
Transcriptional Regulation of the Human 5-HT1A Receptor Gene by Lithium: Role of Deaf1 and GSK3β

Abstract
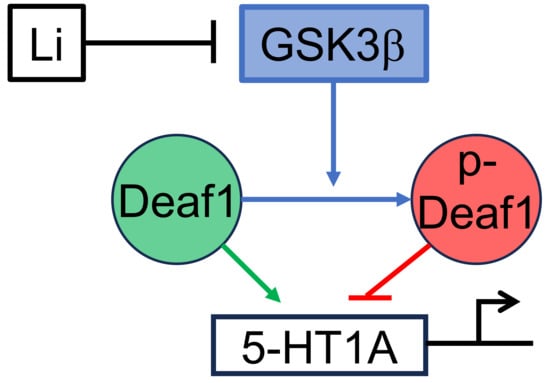
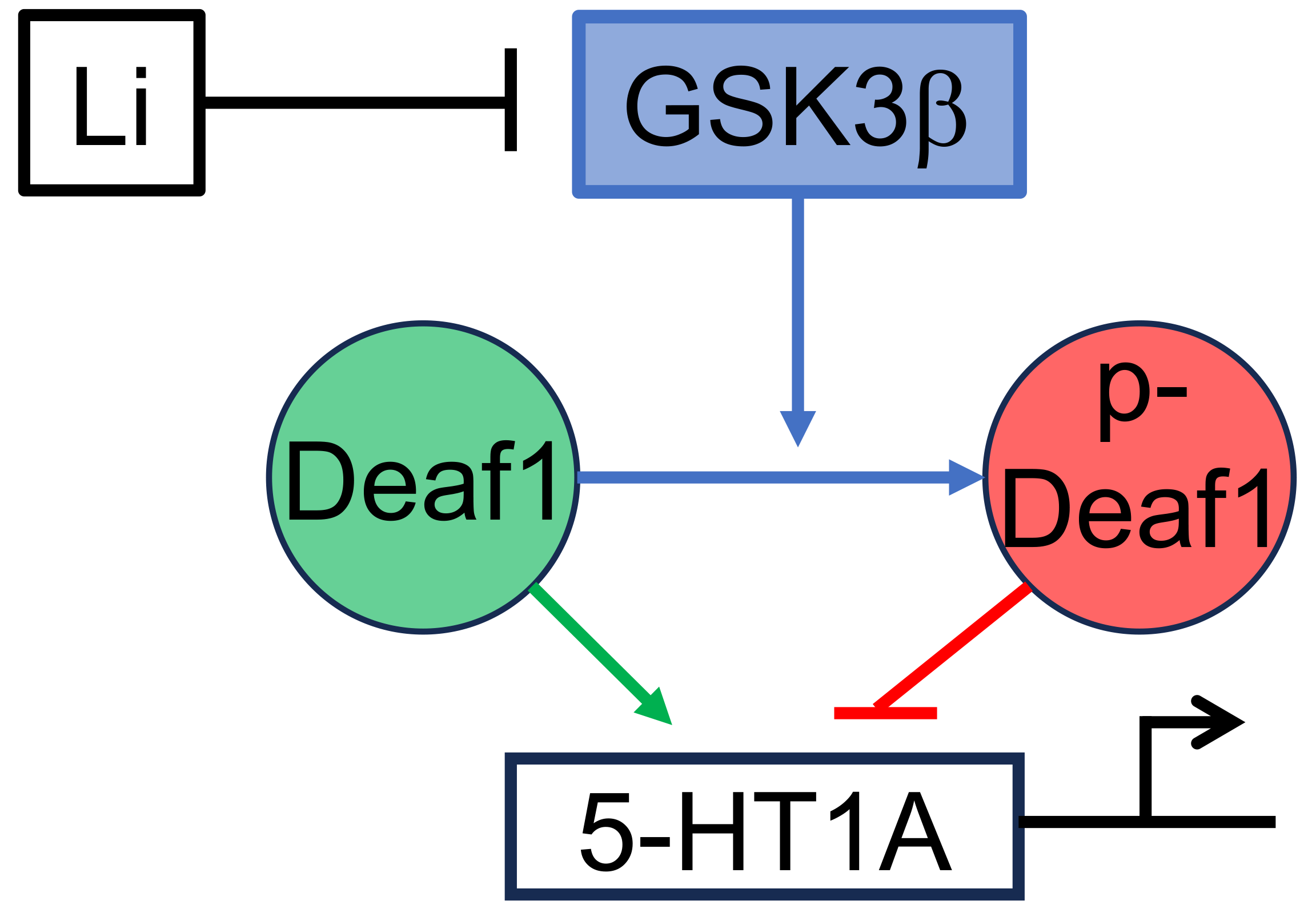
:
1. Introduction
2. Results
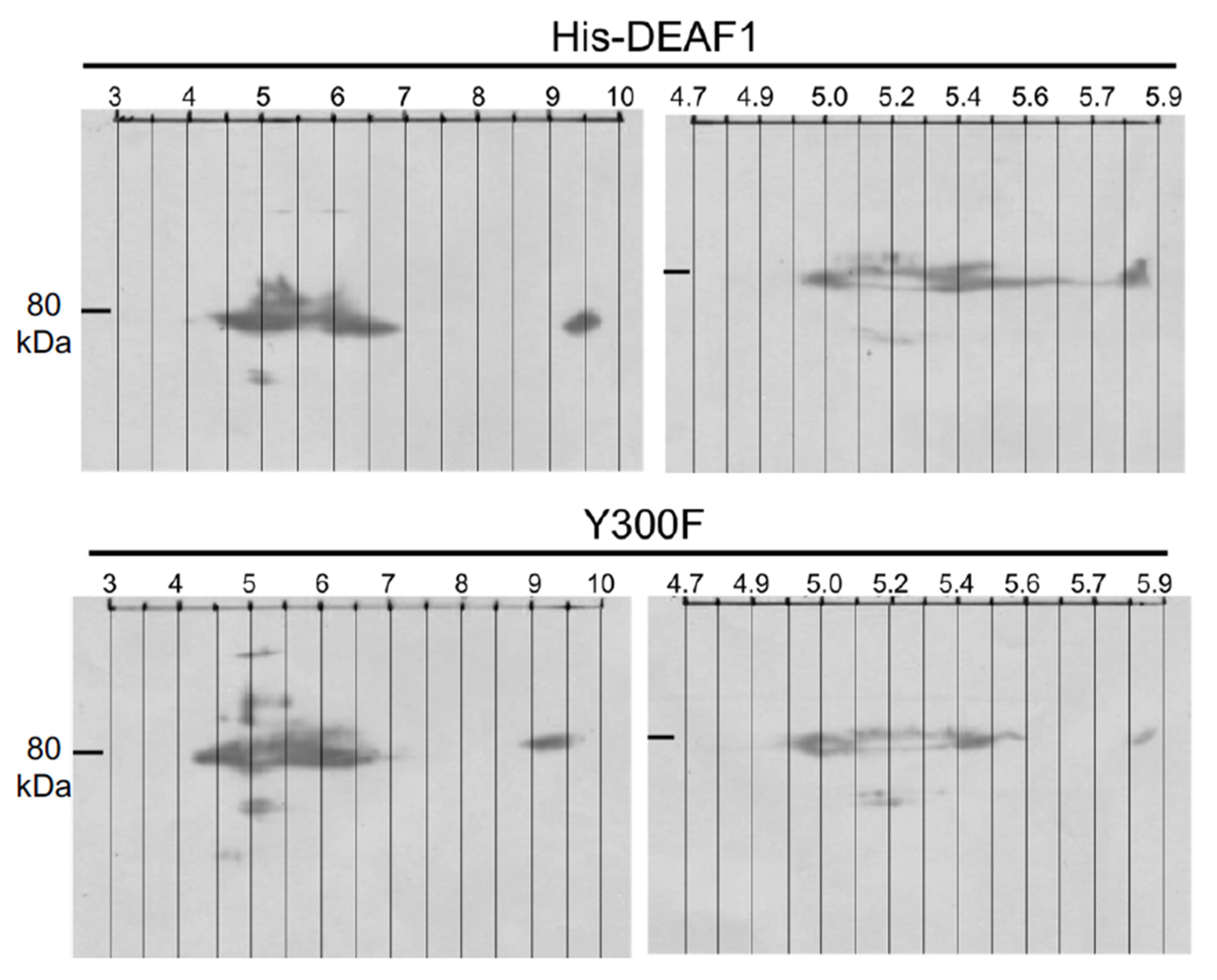
2.1. Deaf1 Phosphorylation Sites
2.2. Effect of GSK-3 Inhibitor Lithium on Deaf1-Mediated Repression of the 5-HT1A Promoter
2.3. Actions of Selective GSK3β Inhibitors on Deaf1-Mediated Repression of the 5-HT1A Promoter
3. Discussion
3.1. Deaf1 Phosphorylation
3.2. Functional Interaction of GSK3β and Deaf-1
3.3. 5-HT1A-GSK3β Signaling in Hippocampus and Antidepressant Response
3.4. Deaf1-Independent Actions of GSK3β Inhibitors
3.5. Therapeutic Relevance: Connection to 5-HT1A Expression in Bipolar Depression
4. Materials and Methods
4.1. Materials
4.2. Phosphorylation Site Prediction
4.3. DNA Constructs
4.4. Cell Culture and Transfection Procedures
4.5. Luciferase Reporter Gene Assays
4.6. Two-Dimensional SDS Polyacrylamide Gel Electrophoresis
4.7. Statistical Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders, 5th ed.; American Psychiatric Association: Washington, DC, USA, 2013. [Google Scholar]
- Kessler, R.C.; Bromet, E.J. The epidemiology of depression across cultures. Annu. Rev. Public Health 2013, 34, 119–138. [Google Scholar] [CrossRef] [PubMed]
- WHO. Depression and Other Common Mental Disorders: Global Health Estimates; World Health Organization: Geneva, Switzerland, 2017. [Google Scholar]
- Albert, P.R.; Benkelfat, C.; Descarries, L. The neurobiology of depression—Revisiting the serotonin hypothesis. I. Cellular and molecular mechanisms. Philos. Trans. R Soc. Lond. B Biol. Sci. 2012, 367, 2378–2381. [Google Scholar] [CrossRef] [PubMed]
- Lesch, K.P.; Araragi, N.; Waider, J.; van den Hove, D.; Gutknecht, L. Targeting brain serotonin synthesis: Insights into neurodevelopmental disorders with long-term outcomes related to negative emotionality, aggression and antisocial behaviour. Philos. Trans. R Soc. Lond. B Biol. Sci. 2012, 367, 2426–2443. [Google Scholar] [CrossRef] [PubMed]
- Warner-Schmidt, J. Treating the brain deep down: Short-circuiting depression. Nat. Med. 2013, 19, 680–681. [Google Scholar] [CrossRef] [PubMed]
- Leyton, M.; Young, S.N.; Blier, P.; Ellenbogen, M.A.; Palmour, R.M.; Ghadirian, A.M.; Benkelfat, C. The effect of tryptophan depletion on mood in medication-free, former patients with major affective disorder. Neuropsychopharmacology 1997, 16, 294–297. [Google Scholar] [CrossRef] [PubMed]
