
Evolution of Cherries (Prunus Subgenus Cerasus) Based on Chloroplast Genomes

 , , ,
, , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
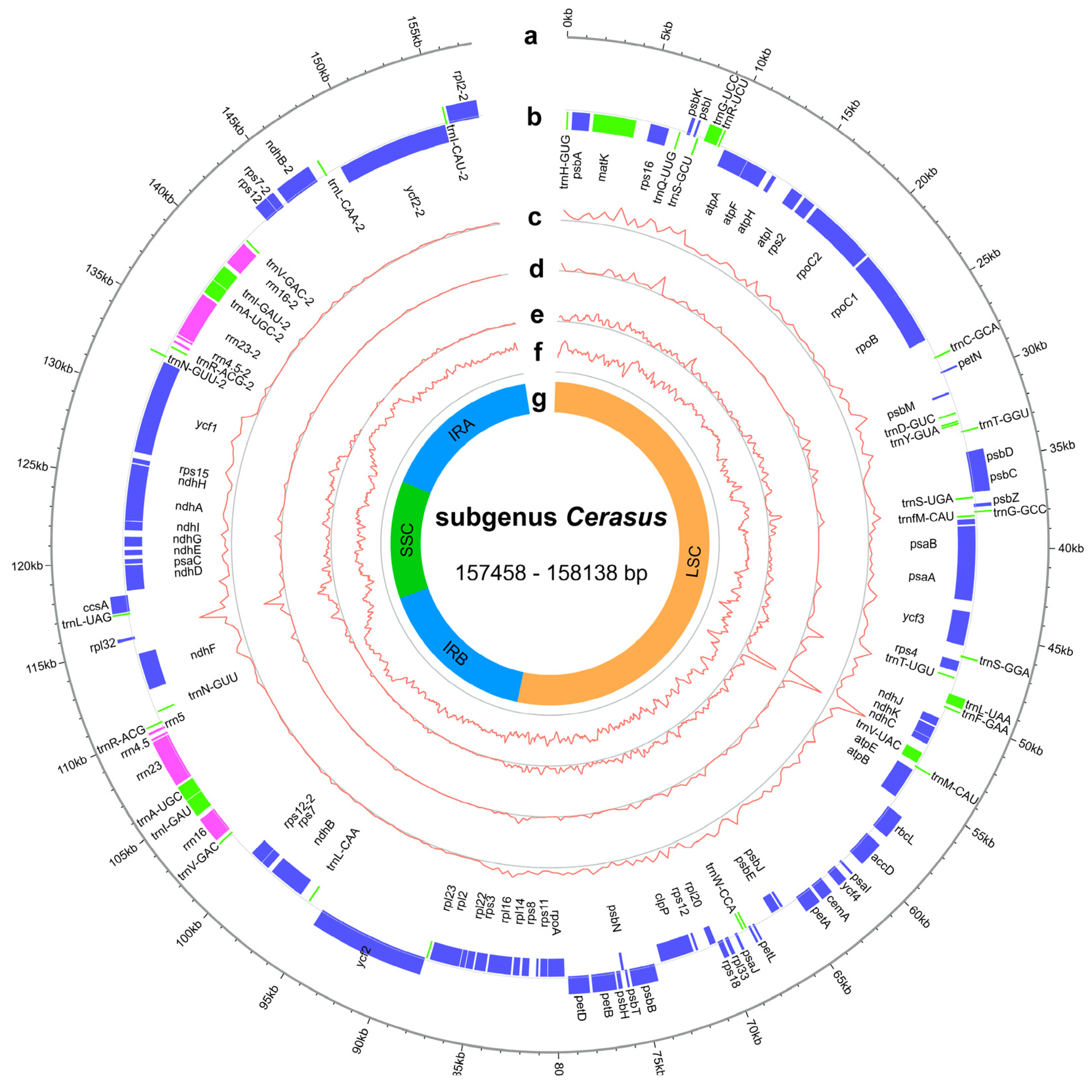
2.1. Chloroplast Genome Features
2.2. Chloroplast Microsatellite, InDel, SNP, and Genome Structure Variation
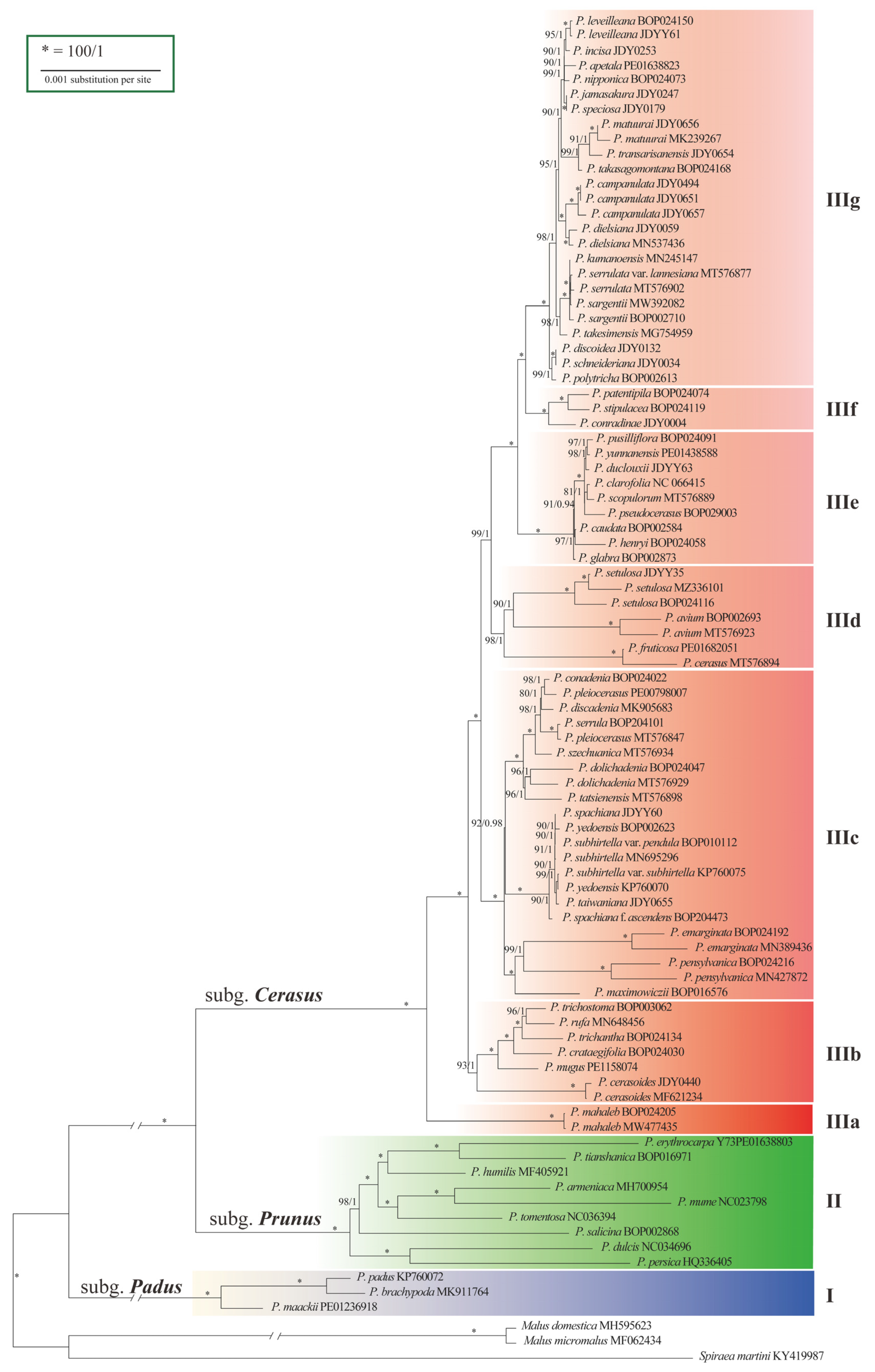
2.3. Phylogenetic Relationships among Lineages
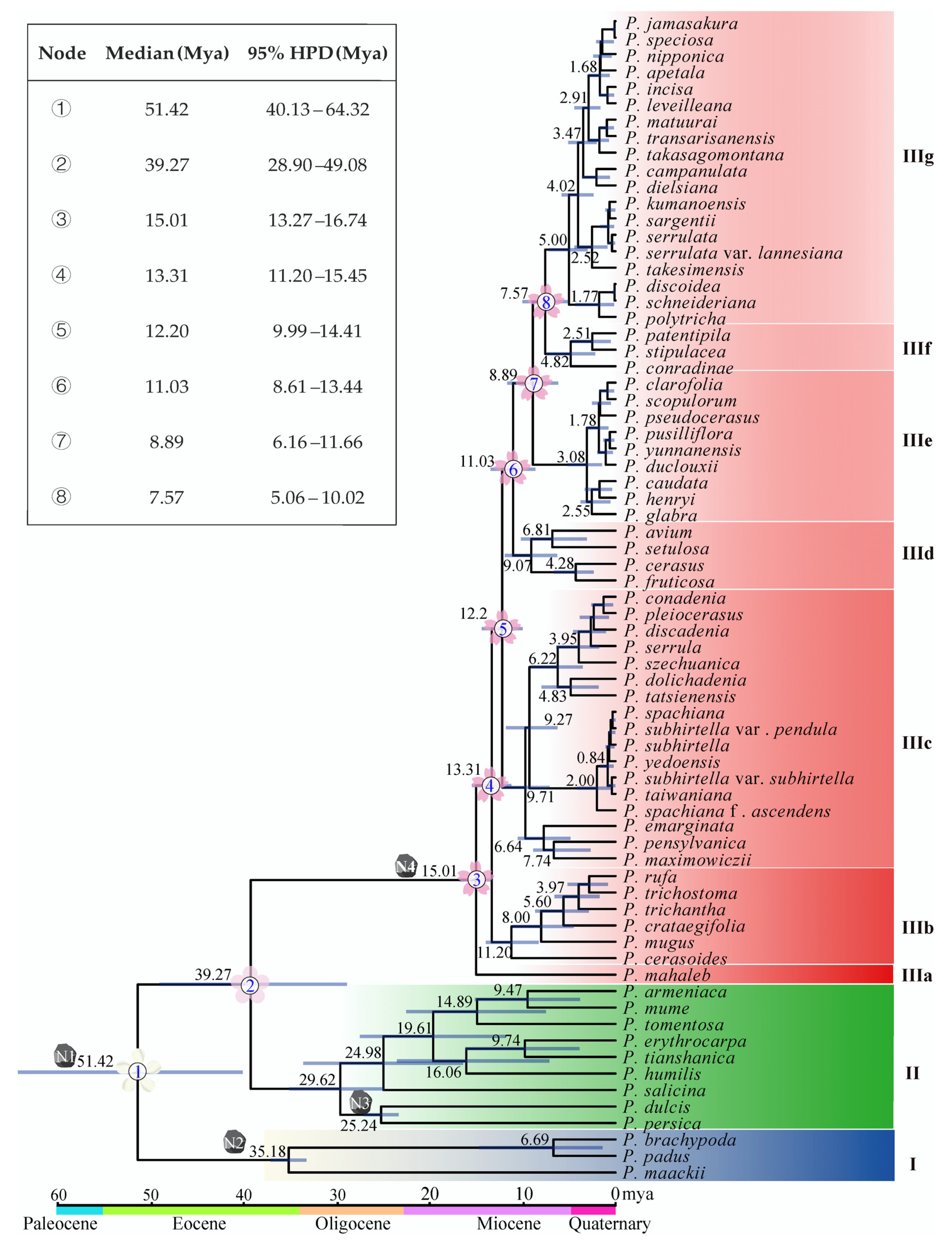
2.4. Divergence Time
3. Discussion
3.1. Phylogenetic Relationship within Subg. Cerasus
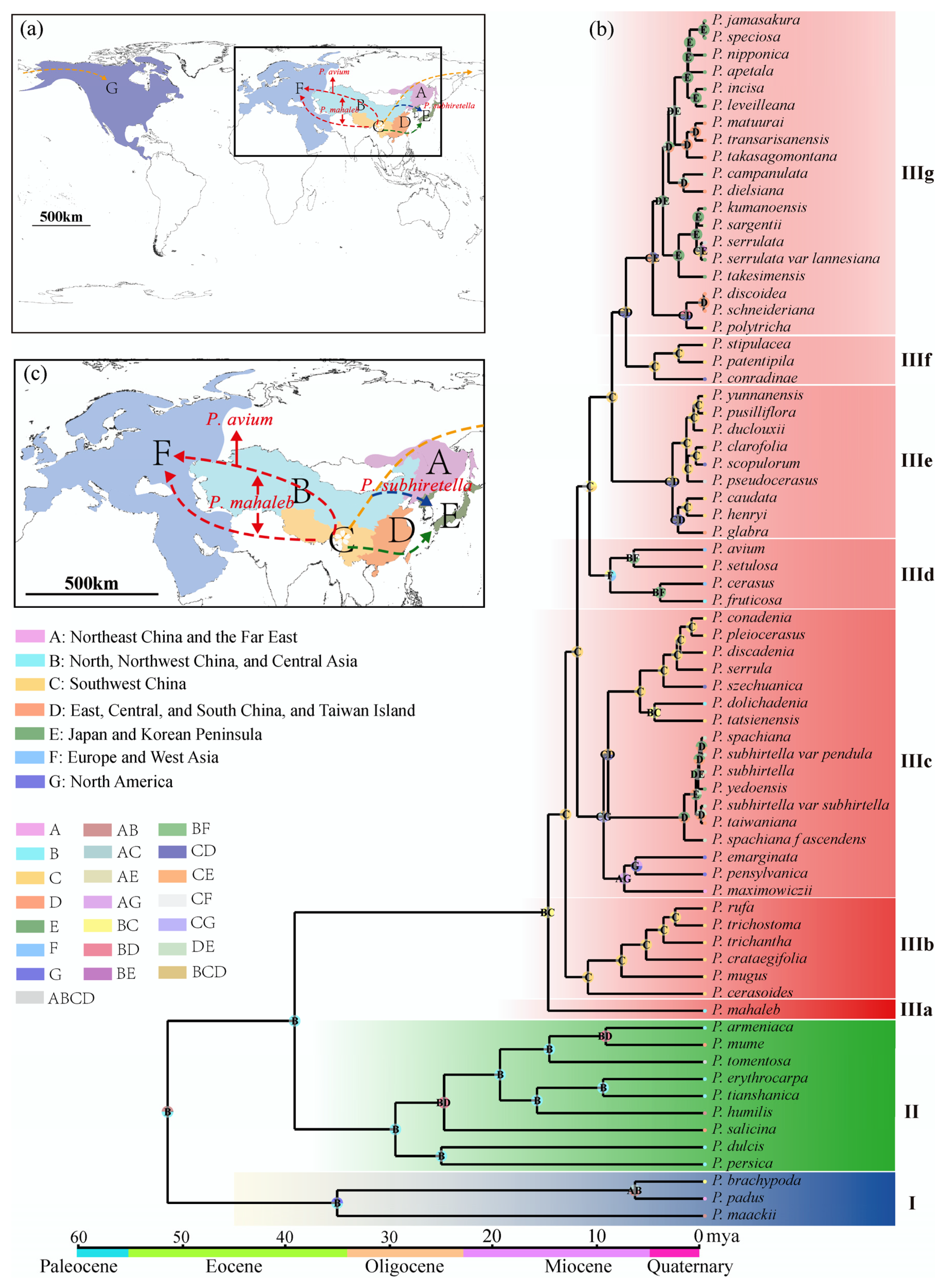
3.2. Origin and Dispersal of Subg. Cerasus Species
3.3. Taxonomic Implications
4. Materials and Methods
4.1. Plant Material and Taxon Sampling
4.2. DNA Extraction and Sequencing
4.3. Chloroplast Genome Assembly and Annotation
4.4. Chloroplast Genome Alignment and Polymorphism Assessment
4.5. Genomic Variation Analyses
4.6. Phylogenetic Analyses
4.7. Divergence Time Estimation and Ancestral Geographic Distribution Inference
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Yu, D.; Li, C.-L. Flora Reipublicae Popularis Sinicae; Science Press: Beijing, China, 1986; Volume 38, pp. 41–87. [Google Scholar]
- Rehder, A. Manual of Cultivated Trees and Shrubs Hardy in North America: Exclusive of the Subtropical and Warmer Temperate Regions, 2nd ed.; Macmillan: New York, NY, USA, 1940; pp. 466–477. [Google Scholar]
- Kawasaki, T. The distribution of Prunus subgenus Cerasus in East-Asia and classification of Japanese wild species. Sakura Sci. 1991, 1, 28–45. [Google Scholar]
- Hodel, G.J.R.; Zimmer, E.; Wen, J. A phylogenomic approach resolves the backbone of Prunus (Rosaceae) and identifies signals of hybridization and allopolyploidy. Mol. Phylogenetics Evol. 2021, 160, 107–118. [Google Scholar] [CrossRef]
- Su, N.; Hodel, R.G.J.; Wang, X.; Wang, J.-R.; Xie, S.-Y.; Gui, C.-X.; Zhang, L.; Chang, Z.-Y.; Zhao, L.; Potter, D.; et al. Molecular phylogeny and inflorescence evolution of Prunus (Rosaceae) based on RAD-Seq and genome skimming analyses. Plant Divers. 2023, 45, 397–408. [Google Scholar] [CrossRef]
