
Nε-Carboxymethyl-Lysine Modification of Extracellular Matrix Proteins Augments Fibroblast Activation

 , and
, and
Abstract
:1. Introduction
2. Results
2.1. Accumulation of CML-Modified ECM in Fibrotic Lesions of IPF Lungs
2.2. CML Modification Attenuates ECM-Dependent Inhibition of Collagen and FN1 Gene Expression in Fibroblasts
2.3. CML Modification Attenuates ECM-Driven Inhibition of Fibroproliferation
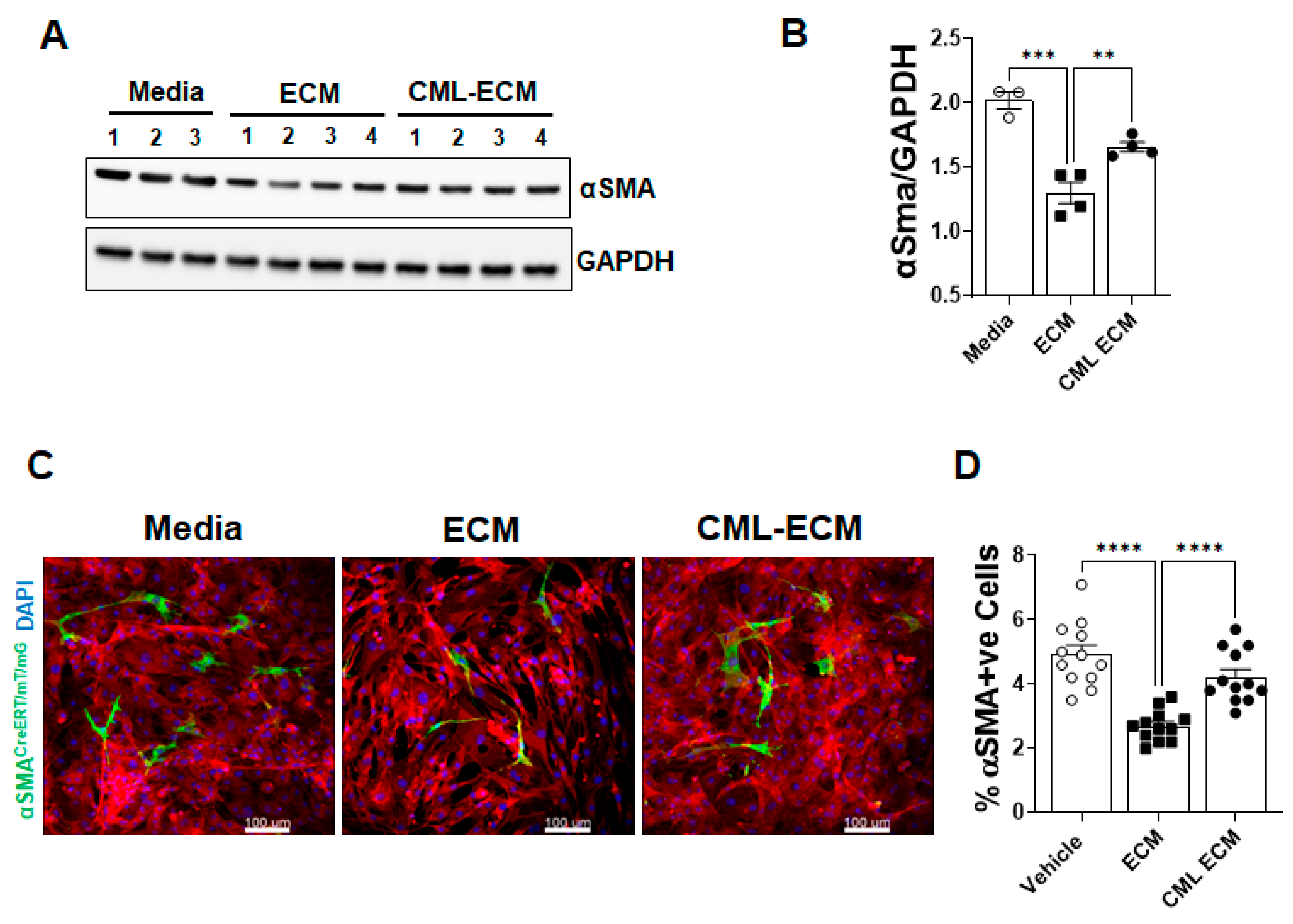
2.4. CML Modification of ECM Attenuates Fibroblast to Myofibroblast Transformation
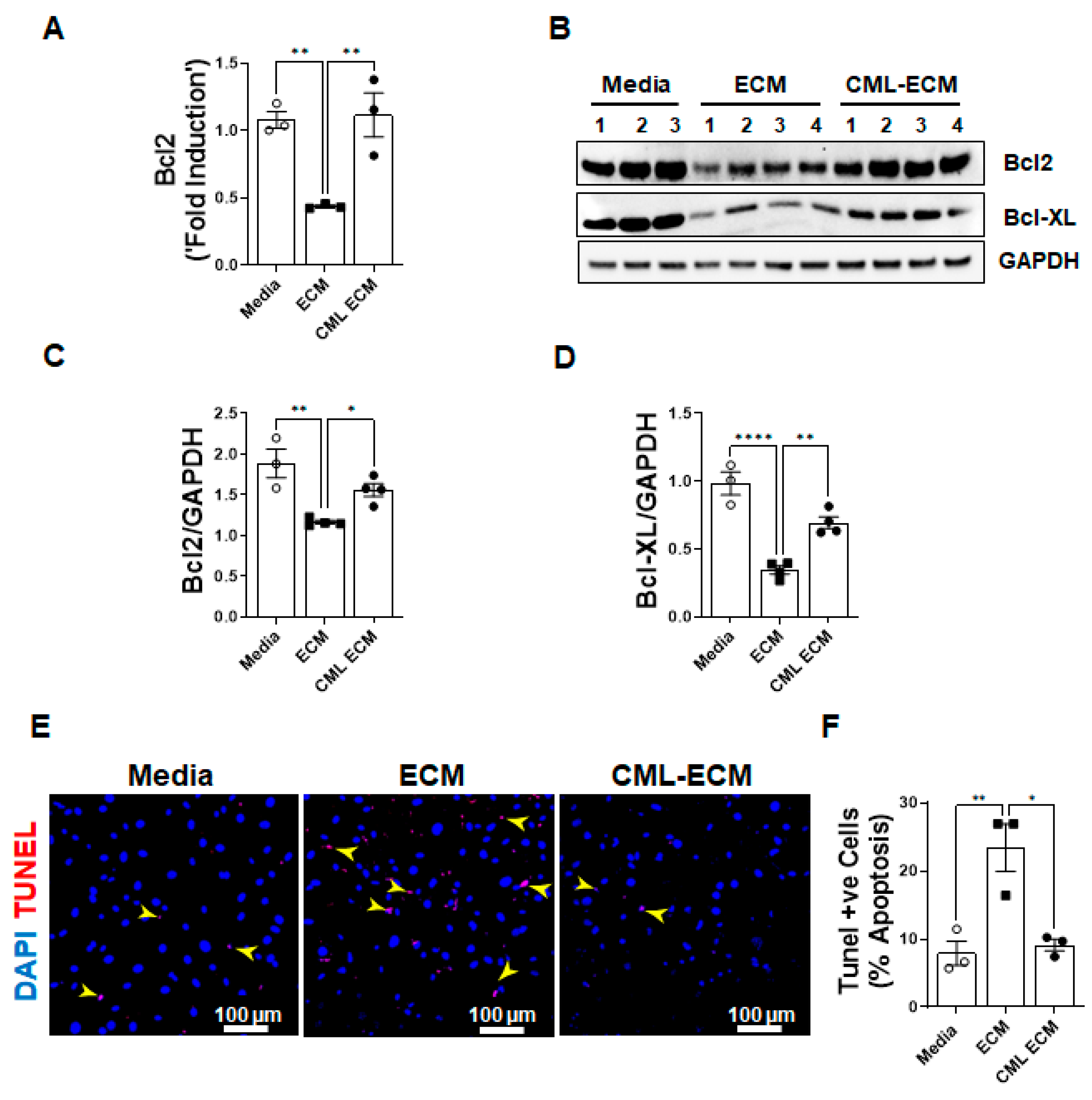
2.5. CML Modification of ECM Attenuates Apoptotic Clearance of Fibroblasts
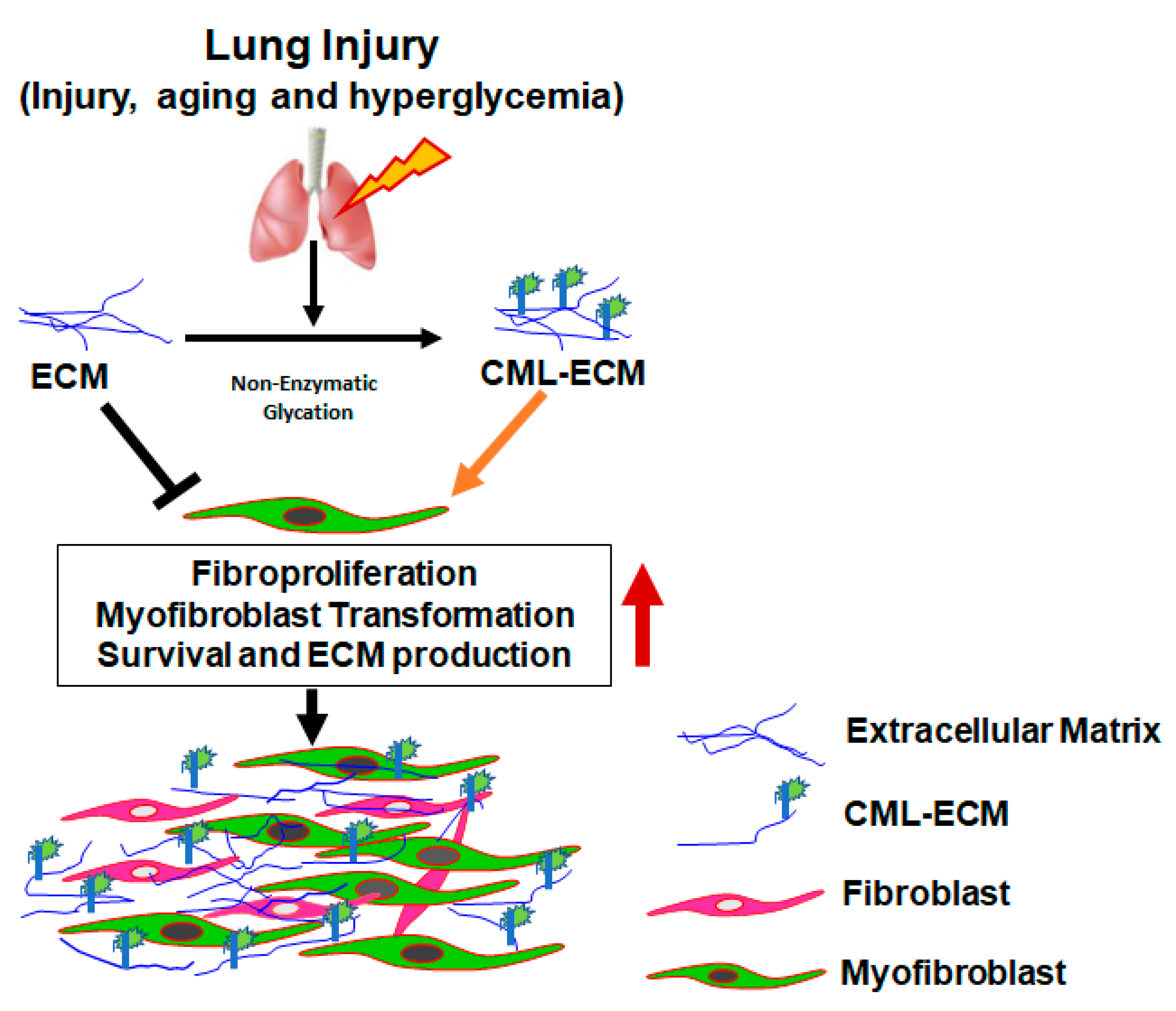
3. Discussion
4. Materials and Methods
4.1. Human Samples
4.2. Extracellular Matrix (ECM) Proteins Extraction
4.3. Nε-Carboxymethyllysine Modification of ECM (CML-ECM)
4.4. Histology and Immunohistochemistry
4.5. Preparation of Lung Resident Fibroblasts
4.6. Immunoblotting
4.7. Quantitative RT-PCR Analysis
4.8. BrdU Cell Proliferation Assay
4.9. TUNEL Assay
4.10. Myofibroblast Transformation Assay
4.11. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Phan, T.H.G.; Paliogiannis, P.; Nasrallah, G.K.; Giordo, R.; Eid, A.H.; Fois, A.G.; Zinellu, A.; Mangoni, A.A.; Pintus, G. Emerging cellular and molecular determinants of idiopathic pulmonary fibrosis. Cell. Mol. Life Sci. 2021, 78, 2031–2057. [Google Scholar] [CrossRef] [PubMed]
- León-Román, F.; Valenzuela, C.; Molina-Molina, M. Idiopathic pulmonary fibrosis. Med. Clín. (Engl. Ed.) 2022, 159, 189–194. [Google Scholar] [CrossRef]
- Gajjala, P.R.; Singh, P.; Odayar, V.; Ediga, H.H.; McCormack, F.X.; Madala, S.K. Wilms Tumor 1-Driven Fibroblast Activation and Subpleural Thickening in Idiopathic Pulmonary Fibrosis. Int. J. Mol. Sci. 2023, 24, 2850. [Google Scholar] [CrossRef]