- Jans, L.A.; Riedel, W.J.; Markus, C.R.; Blokland, A. Serotonergic vulnerability and depression: Assumptions, experimental evidence and implications. Mol. Psychiatry 2007, 12, 522–543. [Google Scholar] [CrossRef] [PubMed]
- Hesselgrave, N.; Parsey, R.V. Imaging the serotonin 1A receptor using [11C]WAY100635 in healthy controls and major depression. Philos. Trans. R Soc. Lond. B Biol. Sci. 2013, 368, 20120004. [Google Scholar] [CrossRef]
- Savitz, J.B.; Drevets, W.C. Imaging phenotypes of major depressive disorder: Genetic correlates. Neuroscience 2009, 164, 300–330. [Google Scholar] [CrossRef]
- Booij, L.; Tremblay, R.E.; Leyton, M.; Seguin, J.R.; Vitaro, F.; Gravel, P.; Perreau-Linck, E.; Levesque, M.L.; Durand, F.; Diksic, M.; et al. Brain serotonin synthesis in adult males characterized by physical aggression during childhood: A 21-year longitudinal study. PLoS ONE 2010, 5, e11255. [Google Scholar] [CrossRef]
- Trivedi, M.H.; Fava, M.; Wisniewski, S.R.; Thase, M.E.; Quitkin, F.; Warden, D.; Ritz, L.; Nierenberg, A.A.; Lebowitz, B.D.; Biggs, M.M.; et al. Medication augmentation after the failure of SSRIs for depression. N. Engl. J. Med. 2006, 354, 1243–1252. [Google Scholar] [CrossRef]
- Cipriani, A.; Furukawa, T.A.; Salanti, G.; Chaimani, A.; Atkinson, L.Z.; Ogawa, Y.; Leucht, S.; Ruhe, H.G.; Turner, E.H.; Higgins, J.P.T.; et al. Comparative efficacy and acceptability of 21 antidepressant drugs for the acute treatment of adults with major depressive disorder: A systematic review and network meta-analysis. Lancet 2018, 391, 1357–1366. [Google Scholar] [CrossRef]
- Cohen, J.Y.; Amoroso, M.W.; Uchida, N. Serotonergic neurons signal reward and punishment on multiple timescales. Elife 2015, 4, e06346. [Google Scholar] [CrossRef] [PubMed]
- Zhong, W.; Li, Y.; Feng, Q.; Luo, M. Learning and Stress Shape the Reward Response Patterns of Serotonin Neurons. J. Neurosci. 2017, 37, 8863–8875. [Google Scholar] [CrossRef] [PubMed]
- Paquelet, G.E.; Carrion, K.; Lacefield, C.O.; Zhou, P.; Hen, R.; Miller, B.R. Single-cell activity and network properties of dorsal raphe nucleus serotonin neurons during emotionally salient behaviors. Neuron 2022, 110, 2664–2679.e8. [Google Scholar] [CrossRef] [PubMed]
- Harkin, E.F.; Lynn, M.B.; Payeur, A.; Boucher, J.F.; Caya-Bissonnette, L.; Cyr, D.; Stewart, C.; Longtin, A.; Naud, R.; Beique, J.C. Temporal derivative computation in the dorsal raphe network revealed by an experimentally-driven augmented integrate-and-fire modeling framework. Elife 2023, 12, e72951. [Google Scholar] [CrossRef] [PubMed]
