgyan, J. Low temperature inhibits RNA silencing-mediated defence by the control of siRNA generation. EMBO J. 2003, 22, 633–640. [Google Scholar] [CrossRef]
- Qiao, L.; Lan, C.; Capriotti, L.; Ah-Fong, A.; Nino Sanchez, J.; Hamby, R.; Heller, J.; Zhao, H.; Glass, N.L.; Judelson, H.S.; et al. Spray-induced gene silencing for disease control is dependent on the efficiency of pathogen RNA uptake. Plant Biotechnol. J. 2021, 19, 1756–1768. [Google Scholar] [CrossRef]
- Werner, B.T.; Gaffar, F.Y.; Schuemann, J.; Biedenkopf, D.; Koch, A.M. RNA-Spray-Mediated silencing of Fusarium graminearum AGO and DCL genes improve barley disease resistance. Front. Plant Sci. 2020, 11, 476. [Google Scholar] [CrossRef]
- Jiao, J.; Peng, D. Wheat microRNA1023 suppresses invasion of Fusarium graminearum via targeting and silencing FGSG_03101. J. Plant Interact. 2018, 13, 514–521. [Google Scholar] [CrossRef]
- Zhou, M.P.; Hayden, M.J.; Zhang, Z.Y.; Lu, W.Z.; Ma, H.X. Saturation and mapping of a major Fusarium head blight resistance QTL on chromosome 3BS of Sumai 3 wheat. J. Appl. Genet. 2010, 51, 19–25. [Google Scholar] [CrossRef]
- Schweiger, W.; Steiner, B.; Ametz, C.; Siegwart, G.; Wiesenberger, G.; Berthiller, F.; Lemmens, M.; Jia, H.; Adam, G.; Muehlbauer, G.J.; et al. Transcriptomic characterization of two major Fusarium resistance quantitative trait loci (QTLs), Fhb1 and Qfhs.ifa-5A, identifies novel candidate genes. Mol. Plant Pathol. 2013, 14, 772–785. [Google Scholar] [CrossRef] [PubMed]
- Choulet, F.; Wicker, T.; Rustenholz, C.; Paux, E.; Salse, J.; Leroy, P.; Schlub, S.; Le Paslier, M.C.; Magdelenat, G.; Gonthier, C.; et al. Megabase level sequencing reveals contrasted organization and evolution patterns of the wheat gene and transposable element spaces. Plant Cell 2010, 22, 1686–1701. [Google Scholar] [CrossRef]
- Ma, H.X.; Bai, G.H.; Zhang, X.; Lu, W.Z. Main effects, epistasis, and environmental interactions of quantitative trait Loci for Fusarium head blight resistance in a recombinant inbred population. Phytopathology 2006, 96, 534–541. [Google Scholar] [CrossRef] [PubMed]
- Bai, G.; Kolb, F.L.; Shaner, G.; Domier, L.L. Amplified fragment length polymorphism markers linked to a major quantitative trait locus controlling scab resistance in wheat. Phytopathology 1999, 89, 343–348. [Google Scholar] [CrossRef] [PubMed]
- Langmead, B.; Trapnell, C.; Pop, M.; Salzberg, S.L. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009, 10, R25. [Google Scholar] [CrossRef] [PubMed]
- Li, R.; Li, Y.; Kristiansen, K.; Wang, J. SOAP: Short oligonucleotide alignment program. Bioinformatics 2008, 24, 713–714. [Google Scholar] [CrossRef]
- Kim, D.; Langmead, B.; Salzberg, S.L. HISAT: A fast spliced aligner with low memory requirements. Nat. Methods 2015, 12, 357–360. [Google Scholar] [CrossRef]
- Li, B.; Dewey, C.N. RSEM: Accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinform. 2011, 12, 323. [Google Scholar] [CrossRef]
- Chen, C.; Chen, H.; Zhang, Y.; Thomas, H.R.; Frank, M.H.; He, Y.; Xia, R. TBtools: An integrative toolkit developed for interactive analyses of big biological data. Mol. Plant 2020, 13, 1194–1202. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| siRNA ID | Length | CSI | CSM | Log2 (CSI/CSM) | p Value |
|---|---|---|---|---|---|
| sir1316 | 24 | NA | 52 | NA | 0.0005 |