- Yan, J.; Li, J.; Yu, L.; Bai, W.; Nie, D.; Xiong, Y.; Wu, S. Comparative chloroplast genomes of Prunus subgenus Cerasus (Rosaceae): Insights into sequence variations and phylogenetic relationships. Tree Genet. Genomes 2021, 17, 50. [Google Scholar] [CrossRef]
- Li, C.; Bartholomew, B. Flora of China: Cerasus Miller; Science Press: Beijing, China, 2003; Volume 9, pp. 404–420. [Google Scholar]
- Koehne, E. Neue chinesische arten und formen von Prunus. Feddes Repert. 1912, 11, 264–267. [Google Scholar] [CrossRef]
- Jiang, D.; Shen, X.; Chen, Y.; Zou, Y.; Wu, F.; Li, Y.; Liu, X. Morphological variation analysis of leaf and branch traits of wild Cerasus spp. in Zhejiang. J. Zhejiang Agric. For. Univ. 2019, 36, 723–732. [Google Scholar]
- Khadivi, A.; Mohammadi, M.; Asgari, K. Morphological and pomological characterizations of sweet cherry (Prunus avium L.), sour cherry (Prunus cerasus L.) and duke cherry (Prunus × gondouinii Rehd.) to choose the promising selections. Sci. Hortic. 2019, 257, 108719. [Google Scholar] [CrossRef]
- Chang, K.; Chang, C.; Park, T.Y.; Roh, M.S. Reconsideration of the Prunus serrulata complex (Rosaceae) and related taxa in Eastern Asia. Bot. J. Linn. Soc. 2007, 154, 35–54. [Google Scholar] [CrossRef]
- Wan, T.; Qiao, B.; Zhou, J.; Shao, K.; Pan, L.; An, F.; He, X.; Liu, T.; Li, P.; Cai, Y. Evolutionary and phylogenetic analyses of 11 Cerasus species based on the complete chloroplast genome. Front. Plant Sci. 2023, 14, 1070600. [Google Scholar] [CrossRef]
- Li, M.; Song, Y.-F.; Sylvester, S.P.; Sylvester, S.P.; Wang, X.-R. Comparative analysis of the complete plastid genomes in Prunus subgenus Cerasus (Rosaceae): Molecular structures and phylogenetic relationships. PLoS ONE 2022, 17, e0266535. [Google Scholar] [CrossRef]
- Zhang, J.; Wang, Y.; Chen, T.; Chen, Q.; Wang, L.; Liu, Z.; Wang, H.; Xie, R.; He, W.; Li, M.; et al. Evolution of Rosaceae plastomes highlights unique Cerasus diversification and independent origins of fruiting cherry. Front. Plant Sci. 2021, 12, 736053. [Google Scholar] [CrossRef]