- Artigas, F.; Bortolozzi, A.; Celada, P. Can we increase speed and efficacy of antidepressant treatments? Part I: General aspects and monoamine-based strategies. Eur. Neuropsychopharmacol. 2018, 28, 445–456. [Google Scholar] [CrossRef] [PubMed]
- Kaufman, J.; Sullivan, G.M.; Yang, J.; Ogden, R.T.; Miller, J.M.; Oquendo, M.A.; Mann, J.J.; Parsey, R.V.; DeLorenzo, C. Quantification of the Serotonin 1A Receptor Using PET: Identification of a Potential Biomarker of Major Depression in Males. Neuropsychopharmacology 2015, 40, 1692–1699. [Google Scholar] [CrossRef] [PubMed]
- Milak, M.S.; Pantazatos, S.; Rashid, R.; Zanderigo, F.; DeLorenzo, C.; Hesselgrave, N.; Ogden, R.T.; Oquendo, M.A.; Mulhern, S.T.; Miller, J.M.; et al. Higher 5-HT1A autoreceptor binding as an endophenotype for major depressive disorder identified in high risk offspring—A pilot study. Psychiatry Res. Neuroimaging 2018, 276, 15–23. [Google Scholar] [CrossRef] [PubMed]
- Sullivan, G.M.; Oquendo, M.A.; Milak, M.; Miller, J.M.; Burke, A.; Ogden, R.T.; Parsey, R.V.; Mann, J.J. Positron Emission Tomography Quantification of Serotonin1A Receptor Binding in Suicide Attempters with Major Depressive Disorder. JAMA Psychiatry 2015, 72, 169–178. [Google Scholar] [CrossRef]
- Miller, J.M.; Brennan, K.G.; Ogden, T.R.; Oquendo, M.A.; Sullivan, G.M.; Mann, J.J.; Parsey, R.V. Elevated serotonin 1A binding in remitted major depressive disorder: Evidence for a trait biological abnormality. Neuropsychopharmacology 2009, 34, 2275–2284. [Google Scholar] [CrossRef]
- Savitz, J.; Lucki, I.; Drevets, W.C. 5-HT(1A) receptor function in major depressive disorder. Prog. Neurobiol. 2009, 88, 17–31. [Google Scholar] [CrossRef] [PubMed]
- Samuels, B.A.; Anacker, C.; Hu, A.; Levinstein, M.R.; Pickenhagen, A.; Tsetsenis, T.; Madronal, N.; Donaldson, Z.R.; Drew, L.J.; Dranovsky, A.; et al. 5-HT1A receptors on mature dentate gyrus granule cells are critical for the antidepressant response. Nat. Neurosci. 2015, 18, 1606–1616. [Google Scholar] [CrossRef] [PubMed]
- Lemonde, S.; Turecki, G.; Bakish, D.; Du, L.; Hrdina, P.D.; Bown, C.D.; Sequeira, A.; Kushwaha, N.; Morris, S.J.; Basak, A.; et al. Impaired repression at a 5-hydroxytryptamine 1A receptor gene polymorphism associated with major depression and suicide. J. Neurosci. 2003, 23, 8788–8799. [Google Scholar] [CrossRef] [PubMed]