- Martinez, F.J.; Collard, H.R.; Pardo, A.; Raghu, G.; Richeldi, L.; Selman, M.; Swigris, J.J.; Taniguchi, H.; Wells, A.U. Idiopathic pulmonary fibrosis. Nat. Rev. Dis. Primers 2017, 3, 17074. [Google Scholar] [CrossRef] [PubMed]
- Moss, B.J.; Ryter, S.W.; Rosas, I.O. Pathogenic mechanisms underlying idiopathic pulmonary fibrosis. Annu. Rev. Pathol. Mech. Dis. 2022, 17, 515–546. [Google Scholar] [CrossRef] [PubMed]
- Steele, M.P.; Schwartz, D.A. Molecular mechanisms in progressive idiopathic pulmonary fibrosis. Annu. Rev. Med. 2013, 64, 265–276. [Google Scholar] [CrossRef]
- Wolters, P.J.; Collard, H.R.; Jones, K.D. Pathogenesis of idiopathic pulmonary fibrosis. Annu. Rev. Pathol. Mech. Dis. 2014, 9, 157–179. [Google Scholar] [CrossRef] [PubMed]
- Mei, Q.; Liu, Z.; Zuo, H.; Yang, Z.; Qu, J. Idiopathic pulmonary fibrosis: An update on pathogenesis. Front. Pharmacol. 2022, 12, 797292. [Google Scholar] [CrossRef]
- Upagupta, C.; Shimbori, C.; Alsilmi, R.; Kolb, M. Matrix abnormalities in pulmonary fibrosis. Eur. Respir. Rev. 2018, 27. [Google Scholar] [CrossRef]
- Gattazzo, F.; Urciuolo, A.; Bonaldo, P. Extracellular matrix: A dynamic microenvironment for stem cell niche. Biochim. Biophys. Acta (BBA)-Gen. Subj. 2014, 1840, 2506–2519. [Google Scholar] [CrossRef]
- Herrera, J.; Henke, C.A.; Bitterman, P.B. Extracellular matrix as a driver of progressive fibrosis. J. Clin. Investig. 2018, 128, 45–53. [Google Scholar] [CrossRef] [PubMed]
- Cho, S.J.; Stout-Delgado, H.W. Aging and lung disease. Annu. Rev. Physiol. 2020, 82, 433–459. [Google Scholar] [CrossRef] [PubMed]
- Kyung, S.Y.; Byun, K.H.; Yoon, J.Y.; Kim, Y.J.; Lee, S.P.; Park, J.-W.; Lee, B.H.; Park, J.S.; Jang, A.S.; Park, C.S. Advanced glycation end-products and receptor for advanced glycation end-products expression in patients with idiopathic pulmonary fibrosis and NSIP. Int. J. Clin. Exp. Pathol. 2014, 7, 221. [Google Scholar]
