
Co-Occurrence of Congenital Aniridia Due to Nonsense PAX6 Variant p.(Cys94*) and Chromosome 21 Trisomy in the Same Patient
 , , , ,
, , , ,
Abstract
:1. Introduction
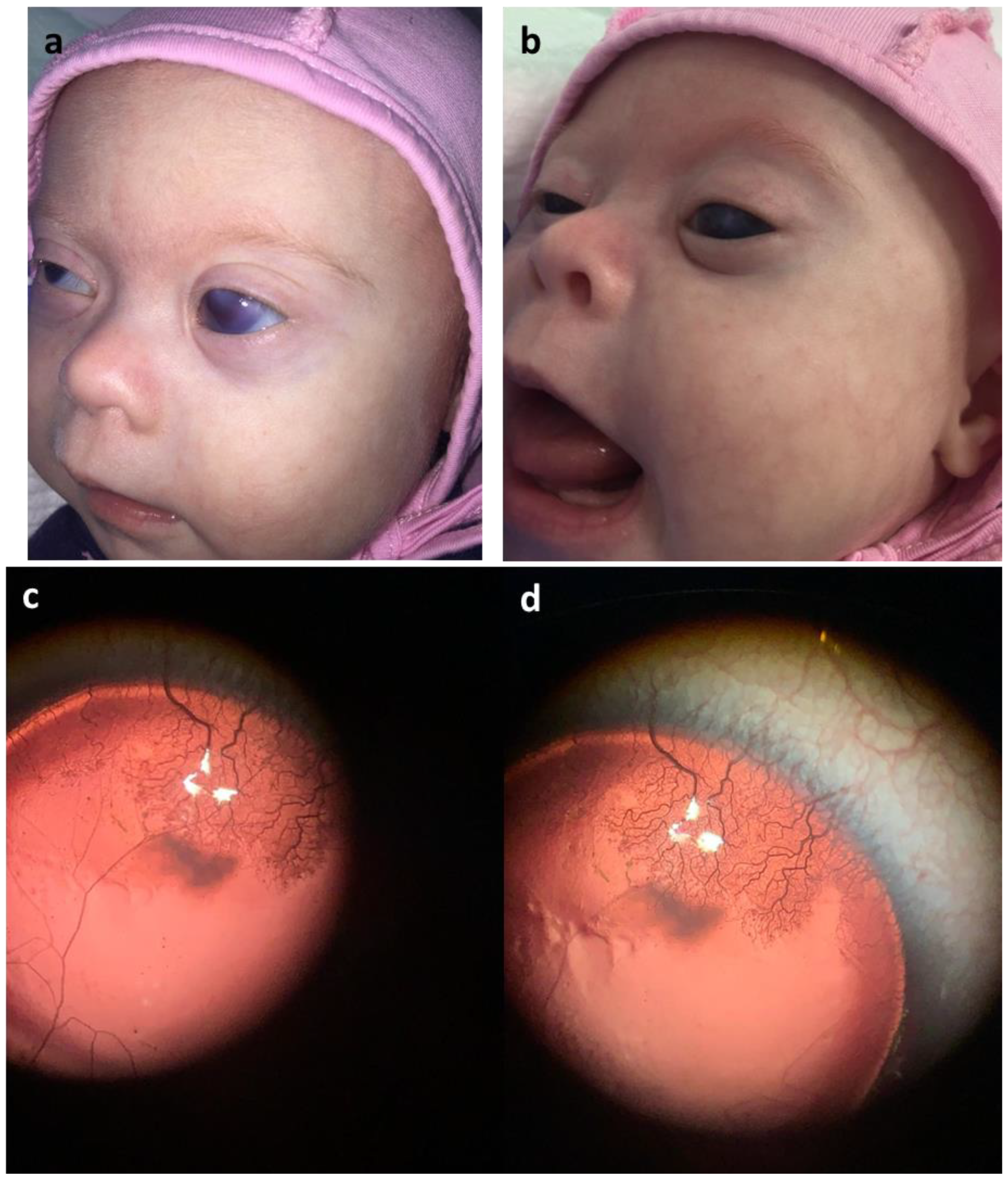
2. Case Presentation
3. Discussion
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hingorani, M.; Hanson, I.; van Heyningen, V. Aniridia. Eur. J. Hum. Genet. 2012, 20, 1011–1017. [Google Scholar] [CrossRef]
- Kasmann-Kellner, B.; Seitz, B. Congenital aniridia or pax6 syndrome. Ophthalmologe 2014, 111, 1144. [Google Scholar] [CrossRef]
- Vasilyeva, T.A.; Voskresenskaya, A.A.; Pozdeyeva, N.A.; Marakhonov, A.V.; Zinchenko, R.A. Pax6 gene characteristic and causative role of pax6 mutations in inherited eye pathologies. Russ. J. Genet. 2018, 54, 995–1002. [Google Scholar] [CrossRef]
- «Prevalence of Rare Diseases: Bibliographic Data», Orphanet Report Series, Rare Diseases Collection, January 2019, Number 2: Diseases Listed by Decreasing Prevalence, Incidence or Number of Published Cases. Available online: http://www.orpha.net/orphacom/cahiers/docs/GB/Prevalence_of_rare_diseases_by_decreasing_prevalence_or_cases.pdf (accessed on 12 January 2019).