- Bottomley, M.J.; Collard, M.W.; Huggenvik, J.I.; Liu, Z.; Gibson, T.J.; Sattler, M. The SAND domain structure defines a novel DNA-binding fold in transcriptional regulation. Nat. Struct. Biol. 2001, 8, 626–633. [Google Scholar] [CrossRef] [PubMed]
- Le Francois, B.; Czesak, M.; Steubl, D.; Albert, P.R. Transcriptional regulation at a HTR1A polymorphism associated with mental illness. Neuropharmacology 2008, 55, 977–985. [Google Scholar] [CrossRef] [PubMed]
- Kishi, T.; Yoshimura, R.; Fukuo, Y.; Okochi, T.; Matsunaga, S.; Umene-Nakano, W.; Nakamura, J.; Serretti, A.; Correll, C.U.; Kane, J.M.; et al. The serotonin 1A receptor gene confer susceptibility to mood disorders: Results from an extended meta-analysis of patients with major depression and bipolar disorder. Eur. Arch. Psychiatry Clin. Neurosci. 2013, 263, 105–118. [Google Scholar] [CrossRef] [PubMed]
- Donaldson, Z.R.; le Francois, B.; Santos, T.L.; Almli, L.M.; Boldrini, M.; Champagne, F.A.; Arango, V.; Mann, J.J.; Stockmeier, C.A.; Galfalvy, H.; et al. The functional serotonin 1a receptor promoter polymorphism, rs6295, is associated with psychiatric illness and differences in transcription. Transl. Psychiatry 2016, 6, e746. [Google Scholar] [CrossRef]
- Czesak, M.; Lemonde, S.; Peterson, E.A.; Rogaeva, A.; Albert, P.R. Cell-specific repressor or enhancer activities of Deaf-1 at a serotonin 1A receptor gene polymorphism. J. Neurosci. 2006, 26, 1864–1871. [Google Scholar] [CrossRef]
- Czesak, M.; Le Francois, B.; Millar, A.M.; Deria, M.; Daigle, M.; Visvader, J.E.; Anisman, H.; Albert, P.R. Increased serotonin-1A (5-HT1A) autoreceptor expression and reduced raphe serotonin levels in deformed epidermal autoregulatory factor-1 (Deaf-1) gene knock-out mice. J. Biol. Chem. 2012, 287, 6615–6627. [Google Scholar] [CrossRef]
- Zanos, P.; Gould, T.D. Mechanisms of ketamine action as an antidepressant. Mol. Psychiatry 2018, 23, 801–811. [Google Scholar] [CrossRef]
- Beaulieu, J.M. A role for Akt and glycogen synthase kinase-3 as integrators of dopamine and serotonin neurotransmission in mental health. J. Psychiatry Neurosci. 2012, 37, 7–16. [Google Scholar] [CrossRef]
- Pilot-Storck, F.; Chopin, E.; Rual, J.F.; Baudot, A.; Dobrokhotov, P.; Robinson-Rechavi, M.; Brun, C.; Cusick, M.E.; Hill, D.E.; Schaeffer, L.; et al. Interactome mapping of the phosphatidylinositol 3-kinase-mammalian target of rapamycin pathway identifies deformed epidermal autoregulatory factor-1 as a new glycogen synthase kinase-3 interactor. Mol. Cell. Proteom. 2010, 9, 1578–1593. [Google Scholar] [CrossRef] [PubMed]