- Machahua, C.; Montes-Worboys, A.; Llatjos, R.; Escobar, I.; Dorca, J.; Molina-Molina, M.; Vicens-Zygmunt, V. Increased AGE-RAGE ratio in idiopathic pulmonary fibrosis. Respir. Res. 2016, 17, 144. [Google Scholar] [CrossRef] [PubMed]
- Matsuse, T.; Ohga, E.; Teramoto, S.; Fukayama, M.; Nagai, R.; Horiuchi, S.; Ouchi, Y. Immunohistochemical localisation of advanced glycation end products in pulmonary fibrosis. J. Clin. Pathol. 1998, 51, 515–519. [Google Scholar] [CrossRef] [PubMed]
- Karsdal, M.A.; Nielsen, M.J.; Sand, J.M.; Henriksen, K.; Genovese, F.; Bay-Jensen, A.-C.; Smith, V.; Adamkewicz, J.I.; Christiansen, C.; Leeming, D.J. Extracellular matrix remodeling: The common denominator in connective tissue diseases possibilities for evaluation and current understanding of the matrix as more than a passive architecture, but a key player in tissue failure. Assay Drug Dev. Technol. 2013, 11, 70–92. [Google Scholar] [CrossRef]
- Schalkwijk, C.G.; Miyata, T. Early-and advanced non-enzymatic glycation in diabetic vascular complications: The search for therapeutics. Amino Acids 2012, 42, 1193–1204. [Google Scholar] [CrossRef]
- Yamagishi, S.-i.; Maeda, S.; Matsui, T.; Ueda, S.; Fukami, K.; Okuda, S. Role of advanced glycation end products (AGEs) and oxidative stress in vascular complications in diabetes. Biochim. Biophys. Acta (BBA)-Gen. Subj. 2012, 1820, 663–671. [Google Scholar] [CrossRef]
- Kumar Pasupulati, A.; Chitra, P.S.; Reddy, G.B. Advanced glycation end products mediated cellular and molecular events in the pathology of diabetic nephropathy. Biomol. Concepts 2016, 7, 293–309. [Google Scholar] [CrossRef]
- Delgado-Andrade, C. Carboxymethyl-lysine: Thirty years of investigation in the field of AGE formation. Food Funct. 2016, 7, 46–57. [Google Scholar] [CrossRef]