| sir7086 | 24 | NA | 56 | NA | 0.0063 |
| sir13238 | 21 | NA | 69 | NA | 0.0023 |
| sir65 | 24 | 19 | 104 | −2.48 | 0.0086 |
| sir422 | 24 | 97 | 275 | −1.49 | 0.0093 |
| sir11652 | 24 | 80 | 173 | −1.12 | 0.0081 |
| sir14089 | 24 | 7 | 78 | −3.54 | 0.0009 |
| sir1202 | 24 | 33 | 104 | −1.64 | 0.0040 |
| sir2890 | 24 | 77 | 188 | −1.28 | 0.0078 |
| sir4944 | 24 | 15 | 93 | −2.61 | 0.0017 |
| sir5351 | 24 | 21 | 192 | −3.22 | 0.0057 |
| sir11759 | 24 | 76 | 409 | −2.43 | 0.0054 |
| sir11357 | 24 | 25 | 62 | −1.33 | 0.0049 |
| sir4897 | 24 | 84 | 237 | −1.50 | 0.0092 |
| sir14609 | 24 | 16 | 79 | −2.30 | 0.0074 |
| sir195 | 24 | 14 | 52 | −1.89 | 0.0070 |
| sir987 | 24 | 17 | 57 | −1.74 | 0.0022 |
| sir6137 | 21 | 47 | 108 | −1.20 | 0.0060 |
| sir655 | 24 | 158 | 675 | −2.1 | 0.0005 |
| sir133 | 24 | 58 | 174 | −1.58 | 0.0080 |
| sir13864 | 24 | 31 | 117 | −1.92 | 0.0069 |
| sir1199 | 24 | 368 | 1205 | −1.71 | 0.0085 |
| sir5670 | 24 | 74 | 259 | −1.81 | 0.0041 |
| sir10341 | 24 | 7 | 51 | −2.93 | 0.0007 |
| sir12766 | 22 | 37 | 141 | −1.92 | 0.0068 |
| siRNA ID | Length | SMI | SMM | Log2 (SMI/SMM) | p Value |
|---|---|---|---|---|---|
| sir13416 | 23 | NA | 52 | NA | 0.0001 |
| sir7183 | 24 | NA | 56 | NA | 0.0001 |
| sir2434 | 24 | 22 | 59 | −1.39 | 0.0016 |
| sir6352 | 24 | 103 | 254 | −1.29 | 0.0064 |
| sir4847 | 24 | 17 | 51 | −1.56 | 0.0027 |
| sir14240 | 23 | 23 | 56 | −1.27 | 0.00877 |
| sir6683 | 24 | 21 | 63 | −1.62 | 0.0012 |
| sir2749 | 24 | 63 | 356 | −2.51 | 0.0006 |
| sir7740 | 24 | 33 | 79 | −1.24 | 0.0073 |
| sir12008 | 24 | 33 | 78 | −1.23 | 0.0075 |
| sir4210 | 24 | 29 | 93 | −1.69 | 0.0049 |
| sir1979 | 24 | 39 | 102 | −1.37 | 0.0016 |
| sir8479 | 23 | 81 | 163 | −1.01 | 0.0066 |
| sir10710 | 24 | 59 | 120 | −1.03 | 0.0059 |
| sir9799 | 24 | 31 | 81 | −1.41 | 0.0036 |
| sir10021 | 24 | 627 | 1346 | −1.10 | 0.0095 |
| sir235 | 24 | 26 | 71 | −1.47 | 0.0029 |
| sir7489 | 21 | 108 | 265 | −1.29 | 0.0011 |
| sir14172 | 24 | 21 | 62 | −1.56 | 0.0059 |
| sir6136 | 21 | 35 | 98 | −1.49 | 0.0062 |
| sir1594 | 21 | 46 | 179 | −1.96 | 0.0001 |
| sir12405 | 22 | 92 | 227 | −1.29 | 0.0011 |
| Name | Forward | Reverse |
|---|---|---|
| TaDLC3-1A | CGGCTCAAAATGGACAAAGG | TCAGCGATGCTGAATCCTGG |
| TaDLC3-1B | AACTTCTCGGTCAAGGGCCT | TATTGCACCGGCAATGCTTT |
| TaDLC3-1D | GTCATTTTCCTCCCCCCAAAC | GGAAGCGCATGTCTGTAGGC |
| TaActin | ACCTTCAGTTGCCCAGCAAT | CAGAGTCGAGCACAATACCAGTTG |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fu, K.; Wu, Q.; Jiang, N.; Hu, S.; Ye, H.; Hu, Y.; Li, L.; Li, T.; Sun, Z. Identification and Expressional Analysis of siRNAs Responsive to Fusarium graminearum Infection in Wheat. Int. J. Mol. Sci. 2023, 24, 16005. https://doi.org/10.3390/ijms242116005
Fu K, Wu Q, Jiang N, Hu S, Ye H, Hu Y, Li L, Li T, Sun Z. Identification and Expressional Analysis of siRNAs Responsive to Fusarium graminearum Infection in Wheat. International Journal of Molecular Sciences. 2023; 24(21):16005. https://doi.org/10.3390/ijms242116005
Chicago/Turabian StyleFu, Kai, Qianhui Wu, Ning Jiang, Sijia Hu, Hongyan Ye, Yi Hu, Lei Li, Tao Li, and Zhengxi Sun. 2023. "Identification and Expressional Analysis of siRNAs Responsive to Fusarium graminearum Infection in Wheat" International Journal of Molecular Sciences 24, no. 21: 16005. https://doi.org/10.3390/ijms242116005