- Severus, E.; Taylor, M.J.; Sauer, C.; Pfennig, A.; Ritter, P.; Bauer, M.; Geddes, J.R. Lithium for prevention of mood episodes in bipolar disorders: Systematic review and meta-analysis. Int. J. Bipolar Disord. 2014, 2, 15. [Google Scholar] [CrossRef] [PubMed]
- Liu, R.J.; Fuchikami, M.; Dwyer, J.M.; Lepack, A.E.; Duman, R.S.; Aghajanian, G.K. GSK-3 inhibition potentiates the synaptogenic and antidepressant-like effects of subthreshold doses of ketamine. Neuropsychopharmacology 2013, 38, 2268–2277. [Google Scholar] [CrossRef] [PubMed]
- Chiu, C.T.; Scheuing, L.; Liu, G.; Liao, H.M.; Linares, G.R.; Lin, D.; Chuang, D.M. The mood stabilizer lithium potentiates the antidepressant-like effects and ameliorates oxidative stress induced by acute ketamine in a mouse model of stress. Int. J. Neuropsychopharmacol. 2015, 18, pyu102. [Google Scholar] [CrossRef]
- Gould, T.D.; Einat, H.; Bhat, R.; Manji, H.K. AR-A014418, a selective GSK-3 inhibitor, produces antidepressant-like effects in the forced swim test. Int. J. Neuropsychopharmacol. 2004, 7, 387–390. [Google Scholar] [CrossRef] [PubMed]
- Keshava Prasad, T.S.; Goel, R.; Kandasamy, K.; Keerthikumar, S.; Kumar, S.; Mathivanan, S.; Telikicherla, D.; Raju, R.; Shafreen, B.; Venugopal, A.; et al. Human Protein Reference Database—2009 update. Nucleic Acids Res. 2009, 37, D767–D772. [Google Scholar] [CrossRef]
- Gauci, S.; Helbig, A.O.; Slijper, M.; Krijgsveld, J.; Heck, A.J.; Mohammed, S. Lys-N and trypsin cover complementary parts of the phosphoproteome in a refined SCX-based approach. Anal. Chem. 2009, 81, 4493–4501. [Google Scholar] [CrossRef]
- Beausoleil, S.A.; Jedrychowski, M.; Schwartz, D.; Elias, J.E.; Villén, J.; Li, J.; Cohn, M.A.; Cantley, L.C.; Gygi, S.P. Large-scale characterization of HeLa cell nuclear phosphoproteins. Proc. Natl. Acad. Sci. USA 2004, 101, 12130–12135. [Google Scholar] [CrossRef]
- Kandasamy, K.; Pandey, A.; Molina, H. Evaluation of several MS/MS search algorithms for analysis of spectra derived from electron transfer dissociation experiments. Anal. Chem. 2009, 81, 7170–7180. [Google Scholar] [CrossRef]
- Ordureau, A.; Enesa, K.; Nanda, S.; Le Francois, B.; Peggie, M.; Prescott, A.; Albert, P.R.; Cohen, P. DEAF1 is a Pellino1-interacting protein required for interferon production by Sendai virus and double stranded RNA. J. Biol. Chem. 2013, 288, 24569–24580. [Google Scholar] [CrossRef] [PubMed]
- Bain, J.; Plater, L.; Elliott, M.; Shpiro, N.; Hastie, C.J.; McLauchlan, H.; Klevernic, I.; Arthur, J.S.; Alessi, D.R.; Cohen, P. The selectivity of protein kinase inhibitors: A further update. Biochem. J. 2007, 408, 297–315. [Google Scholar] [CrossRef] [PubMed]