- Chin, S.-W.; Shaw, J.; Haberle, R.; Wen, J.; Potter, D. Diversification of almonds, peaches, plums and cherries—Molecular systematics and biogeographic history of Prunus (Rosaceae). Mol. Phylogenet. Evol. 2014, 76, 34–48. [Google Scholar] [CrossRef]
- Shi, S.; Li, J.; Sun, J.; Yu, J.; Zhou, S. Phylogeny and classification of Prunus sensu lato (Rosaceae). J. Integr. Plant Biol. 2013, 55, 1069–1079. [Google Scholar] [CrossRef]
- Wen, J.; Berggren, S.T.; Lee, C.-H.; Ickert-Bond, S.; Yi, T.-S.; Yoo, K.-O.; Xie, L.; Shaw, J.; Potter, D. Phylogenetic inferences in Prunus (Rosaceae) using chloroplast ndhF and nuclear ribosomal ITS sequences. J. Syst. Evol. 2008, 46, 322. [Google Scholar] [CrossRef]
- Bortiri, E.; Heuvel, B.V.; Potter, D. Phylogenetic analysis of morphology in Prunus reveals extensive homoplasy. Plant Syst. Evol. 2006, 259, 53–71. [Google Scholar] [CrossRef]
- Kato, S.; Matsumoto, A.; Yoshimura, K.; Katsuki, T.; Iwamoto, K.; Kawahara, T.; Mukai, Y.; Tsuda, Y.; Ishio, S.; Nakamura, K.; et al. Origins of Japanese flowering cherry (Prunus subgenus Cerasus) cultivars revealed using nuclear SSR markers. Tree Genet. Genomes 2014, 10, 477–487. [Google Scholar] [CrossRef]
- Ohta, S.; Yamamoto, T.; Nishitani, C.; Katsuki, T.; Iketani, H.; Omura, M. Phylogenetic relationships among Japanese flowering cherries (Prunus subgenus Cerasus) based on nucleotide sequences of chloroplast DNA. Plant Syst. Evol. 2007, 263, 209–225. [Google Scholar] [CrossRef]
- Zhu, H.; Yi, X.-G.; Li, Y.-F.; Zhu, S.-X.; Li, M.; Duan, Y.-F.; Wang, X.-R. Phylogeography and population genetic structure of flowering cherry species Cerasus dielsiana in subtropical China. Syst. Biodivers. 2019, 17, 622–633. [Google Scholar] [CrossRef]