- Reiser, K.; Whitcomb, C.; Robinson, K.; MacKenzie, M.R. T and b lymphocytes in patients with down’s syndrome. Am. J. Ment. Defic. 1976, 80, 613–619. [Google Scholar]
- Lagan, N.; Huggard, D.; Mc Grane, F.; Leahy, T.R.; Franklin, O.; Roche, E.; Webb, D.; O’Marcaigh, A.; Cox, D.; El-Khuffash, A.; et al. Multiorgan involvement and management in children with down syndrome. Acta Paediatr. 2020, 109, 1096–1111. [Google Scholar] [CrossRef]
- Rosca, I.; Turenschi, A.; Nicolescu, A.; Constantin, A.T.; Canciu, A.M.; Dica, A.D.; Bratila, E.; Coroleuca, C.A.; Nastase, L. Endocrine disorders in a newborn with heterozygous galactosemia, down syndrome and complex cardiac malformation: Case report. Medicina 2023, 59, 856. [Google Scholar] [CrossRef] [PubMed]
- Haseeb, A.; Huynh, E.; ElSheikh, R.H.; ElHawary, A.S.; Scelfo, C.; Ledoux, D.M.; Maidana, D.E.; Elhusseiny, A.M. Down syndrome: A review of ocular manifestations. Ther. Adv. Ophthalmol. 2022, 14, 25158414221101718. [Google Scholar] [CrossRef] [PubMed]
- Cleves, M.A.; Hobbs, C.A.; Cleves, P.A.; Tilford, J.M.; Bird, T.M.; Robbins, J.M. Congenital defects among liveborn infants with down syndrome. Birth Defects Res. A Clin. Mol. Teratol. 2007, 79, 657–663. [Google Scholar] [CrossRef]
- Fries, F.N.; Naray, A.; Munteanu, C.; Stachon, T.; Lagali, N.; Seitz, B.; Szentmary, N.; Kasmann-Kellner, B. A cross-sectional analysis of 556 eyes entering the homburg aniridia centre. Klin. Monatsblätter Augenheilkd. 2023. online ahead of print. [Google Scholar] [CrossRef]
- You, B.; Zhang, X.; Xu, K.; Xie, Y.; Ye, H.; Li, Y. Mutation spectrum of pax6 and clinical findings in 95 chinese patients with aniridia. Mol. Vis. 2020, 26, 226–234. [Google Scholar] [PubMed]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the american college of medical genetics and genomics and the association for molecular pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef] [PubMed]
- Sommer, A.; Eaton, A.P. Achondroplasia and down’s syndrome. J. Med. Genet. 1970, 7, 63–66. [Google Scholar] [CrossRef] [PubMed]
- Cela, E.; BenEzra, D. Aniridia and down syndrome. J. Pediatr. Ophthalmol. Strabismus 2000, 37, 369–370. [Google Scholar] [CrossRef]
- Solomon, B.D.; Pineda-Alvarez, D.E.; Balog, J.Z.; Hadley, D.; Gropman, A.L.; Nandagopal, R.; Han, J.C.; Hahn, J.S.; Blain, D.; Brooks, B.; et al. Compound heterozygosity for mutations in pax6 in a patient with complex brain anomaly, neonatal diabetes mellitus, and microophthalmia. Am. J. Med. Genet. A 2009, 149A, 2543–2546. [Google Scholar] [CrossRef] [PubMed]
- Henderson, R.A.; Williamson, K.; Cumming, S.; Clarke, M.P.; Lynch, S.A.; Hanson, I.M.; FitzPatrick, D.R.; Sisodiya, S.; van Heyningen, V. Inherited pax6, nf1 and otx2 mutations in a child with microphthalmia and aniridia. Eur. J. Hum. Genet. 2007, 15, 898–901. [Google Scholar] [CrossRef] [PubMed]
- Wilson, D.S.; Guenther, B.; Desplan, C.; Kuriyan, J. High resolution crystal structure of a paired (pax) class cooperative homeodomain dimer on DNA. Cell 1995, 82, 709–719. [Google Scholar] [CrossRef] [PubMed]