- Reddy, S.; Bichler, J.; Wells-Knecht, K.J.; Thorpe, S.R.; Baynes, J.W. N.epsilon.-(Carboxymethyl) lysine is a dominant advanced glycation end product (AGE) antigen in tissue proteins. Biochemistry 1995, 34, 10872–10878. [Google Scholar] [CrossRef] [PubMed]
- Brownlee, M. Michael, Advanced protein glycosylation in diabetes and aging. Annu. Rev. Med. 1995, 46, 223–234. [Google Scholar] [CrossRef] [PubMed]
- Fontaine-Delaruelle, C.; Viart-Ferber, C.; Luyton, C.; Couraud, S. Lung function in patients with diabetes mellitus. Rev. Pneumol. Clin. 2015, 72, 10–16. [Google Scholar] [CrossRef] [PubMed]
- Anandhalakshmi, S.; Manikandan, S.; Ganeshkumar, P.; Ramachandran, C. Alveolar gas exchange and pulmonary functions in patients with type II diabetes mellitus. J. Clin. Diagn. Res. JCDR 2013, 7, 1874. [Google Scholar]
- Zawada, A.; Machowiak, A.; Rychter, A.M.; Ratajczak, A.E.; Szymczak-Tomczak, A.; Dobrowolska, A.; Krela-Kaźmierczak, I. Accumulation of Advanced Glycation End-Products in the Body and Dietary Habits. Nutrients 2022, 14, 3982. [Google Scholar] [CrossRef] [PubMed]
- Zieman, S.J.; Kass, D.A. Advanced glycation end product cross-linking: Pathophysiologic role and therapeutic target in cardiovascular disease. Congest. Heart Fail. 2004, 10, 144–151. [Google Scholar] [CrossRef]
- Nishad, R.; Tahaseen, V.; Kavvuri, R.; Motrapu, M.; Singh, A.K.; Peddi, K.; Pasupulati, A.K. Advanced-glycation end-products induce podocyte injury and contribute to proteinuria. Front. Med. 2021, 8, 685447. [Google Scholar] [CrossRef]
- Lee, J.; Hyon, J.-Y.; Min, J.Y.; Huh, Y.H.; Kim, H.J.; Lee, H.; Yun, S.H.; Choi, C.-W.; Ha, S.J.; Park, J. Mitochondrial carnitine palmitoyltransferase 2 is involved in Nε-(carboxymethyl)-lysine-mediated diabetic nephropathy. Pharmacol. Res. 2020, 152, 104600. [Google Scholar] [CrossRef]