- Cho, M.-S.; Kim, C.-S.; Kim, S.-H.; Kim, T.O.; Heo, K.-I.; Jun, J.; Kim, S.-C. Molecular and morphological data reveal hybrid origin of wild Prunus yedoensis (Rosaceae) from Jeju island, Korea: Implications for the origin of the flowering cherry. Am. J. Bot. 2014, 101, 1976–1986. [Google Scholar] [CrossRef]
- Donoghue, M.J.; Smith, S.A. Patterns in the assembly of temperate forests around the Northern hemisphere. Phil. Trans. R. Soc. Lond. B 2004, 359, 1633–1644. [Google Scholar] [CrossRef]
- Qiu, Y.-X.; Fu, C.-X.; Comes, H.P. Plant molecular phylogeography in China and adjacent regions: Tracing the genetic imprints of Quaternary climate and environmental change in the world’s most diverse temperate flora. Mol. Phylogenet. Evol. 2011, 59, 225–244. [Google Scholar] [CrossRef]
- Qian, H.; Ricklefs, R.E. Large-Scale processes and the Asian bias in species diversity of temperate plants. Nature 2000, 407, 180–182. [Google Scholar] [CrossRef]
- Favre, A.; Päckert, M.; Pauls, S.U.; Jähnig, S.C.; Uhl, D.; Michalak, I.; Muellner-Riehl, A.N. The role of the uplift of the Qinghai-Tibetan plateau for the evolution of Tibetan biotas. Biol. Rev. 2015, 90, 236–253. [Google Scholar] [CrossRef]
- Jiang, D.; Li, X.; Li, Y.; Zhou, S.; Zhou, Q.; Liu, X.; Shen, X. Chromosome-Level Assembly of Flowering Cherry (Prunus campanulata) Provides Insight into Anthocyanin Accumulation. Genes 2023, 14, 389. [Google Scholar] [CrossRef]
- Das, B.; Ahmed, N.; Singh, P. Prunus diversity- early and present development: A review. Int. J. Biodvers. Conserv. 2011, 3, 721–734. [Google Scholar] [CrossRef]
- Bufford, D.; Hsieh, C.; Huang, T. Flora of Taiwan; Epoch Pub. Co.: Taipei, Taiwan, 1993. [Google Scholar]
- Lee, Y.N. New Flora of Korea; Kyo-Hak Publ.: Seoul, Republic of Korea, 2006; ISBN 89-09-11802-4. [Google Scholar]
- Moore, D.M. Flora Europaea Check-List and Chromosome Index; Cambridge University Press: Cambridge, UK, 1982; Volume 1, ISBN 0-521-23759-9. [Google Scholar]
- Rechinger, K.H. Flora Iranica; Graz: Vienna, Austria, 1963; Volume 150, pp. 148–149. [Google Scholar]