- Korenberg, J.R.; Kawashima, H.; Pulst, S.M.; Ikeuchi, T.; Ogasawara, N.; Yamamoto, K.; Schonberg, S.A.; West, R.; Allen, L.; Magenis, E.; et al. Molecular definition of a region of chromosome 21 that causes features of the down syndrome phenotype. Am. J. Hum. Genet. 1990, 47, 236–246. [Google Scholar] [PubMed]
- Letourneau, A.; Santoni, F.A.; Bonilla, X.; Sailani, M.R.; Gonzalez, D.; Kind, J.; Chevalier, C.; Thurman, R.; Sandstrom, R.S.; Hibaoui, Y.; et al. Domains of genome-wide gene expression dysregulation in down’s syndrome. Nature 2014, 508, 345–350. [Google Scholar] [CrossRef]
- Martin, K.R.; Corlett, A.; Dubach, D.; Mustafa, T.; Coleman, H.A.; Parkington, H.C.; Merson, T.D.; Bourne, J.A.; Porta, S.; Arbones, M.L.; et al. Over-expression of rcan1 causes down syndrome-like hippocampal deficits that alter learning and memory. Hum. Mol. Genet. 2012, 21, 3025–3041. [Google Scholar] [CrossRef]
- Maslen, C.L.; Babcock, D.; Robinson, S.W.; Bean, L.J.; Dooley, K.J.; Willour, V.L.; Sherman, S.L. Creld1 mutations contribute to the occurrence of cardiac atrioventricular septal defects in down syndrome. Am. J. Med. Genet. A 2006, 140, 2501–2505. [Google Scholar] [CrossRef]
- de Azevedo Moreira, L.M.; Matos, M.A.; Schiper, P.P.; Carvalho, A.F.; Gomes, I.C.; Rolemberg, J.C.; Ferreira de Lima, R.L.; Toralles, M.B. Co-occurrence of achondroplasia and down syndrome: Genotype/phenotype association. Birth Defects Res. A Clin. Mol. Teratol. 2010, 88, 228–231. [Google Scholar] [CrossRef] [PubMed]
- Antonarakis, S.E.; Skotko, B.G.; Rafii, M.S.; Strydom, A.; Pape, S.E.; Bianchi, D.W.; Sherman, S.L.; Reeves, R.H. Down syndrome. Nat. Rev. Dis. Primers 2020, 6, 9. [Google Scholar] [CrossRef] [PubMed]
- Glaser, T.; Jepeal, L.; Edwards, J.G.; Young, S.R.; Favor, J.; Maas, R.L. Pax6 gene dosage effect in a family with congenital cataracts, aniridia, anophthalmia and central nervous system defects. Nat. Genet. 1994, 7, 463–471. [Google Scholar] [CrossRef] [PubMed]
- Vasilyeva, T.A.; Voskresenskaya, A.A.; Käsmann-Kellner, B.; Khlebnikova, O.V.; Pozdeyeva, N.A.; Bayazutdinova, G.M.; Kutsev, S.I.; Ginter, E.K.; Semina, E.V.; Marakhonov, A.V.; et al. Molecular analysis of patients with aniridia in russian federation broadens the spectrum of pax6 mutations. Clin. Genet. 2017, 92, 639–644. [Google Scholar] [CrossRef]
- Sun, E.; Kraus, C.L. The ophthalmic manifestations of down syndrome. Children 2023, 10, 341. [Google Scholar] [CrossRef]
- Mazzoni, D.S.; Ackley, R.S.; Nash, D.J. Abnormal pinna type and hearing loss correlations in down’s syndrome. J. Intellect. Disabil. Res. 1994, 38 Pt 6, 549–560. [Google Scholar] [CrossRef]
- Bamiou, D.E.; Campbell, N.G.; Musiek, F.E.; Taylor, R.; Chong, W.K.; Moore, A.; van Heyningen, V.; Free, S.; Sisodiya, S.; Luxon, L.M. Auditory and verbal working memory deficits in a child with congenital aniridia due to a pax6 mutation. Int. J. Audiol. 2007, 46, 196–202. [Google Scholar] [CrossRef]
- Grangeon, L.; Cassinari, K.; Rousseau, S.; Croisile, B.; Formaglio, M.; Moreaud, O.; Boutonnat, J.; Le Meur, N.; Mine, M.; Coste, T.; et al. Early-onset cerebral amyloid angiopathy and alzheimer disease related to an app locus triplication. Neurol. Genet. 2021, 7, e609. [Google Scholar] [CrossRef]