- Lyu, C.; Kong, W.; Liu, Z.; Wang, S.; Zhao, P.; Liang, K.; Niu, Y.; Yang, W.; Xiang, C.; Hu, X. Advanced glycation end-products as mediators of the aberrant crosslinking of extracellular matrix in scarred liver tissue. Nat. Biomed. Eng. 2023, 1–18. [Google Scholar] [CrossRef]
- Jang, M.; Oh, S.W.; Lee, Y.; Kim, J.Y.; Ji, E.S.; Kim, P. Targeting extracellular matrix glycation to attenuate fibroblast activation. Acta Biomater. 2022, 141, 255–263. [Google Scholar] [CrossRef]
- Sontake, V.; Wang, Y.; Kasam, R.K.; Sinner, D.; Reddy, G.B.; Naren, A.P.; McCormack, F.X.; White, E.S.; Jegga, A.G.; Madala, S.K. Hsp90 regulation of fibroblast activation in pulmonary fibrosis. JCI Insight 2017, 2, e91454. [Google Scholar] [CrossRef] [PubMed]
- Thomas, M.C. Advanced glycation end products. Diabetes Kidney 2011, 170, 66–74. [Google Scholar]
- Yuan, Y.; Sun, H.; Sun, Z. Advanced glycation end products (AGEs) increase renal lipid accumulation: A pathogenic factor of diabetic nephropathy (DN). Lipids Health Dis. 2017, 16, 126. [Google Scholar] [CrossRef] [PubMed]
- Lee, E.-J.; Kang, M.-K.; Kim, D.Y.; Kim, Y.-H.; Oh, H.; Kang, Y.-H. Chrysin inhibits advanced glycation end products-induced kidney fibrosis in renal mesangial cells and diabetic kidneys. Nutrients 2018, 10, 882. [Google Scholar] [CrossRef] [PubMed]
- Kumar, P.A.; Welsh, G.I.; Raghu, G.; Menon, R.K.; Saleem, M.A.; Reddy, G.B. Carboxymethyl lysine induces EMT in podocytes through transcription factor ZEB2: Implications for podocyte depletion and proteinuria in diabetes mellitus. Arch. Biochem. Biophys. 2016, 590, 10–19. [Google Scholar] [CrossRef] [PubMed]
- Oldfield, M.D.; Bach, L.A.; Forbes, J.M.; Nikolic-Paterson, D.; McRobert, A.; Thallas, V.; Atkins, R.C.; Osicka, T.; Jerums, G.; Cooper, M.E. Advanced glycation end products cause epithelial-myofibroblast transdifferentiation via the receptor for advanced glycation end products (RAGE). J. Clin. Investig. 2001, 108, 1853–1863. [Google Scholar] [CrossRef]