- Burrows, G.E.; Tyrl, R.J. Toxic Plants of North America; John Wiley & Sons: Hoboken, NJ, USA, 2013; ISBN 0-8138-2034-0. [Google Scholar]
- Rehder, A. Manual of Cultivated Trees and Shrubs; Macmillan: New York, NY, USA, 1949; p. 996. [Google Scholar]
- Schneider, C.K. Illustriertes Handbuch Der Laubholzkunde; Verlag von Gustav Fischer: Jena, Germany, 1912; pp. 589–590. [Google Scholar]
- Brettin, T.; Karle, R.; Crowe, E.; Lezzoni, A. Chloroplast inheritance and DNA variation in sweet, sour, and ground cherry. J. Hered. 2000, 91, 75–79. [Google Scholar] [CrossRef]
- Cho, M.-S.; Yang, J.Y.; Kim, S.-C. Complete chloroplast genome of Ulleung island endemic flowering cherry, Prunus takesimensis (Rosaceae), in Korea. Mitochondrial DNA Part B 2018, 3, 274–275. [Google Scholar] [CrossRef]
- Ohba, H.; Iwatsuki, K.; Boufford, D.E. Genus Cerasus; Kodasha: Tokyo, Japan, 2001; pp. 128–144. [Google Scholar]
- Zachos, J.; Pagani, M.; Sloan, L.; Thomas, E.; Billups, K. Trends, rhythms, and aberrations in global climate 65 Ma to present. Science 2001, 292, 686–693. [Google Scholar] [CrossRef]
- Deng, W.; Su, T.; Wappler, T.; Liu, J.; Li, S.; Huang, J.; Tang, H.; Low, S.L.; Wang, T.; Xu, H.; et al. Sharp changes in plant diversity and plant-herbivore interactions during the Eocene–Oligocene transition on the southeastern Qinghai-Tibetan plateau. Glob. Planet. Chang. 2020, 194, 103293. [Google Scholar] [CrossRef]
- Prothero, D.R. The late Eocene-Oligocene extinctions. Annu. Rev. Earth Planet. Sci. 1994, 22, 145–165. [Google Scholar] [CrossRef]
- Sun, J.; Ni, X.; Bi, S.; Wu, W.; Ye, J.; Meng, J.; Windley, B.F. Synchronous turnover of flora, fauna and climate at the Eocene–Oligocene boundary in Asia. Sci. Rep. 2014, 4, 7463. [Google Scholar] [CrossRef]
- Jiang, X.; Hipp, A.L.; Deng, M.; Su, T.; Zhou, Z.; Yan, M. East Asian origins of European holly oaks (Quercus section Ilex Loudon) via the Tibet-Himalaya. J. Biogeogr. 2019, 46, 2203–2214. [Google Scholar] [CrossRef]