- Pan, J.Q.; Lewis, M.C.; Ketterman, J.K.; Clore, E.L.; Riley, M.; Richards, K.R.; Berry-Scott, E.; Liu, X.; Wagner, F.F.; Holson, E.B.; et al. AKT kinase activity is required for lithium to modulate mood-related behaviors in mice. Neuropsychopharmacology 2011, 36, 1397–1411. [Google Scholar] [CrossRef] [PubMed]
- Roskoski, R., Jr. VEGF receptor protein-tyrosine kinases: Structure and regulation. Biochem. Biophys. Res. Commun. 2008, 375, 287–291. [Google Scholar] [CrossRef] [PubMed]
- Greene, J.; Banasr, M.; Lee, B.; Warner-Schmidt, J.; Duman, R.S. Vascular endothelial growth factor signaling is required for the behavioral actions of antidepressant treatment: Pharmacological and cellular characterization. Neuropsychopharmacology 2009, 34, 2459–2468. [Google Scholar] [CrossRef] [PubMed]
- Warner-Schmidt, J.L.; Duman, R.S. VEGF as a potential target for therapeutic intervention in depression. Curr. Opin. Pharmacol. 2008, 8, 14–19. [Google Scholar] [CrossRef] [PubMed]
- McGee, S.R.; Rose, G.M.; Jensik, P.J. Impaired memory and marble burying activity in deformed epidermal autoregulatory factor 1 (Deaf1) conditional knockout mice. Behav. Brain Res. 2020, 380, 112383. [Google Scholar] [CrossRef]
- Cubeddu, L.; Joseph, S.; Richard, D.J.; Matthews, J.M. Contribution of DEAF1 structural domains to the interaction with the breast cancer oncogene LMO4. PLoS ONE 2012, 7, e39218. [Google Scholar] [CrossRef]
- Philippe, T.J.; Vahid-Ansari, F.; Donaldson, Z.R.; Le Francois, B.; Zahrai, A.; Turcotte-Cardin, V.; Daigle, M.; James, J.; Hen, R.; Merali, Z.; et al. Loss of MeCP2 in adult 5-HT neurons induces 5-HT1A autoreceptors, with opposite sex-dependent anxiety and depression phenotypes. Sci. Rep. 2018, 8, 5788. [Google Scholar] [CrossRef]
- Jensik, P.J.; Huggenvik, J.I.; Collard, M.W. Deformed epidermal autoregulatory factor-1 (DEAF1) interacts with the Ku70 subunit of the DNA-dependent protein kinase complex. PLoS ONE 2012, 7, e33404. [Google Scholar] [CrossRef]
- Graca, F.A.; Sheffield, N.; Puppa, M.; Finkelstein, D.; Hunt, L.C.; Demontis, F. A large-scale transgenic RNAi screen identifies transcription factors that modulate myofiber size in Drosophila. PLoS Genet. 2021, 17, e1009926. [Google Scholar] [CrossRef]