- Ichimata, S.; Martinez-Valbuena, I.; Lee, S.; Li, J.; Karakani, A.M.; Kovacs, G.G. Distinct molecular signatures of amyloid-beta and tau in alzheimer’s disease associated with down syndrome. Int. J. Mol. Sci. 2023, 24, 11596. [Google Scholar] [CrossRef]
- Maurya, S.K.; Mishra, R. Pax6 binds to promoter sequence elements associated with immunological surveillance and energy homeostasis in brain of aging mice. Ann. Neurosci. 2017, 24, 20–25. [Google Scholar] [CrossRef]
- Grant, M.K.; Bobilev, A.M.; Branch, A.; Lauderdale, J.D. Structural and functional consequences of pax6 mutations in the brain: Implications for aniridia. Brain Res. 2021, 1756, 147283. [Google Scholar] [CrossRef]
- Voskresenskaya, A.; Pozdeyeva, N.; Vasilyeva, T.; Batkov, Y.; Shipunov, A.; Gagloev, B.; Zinchenko, R. Clinical and morphological manifestations of aniridia-associated keratopathy on anterior segment optical coherence tomography and in vivo confocal microscopy. Ocul. Surf. 2017, 15, 759–769. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.K.; Kim, M.K.; Oh, J.Y. Corneal abnormalities in congenital aniridia: Congenital central corneal opacity versus aniridia-associated keratopathy. Am. J. Ophthalmol. 2018, 185, 75–80. [Google Scholar] [CrossRef] [PubMed]
- Landsend, E.C.S.; Lagali, N.; Utheim, T.P. Congenital aniridia—A comprehensive review of clinical features and therapeutic approaches. Surv. Ophthalmol. 2021, 66, 1031–1050. [Google Scholar] [CrossRef] [PubMed]
- Moshirfar, M.; Hastings, J.; Ronquillo, Y. Aniridic fibrosis syndrome. In Statpearls; Ineligible Companies: Treasure Island, FL, USA, 2023. [Google Scholar]
- Gimenez-Barcons, M.; Casteras, A.; Armengol Mdel, P.; Porta, E.; Correa, P.A.; Marin, A.; Pujol-Borrell, R.; Colobran, R. Autoimmune predisposition in down syndrome may result from a partial central tolerance failure due to insufficient intrathymic expression of aire and peripheral antigens. J. Immunol. 2014, 193, 3872–3879. [Google Scholar] [CrossRef] [PubMed]
- Eissa, E.; Afifi, H.H.; Abo-Shanab, A.M.; Thomas, M.M.; Taher, M.B.; Kandil, R.; Kholoussi, N.M. Importance of trec and krec as molecular markers for immunological evaluation of down syndrome children. Sci. Rep. 2023, 13, 15445. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Patients | Patient 1 | Patient 2 | Patient 3 |
|---|---|---|---|
| Age, gender | A 4-year-old girl | A 4.5-month-old boy | A 4-year-old boy |
| Reference | (this study) | [14] | [15] |
| Karyotype | 47,XX,+21 | 47,XY,+21 | 47,XY,+21 |
| PAX6 genotype | c.[282C>A];[=] | not tested | c.[112C>T];[718C>T] |
| Facial features consistent with diagnosis of trisomy 21 | Facial dysmorphism, depressed nasal bridge, hypertelorism, antimongoloid slant and eyelid epicanthal folds, asymmetric eyes due to the left eyeball buphthalmos, open mouth, macroglossia, hypersalivation | Facial dysmorphism, depressed nasal bridge, hypertelorism, antimongoloid slant and eyelid epicanthal folds | Facial dysmorphism, depressed nasal bridge, hypertelorism, antimongoloid slant and eyelid epicanthal folds, asymmetric microphthalmia, open mouth, macroglossia, hypersalivation |
| Neurologic status | Myotonic syndrome, reduced tendon reflexes, postural kyphosis abnormality, weakened back muscles, valgus right foot and varus left foot deformities | not tested | Extreme microcephaly, a smooth philtrum |