- Sun, X.; Wang, P. How old is the Asian monsoon system?—Palaeobotanical records from China. Palaeogeogr. Palaeoclimatol. Palaeoecol. 2005, 222, 181–222. [Google Scholar] [CrossRef]
- Lewis, A.R.; Marchant, D.R.; Ashworth, A.C.; Hemming, S.R.; Machlus, M.L. Major middle Miocene global climate change: Evidence from East Antarctica and the Transantarctic Mountains. GSA Bull. 2007, 119, 1449–1461. [Google Scholar] [CrossRef]
- Spicer, R.A.; Harris, N.B.W.; Widdowson, M.; Herman, A.B.; Guo, S.; Valdes, P.J.; Wolfe, J.A.; Kelley, S.P. Constant elevation of Southern Tibet over the past 15 million years. Nature 2003, 421, 622–624. [Google Scholar] [CrossRef]
- Wen, J.; Ickert-Bond, S.; Nie, Z.-L.; Li, R. Timing and modes of evolution of Eastern Asian-North American biogeographic disjunctions in seed plants. In Darwin’s Heritage Today: Proceedings of the Darwin 200 Beijing International Conference; High Education Press: Beijing, China, 2010; pp. 252–269. [Google Scholar]
- Lu, H.; Guo, Z. Evolution of the monsoon and dry climate in East Asia during Late Cenozoic: A review. Sci. China Earth Sci. 2014, 57, 70–79. [Google Scholar] [CrossRef]
- Zheng, H.; Powell, C.M.; Rea, D.K.; Wang, J.; Wang, P. Late Miocene and Mid-Pliocene Enhancement of the East Asian monsoon as viewed from the land and sea. Glob. Planet. Chang. 2004, 41, 147–155. [Google Scholar] [CrossRef]
- Kimura, M. Paleogeography of the Ryukyu Islands. Tropics 2000, 10, 5–24. [Google Scholar] [CrossRef]
- Lambeck, K.; Chappell, J. Sea level change through the last glacial cycle. Science 2001, 292, 679–686. [Google Scholar] [CrossRef]
- Chou, Y.-W.; Thomas, P.I.; Ge, X.-J.; LePage, B.A.; Wang, C.-N. Refugia and phylogeography of Taiwania in East Asia. J. Biogeogr. 2011, 38, 1992–2005. [Google Scholar] [CrossRef]
- Keally, C.T. Japanese Pleistocene Landbridges and the Earliest Watercraft. Japanese Archaeology. [Homepage on the Internet] Tama Cable Network, Tokyo, Japan. [Cited 21 July 2010]. 2005. Available online: http://www.tnet.ne.jp/~keally/MiddlePalaeol/landbridges.html (accessed on 8 May 2023).