- Gorlova, A.; Ortega, G.; Waider, J.; Bazhenova, N.; Veniaminova, E.; Proshin, A.; Kalueff, A.V.; Anthony, D.C.; Lesch, K.P.; Strekalova, T. Stress-induced aggression in heterozygous TPH2 mutant mice is associated with alterations in serotonin turnover and expression of 5-HT6 and AMPA subunit 2A receptors. J. Affect. Disord. 2020, 272, 440–451. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.S.; Long, N.; Pigino, G.; Brady, S.T.; Lazarov, O. Molecular mechanisms of environmental enrichment: Impairments in Akt/GSK3β, neurotrophin-3 and CREB signaling. PLoS ONE 2013, 8, e64460. [Google Scholar] [CrossRef] [PubMed]
- Trazzi, S.; Fuchs, C.; De Franceschi, M.; Mitrugno, V.M.; Bartesaghi, R.; Ciani, E. APP-dependent alteration of GSK3beta activity impairs neurogenesis in the Ts65Dn mouse model of Down syndrome. Neurobiol. Dis. 2014, 67c, 24–36. [Google Scholar] [CrossRef] [PubMed]
- Polter, A.M.; Yang, S.; Jope, R.S.; Li, X. Functional significance of glycogen synthase kinase-3 regulation by serotonin. Cell. Signal. 2012, 24, 265–271. [Google Scholar] [CrossRef] [PubMed]
- Hui, J.; Zhang, J.; Kim, H.; Tong, C.; Ying, Q.; Li, Z.; Mao, X.; Shi, G.; Yan, J.; Zhang, Z.; et al. Fluoxetine regulates neurogenesis in vitro through modulation of GSK-3beta/beta-catenin signaling. Int. J. Neuropsychopharmacol. 2015, 18, pyu099. [Google Scholar] [CrossRef] [PubMed]
- Zhou, W.; Chen, L.; Paul, J.; Yang, S.; Li, F.; Sampson, K.; Woodgett, J.R.; Beaulieu, J.M.; Gamble, K.L.; Li, X. The effects of glycogen synthase kinase-3beta in serotonin neurons. PLoS ONE 2012, 7, e43262. [Google Scholar] [CrossRef] [PubMed]
- Smagin, D.A.; Kovalenko, I.L.; Galyamina, A.G.; Belozertseva, I.V.; Tamkovich, N.V.; Baranov, K.O.; Kudryavtseva, N.N. Chronic Lithium Treatment Affects Anxious Behaviors and the Expression of Serotonergic Genes in Midbrain Raphe Nuclei of Defeated Male Mice. Biomedicines 2021, 9, 1293. [Google Scholar] [CrossRef] [PubMed]
- Nugent, A.C.; Bain, E.E.; Carlson, P.J.; Neumeister, A.; Bonne, O.; Carson, R.E.; Eckelman, W.; Herscovitch, P.; Zarate, C.A., Jr.; Charney, D.S.; et al. Reduced post-synaptic serotonin type 1A receptor binding in bipolar depression. Eur. Neuropsychopharmacol. 2013, 23, 822–829. [Google Scholar] [CrossRef]
- Ananth, M.; Bartlett, E.A.; DeLorenzo, C.; Lin, X.; Kunkel, L.; Vadhan, N.P.; Perlman, G.; Godstrey, M.; Holzmacher, D.; Ogden, R.T.; et al. Prediction of lithium treatment response in bipolar depression using 5-HTT and 5-HT1A PET. Eur. J. Nucl. Med. Mol. Imaging 2020, 47, 2417–2428. [Google Scholar] [CrossRef]
- Blom, N.; Gammeltoft, S.; Brunak, S. Sequence and structure-based prediction of eukaryotic protein phosphorylation sites. J. Mol. Biol. 1999, 294, 1351–1362. [Google Scholar] [CrossRef]