| Developmental delay | Developmental delay | not tested | Severe developmental delay |
| Brain structure | Normal brain structure | not tested | Complex structural brain anomaly, corpus callosum agenesis, midline interhemispheric cyst, hypoplastic pons and vermis (absent inferiorly), dysplastic tectum, pituitary and hypothalamic hypoplasia, and a globular (though not fused) basal ganglia |
| Eyes | Congenital bilateral complete aniridia | Congenital bilateral complete aniridia | Bilateral extreme microphthalmia without visual function |
| Congenital bilateral glaucoma | No glaucoma, normal eye pressure | No data | |
| Clear lenses with first signs of developing opacities | Clear lenses | No data | |
| Vitreous bodies with first signs of degeneration | Normal vitreous bodies | No data | |
| Keratopathy OSCornea opacity OD | Clear cornea without vascularization or other signs of keratopathy | No data | |
| Normal anterior chambers | Normal anterior chambers | No data | |
| Horizontal nystagmus | Horizontal nystagmus | No data | |
| Mild hypoplasia of the optic nerve disks | Mild hypoplasia of the optic nerve disks | No data | |
| Foveal hypoplasia | Foveal hypoplasia | No data | |
| Albinotic color of both fundi | Subalbinotic changes in both fundi | No data | |
| Strabismus | No strabismus | No data | |
| Myopia | not tested | No data | |
| Hearing | Unilateral hearing loss | Normal hearing | No data |
| Heart | Persistent ductus arteriosus, atrial septal defect | Ventricular septal defect, aortic coarctation | No data |
| Airway anomalies | Laryngomalacia, breath failure on the first week after birth | Bronchopulmonary dysplasia, breath failure | Choanal atresia |
| Renal anomalies | Megaureter | No anomalies | Renal dysplasia with recurrent urinary tract infections |
| Endocrine status | Increased TTH level | not tested | Central hypothyroidism Gonadotropin deficiency Cryptorchidism Secondary adrenal insufficiency Insulin-dependent diabetes mellitus without pancreatic anomalies |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vasilyeva, T.A.; Sukhanova, N.V.; Marakhonov, A.V.; Kuzina, N.Y.; Shilova, N.V.; Kadyshev, V.V.; Kutsev, S.I.; Zinchenko, R.A. Co-Occurrence of Congenital Aniridia Due to Nonsense PAX6 Variant p.(Cys94*) and Chromosome 21 Trisomy in the Same Patient. Int. J. Mol. Sci. 2023, 24, 15527. https://doi.org/10.3390/ijms242115527
Vasilyeva TA, Sukhanova NV, Marakhonov AV, Kuzina NY, Shilova NV, Kadyshev VV, Kutsev SI, Zinchenko RA. Co-Occurrence of Congenital Aniridia Due to Nonsense PAX6 Variant p.(Cys94*) and Chromosome 21 Trisomy in the Same Patient. International Journal of Molecular Sciences. 2023; 24(21):15527. https://doi.org/10.3390/ijms242115527
Chicago/Turabian StyleVasilyeva, Tatyana A., Natella V. Sukhanova, Andrey V. Marakhonov, Natalia Yu. Kuzina, Nadezhda V. Shilova, Vitaly V. Kadyshev, Sergey I. Kutsev, and Rena A. Zinchenko. 2023. "Co-Occurrence of Congenital Aniridia Due to Nonsense PAX6 Variant p.(Cys94*) and Chromosome 21 Trisomy in the Same Patient" International Journal of Molecular Sciences 24, no. 21: 15527. https://doi.org/10.3390/ijms242115527