- Li, J.; Wang, S.; Yu, J.; Wang, L.; Zhou, S. A modified CTAB protocol for plant DNA extraction. Chin. Bull. Bot. 2013, 48, 72. [Google Scholar] [CrossRef]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef]
- Bankevich, A.; Nurk, S.; Antipov, D.; Gurevich, A.A.; Dvorkin, M.; Kulikov, A.S.; Lesin, V.M.; Nikolenko, S.I.; Pham, S.; Prjibelski, A.D.; et al. SPAdes: A new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 2012, 19, 455–477. [Google Scholar] [CrossRef] [PubMed]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Xu, X.; Wen, J.; Wang, W.; Zheng, W. The complete chloroplast genome of the threatened Prunus cerasoides, a rare winter blooming cherry in the Himalayan region. Conserv. Genet. Resour. 2018, 10, 499–502. [Google Scholar] [CrossRef]
- Kearse, M.; Moir, R.; Wilson, A.; Stones-Havas, S.; Cheung, M.; Sturrock, S.; Buxton, S.; Cooper, A.; Markowitz, S.; Duran, C.; et al. Geneious basic: An integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 2012, 28, 1647–1649. [Google Scholar] [CrossRef]
- Huang, D.I.; Cronk, Q.C.B. Plann: A command-line application for annotating plastome sequences. Appl. Plant Sci. 2015, 3, 1500026. [Google Scholar] [CrossRef]
- Krzywinski, M.; Schein, J.; Birol, İ.; Connors, J.; Gascoyne, R.; Horsman, D.; Jones, S.J.; Marra, M.A. Circos: An Information Aesthetic for Comparative Genomics. Genome Res. 2009, 19, 1639–1645. [Google Scholar] [CrossRef]
- Katoh, K.; Standley, D.M. Mafft multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef]
- Rambaut, A. Se-Al: Sequence Alignment Editor, Version 2.0 A11. 2007. Available online: http://tree.bio.ed.ac.uk/software/seal/ (accessed on 1 March 2023).
- Frazer, K.A.; Pachter, L.; Poliakov, A.; Rubin, E.M.; Dubchak, I. VISTA: Computational tools for comparative genomics. Nucleic Acids Res. 2004, 32, W273–W279. [Google Scholar] [CrossRef]
- Amiryousefi, A.; Hyvönen, J.; Poczai, P. IRscope: An online program to visualize the junction sites of chloroplast genomes. Bioinformatics 2018, 34, 3030–3031. [Google Scholar] [CrossRef]
- Librado, P.; Rozas, J. DnaSP v5: A software for comprehensive analysis of DNA polymorphism data. Bioinformatics 2009, 25, 1451–1452. [Google Scholar] [CrossRef]
- Kumar, S.; Stecher, G.; Tamura, K. MEGA7: Molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 2016, 33, 1870–1874. [Google Scholar] [CrossRef] [PubMed]
- Marçais, G.; Delcher, A.L.; Phillippy, A.M.; Coston, R.; Salzberg, S.L.; Zimin, A. MUMmer4: A fast and versatile genome alignment system. PLoS Comput. Biol. 2018, 14, e1005944. [Google Scholar] [CrossRef] [PubMed]
- Goel, M.; Sun, H.; Jiao, W.; Schneeberger, K. SyRI: Finding Genomic Rearrangements and Local Sequence Differences from Whole-Genome Assemblies. Genome Biol. 2019, 20, 1–13. [Google Scholar] [CrossRef]
- Danecek, P.; Bonfield, J.K.; Liddle, J.; Marshall, J.; Ohan, V.; Pollard, M.O.; Whitwham, A.; Keane, T.; McCarthy, S.A.; Davies, R.M.; et al. Twelve years of SAMtools and BCFtools. GigaScience 2021, 10, giab008. [Google Scholar] [CrossRef]
- Quinlan, A.R.; Hall, I.M. BEDTools: A flexible suite of utilities for comparing genomic features. Bioinformatics 2010, 26, 841–842. [Google Scholar] [CrossRef]
- Cingolani, P.; Platts, A.; Wang, L.L.; Coon, M.; Nguyen, T.; Wang, L.; Land, S.J.; Lu, X.; Ruden, D.M. A program for annotating and predicting the effects of single nucleotide polymorphisms, snpeff. Fly 2012, 6, 80–92. [Google Scholar] [CrossRef]
- Thiel, T.; Michalek, W.; Varshney, R.; Graner, A. Exploiting EST databases for the development and characterization of gene-derived SSR-markers in Barley (Hordeum vulgare L.). Theor. Appl. Genet. 2003, 106, 411–422. [Google Scholar] [CrossRef] [PubMed]
- Gao, F.; Chen, C.; Arab, D.A.; Du, Z.; He, Y.; Ho, S.Y.W. EasyCodeML: A visual tool for analysis of selection using CodeML. Ecol. Evol. 2019, 9, 3891–3898. [Google Scholar] [CrossRef]
- Xia, X. DAMBE7: New and improved tools for data analysis in molecular biology and evolution. Mol. Biol. Evol. 2018, 35, 1550–1552. [Google Scholar] [CrossRef]