- Blom, N.; Sicheritz-Pontén, T.; Gupta, R.; Gammeltoft, S.; Brunak, S. Prediction of post-translational glycosylation and phosphorylation of proteins from the amino acid sequence. Proteomics 2004, 4, 1633–1649. [Google Scholar] [CrossRef]
- Iakoucheva, L.M.; Radivojac, P.; Brown, C.J.; O’Connor, T.R.; Sikes, J.G.; Obradovic, Z.; Dunker, A.K. The importance of intrinsic disorder for protein phosphorylation. Nucleic Acids Res. 2004, 32, 1037–1049. [Google Scholar] [CrossRef]
- Xue, Y.; Ren, J.; Gao, X.; Jin, C.; Wen, L.; Yao, X. GPS 2.0, a tool to predict kinase-specific phosphorylation sites in hierarchy. Mol. Cell. Proteom. 2008, 7, 1598–1608. [Google Scholar] [CrossRef]
- Lemonde, S.; Rogaeva, A.; Albert, P.R. Cell type-dependent recruitment of trichostatin A-sensitive repression of the human 5-HT1A receptor gene. J. Neurochem. 2004, 88, 857–868. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Mutant | Sense Primer (from 5′ to 3′) | Anti-Sense Primer (from 5′ to 3′) |
|---|---|---|
| S43A | GAGCCGGTGCTGGCCAGGGACGAGGAC | GTCCTCGTCCCTGGCCAGCACCGGCTC |
| S48A | CAGGGACGAGGACGCGGAGGAGGACGCAGACTC | GAGTCTGCGTCCTCCTCCGCGTCCTCGTCCCTG |
| S54A | GAGGAGGACGCAGACGCGGAGGCGGAGCGGGAGAC | GTCTCCCGCTCCGCCTCCGCGTCTGCGTCCTCCTC |
| T60A | GAGGCGGAGCGGGAGGCGCCGCGGGTCAC | GTGACCCGCGGCGCCTCCCGCTCCGCCTC |
| S176A | ACCCCAGGTCCTCAGGCTCCTCCAACCCCTCTG | CAGAGGGGTTGGAGGAGCCTGAGGACCTGGGGT |
| T179A | TCAGTCTCCTCCAGCCCCTCTGGCTCCC | GGGAGCCAGAGGGGCTGGAGGAGACTGA |
| Y300F | TCAGGCTTTTTGTGCCTTTCAAAAGGCGCAAGAAGG | CCTTCTTGCGCCTTTTGAAAGGCACAAAAAGCCTGA |
| T482A | CAAGCCAAGCATGCCAGCGCCTACCGAGAAGCTG | CAGCTTCTCGGTAGGCGCTGGCATGCTTGGCTTG |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Harkin, E.F.; Nasrallah, G.; Le François, B.; Albert, P.R. Transcriptional Regulation of the Human 5-HT1A Receptor Gene by Lithium: Role of Deaf1 and GSK3β. Int. J. Mol. Sci. 2023, 24, 15620. https://doi.org/10.3390/ijms242115620
Harkin EF, Nasrallah G, Le François B, Albert PR. Transcriptional Regulation of the Human 5-HT1A Receptor Gene by Lithium: Role of Deaf1 and GSK3β. International Journal of Molecular Sciences. 2023; 24(21):15620. https://doi.org/10.3390/ijms242115620
Chicago/Turabian StyleHarkin, Emerson F., Georges Nasrallah, Brice Le François, and Paul R. Albert. 2023. "Transcriptional Regulation of the Human 5-HT1A Receptor Gene by Lithium: Role of Deaf1 and GSK3β" International Journal of Molecular Sciences 24, no. 21: 15620. https://doi.org/10.3390/ijms242115620