- Li, X.; Huang, K.; Liu, X.; Ruan, H.; Ma, L.; Liang, J.; Cui, Y.; Wang, Y.; Wu, S.; Li, H. Ellagic acid attenuates BLM-induced pulmonary fibrosis via inhibiting Wnt signaling pathway. Front. Pharmacol. 2021, 12, 639574. [Google Scholar] [CrossRef]
- Walter, K.R.; Ford, M.E.; Gregoski, M.J.; Kramer, R.M.; Knight, K.D.; Spruill, L.; Nogueira, L.M.; Krisanits, B.A.; Phan, V.; La Rue, A.C. Advanced glycation end products are elevated in estrogen receptor-positive breast cancer patients, alter response to therapy, and can be targeted by lifestyle intervention. Breast Cancer Res. Treat. 2019, 173, 559–571. [Google Scholar] [CrossRef]
- Chauveau, P.; Lasseur, C.; Azar, R.; Niu, W.; Combe, C.; Aparicio, M. Hygieno-dietetic recommendations in the prevention of accumulation of advanced glycation products. Néphrologie Thérapeutique 2019, 15, 485–490. [Google Scholar] [CrossRef]
- Cheresh, P.; Kim, S.-J.; Tulasiram, S.; Kamp, D.W. Oxidative stress and pulmonary fibrosis. Biochim. Biophys. Acta (BBA)-Mol. Basis Dis. 2013, 1832, 1028–1040. [Google Scholar] [CrossRef]
- Kliment, C.R.; Englert, J.M.; Gochuico, B.R.; Yu, G.; Kaminski, N.; Rosas, I.; Oury, T.D. Oxidative stress alters syndecan-1 distribution in lungs with pulmonary fibrosis. J. Biol. Chem. 2009, 284, 3537–3545. [Google Scholar] [CrossRef] [PubMed]
- Booth, A.J.; Hadley, R.; Cornett, A.M.; Dreffs, A.A.; Matthes, S.A.; Tsui, J.L.; Weiss, K.; Horowitz, J.C.; Fiore, V.F.; Barker, T.H. Acellular normal and fibrotic human lung matrices as a culture system for in vitro investigation. Am. J. Respir. Crit. Care Med. 2012, 186, 866–876. [Google Scholar] [CrossRef] [PubMed]
- Sontake, V.; Shanmukhappa, S.K.; DiPasquale, B.A.; Reddy, G.B.; Medvedovic, M.; Hardie, W.D.; White, E.S.; Madala, S.K. Fibrocytes regulate Wilms tumor 1–positive cell accumulation in severe fibrotic lung disease. J. Immunol. 2015, 195, 3978–3991. [Google Scholar] [CrossRef]