- Nguyen, L.-T.; Schmidt, H.A.; von Haeseler, A.; Minh, B.Q. IQ-TREE: A fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 2015, 32, 268–274. [Google Scholar] [CrossRef]
- Kalyaanamoorthy, S.; Minh, B.Q.; Wong, T.K.F.; von Haeseler, A.; Jermiin, L.S. ModelFinder: Fast model selection for accurate phylogenetic estimates. Nat. Methods 2017, 14, 587–589. [Google Scholar] [CrossRef]
- Zhang, D.; Gao, F.; Jakovlić, I.; Zou, H.; Zhang, J.; Li, W.X.; Wang, G.T. PhyloSuite: An integrated and scalable desktop platform for streamlined molecular sequence data management and evolutionary phylogenetics studies. Mol. Ecol. Resour. 2020, 20, 348–355. [Google Scholar] [CrossRef] [PubMed]
- Ronquist, F.; Huelsenbeck, J.P. MrBayes 3: Bayesian phylogenetic inference under mixed models. Bioinformatics 2003, 19, 1572–1574. [Google Scholar] [CrossRef] [PubMed]
- Miller, M.A.; Pfeiffer, W.; Schwartz, T. Creating the CIPRES science gateway for inference of large phylogenetic trees. In Proceedings of the 2010 Gateway Computing Environments Workshop (GCE), New Orleans, LA, USA, 14 November 2010; pp. 1–8. [Google Scholar]
- Pritchard, J.K.; Stephens, M.; Donnelly, P. Inference of population structure using multilocus genotype data. Genetics 2000, 155, 945–959. [Google Scholar] [CrossRef] [PubMed]
- Porras-Hurtado, L.; Ruiz, Y.; Santos, C.; Phillips, C.; Carracedo, Á.; Lareu, M. An overview of STRUCTURE: Applications, parameter settings, and supporting software. Front. Genet. 2013, 4, 98. [Google Scholar] [CrossRef]
- Evanno, G.; Regnaut, S.; Goudet, J. Detecting the number of clusters of individuals using the software structure: A simulation study. Mol. Ecol. 2005, 14, 2611–2620. [Google Scholar] [CrossRef]
- Drummond, A.J.; Rambaut, A. BEAST: Bayesian evolutionary analysis by sampling trees. BMC Evol. Biol. 2007, 7, 214. [Google Scholar] [CrossRef]
- Li, Y.; Smith, T.; Liu, C.-J.; Awasthi, N.; Yang, J.; Wang, Y.-F.; Li, C.-S. Endocarps of Prunus (Rosaceae: Prunoideae) from the early Eocene of Wutu, Shandong Province, China. TAXON 2011, 60, 555–564. [Google Scholar] [CrossRef]
- Dorofeev, P.I. Tretichnye Flory Zapadnoi Sibiri; Akad. Nauk: Moscow, Russia, 1963. [Google Scholar]
- Sokoloff, D.D.; Ignatov, M.S.; Remizowa, M.V.; Nuraliev, M.S.; Blagoderov, V.; Garbout, A.; Perkovsky, E.E. Staminate flower of Prunus s. l. (Rosaceae) from Eocene rovno amber (Ukraine). J. Plant Res. 2018, 131, 925–943. [Google Scholar] [CrossRef] [PubMed]
- Szafer, W. Miocene Flora from Stare Gliwice in Upper Silesia: Instytut Geologiczny; Prace: Avenue Fenton, MI, USA, 1961. [Google Scholar]
- Tiffney, B.H. Dicotyledonous angiosperm flower from the upper cretaceous of Martha’s Vineyard, Massachusetts. Nature 1977, 265, 136–137. [Google Scholar] [CrossRef]
- Rambaut, A.; Drummond, A.J.; Xie, D.; Baele, G.; Suchard, M.A. Posterior summarization in bayesian phylogenetics using Tracer 1.7. Syst. Biol. 2018, 67, 901–904. [Google Scholar] [CrossRef]
- Yu, Y.; Harris, A.J.; Blair, C.; He, X. RASP (Reconstruct Ancestral State in Phylogenies): A tool for historical biogeography. Mol. Phylogenetics Evol. 2015, 87, 46–49. [Google Scholar] [CrossRef] [PubMed]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shen, X.; Zong, W.; Li, Y.; Liu, X.; Zhuge, F.; Zhou, Q.; Zhou, S.; Jiang, D. Evolution of Cherries (Prunus Subgenus Cerasus) Based on Chloroplast Genomes. Int. J. Mol. Sci. 2023, 24, 15612. https://doi.org/10.3390/ijms242115612
Shen X, Zong W, Li Y, Liu X, Zhuge F, Zhou Q, Zhou S, Jiang D. Evolution of Cherries (Prunus Subgenus Cerasus) Based on Chloroplast Genomes. International Journal of Molecular Sciences. 2023; 24(21):15612. https://doi.org/10.3390/ijms242115612
Chicago/Turabian StyleShen, Xin, Wenjin Zong, Yingang Li, Xinhong Liu, Fei Zhuge, Qi Zhou, Shiliang Zhou, and Dongyue Jiang. 2023. "Evolution of Cherries (Prunus Subgenus Cerasus) Based on Chloroplast Genomes" International Journal of Molecular Sciences 24, no. 21: 15612. https://doi.org/10.3390/ijms242115612