- Madala, S.K.; Thomas, G.; Edukulla, R.; Davidson, C.; Schmidt, S.; Schehr, A.; Hardie, W.D. Translational Research in Acute Lung Injury and Pulmonary Fibrosis: p70 ribosomal S6 kinase regulates subpleural fibrosis following transforming growth factor-α expression in the lung. Am. J. Physiol.-Lung Cell Mol. Physiol. 2016, 310, L175. [Google Scholar] [CrossRef] [PubMed]
- Gajjala, P.R.; Kasam, R.K.; Soundararajan, D.; Sinner, D.; Huang, S.K.; Jegga, A.G.; Madala, S.K. Dysregulated overexpression of Sox9 induces fibroblast activation in pulmonary fibrosis. JCI Insight 2021, 6, e152503. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Clinical Parameters | Normal | IPF | |
|---|---|---|---|
| Young | Old | ||
| Age (years) Median (range) | 25 (19–33) | 58.5 (52–70) | 62 (59–70) |
| Gender (male/female) | 5/0 | 4/0 | 8/0 |
| CML-AGE immunostaining (%) | 1.4 ± 0.4 | 6.5 ± 0.5 | 19.9 ± 3.5 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ediga, H.H.; Hester, P.; Yepuri, A.; Reddy, G.B.; Madala, S.K. Nε-Carboxymethyl-Lysine Modification of Extracellular Matrix Proteins Augments Fibroblast Activation. Int. J. Mol. Sci. 2023, 24, 15811. https://doi.org/10.3390/ijms242115811
Ediga HH, Hester P, Yepuri A, Reddy GB, Madala SK. Nε-Carboxymethyl-Lysine Modification of Extracellular Matrix Proteins Augments Fibroblast Activation. International Journal of Molecular Sciences. 2023; 24(21):15811. https://doi.org/10.3390/ijms242115811
Chicago/Turabian StyleEdiga, Harshavardhana H., Patrick Hester, Adithi Yepuri, Geereddy Bhanuprakash Reddy, and Satish K. Madala. 2023. "Nε-Carboxymethyl-Lysine Modification of Extracellular Matrix Proteins Augments Fibroblast Activation" International Journal of Molecular Sciences 24, no. 21: 15811. https://doi.org/10.3390/ijms242115811