
Metabolic-Dysfunction-Associated Steatotic Liver Disease—Its Pathophysiology, Association with Atherosclerosis and Cardiovascular Disease, and Treatments

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
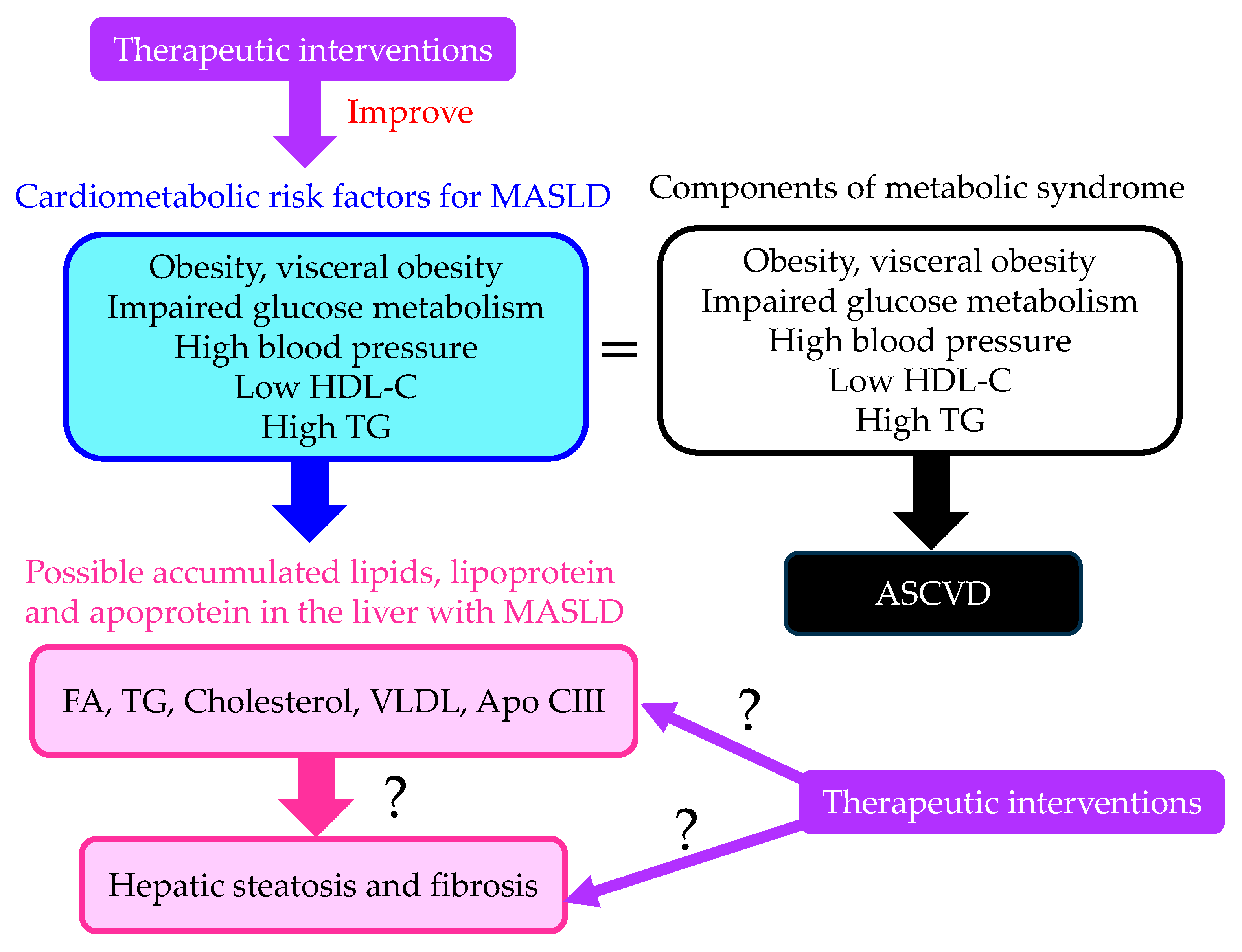
:1. Introduction
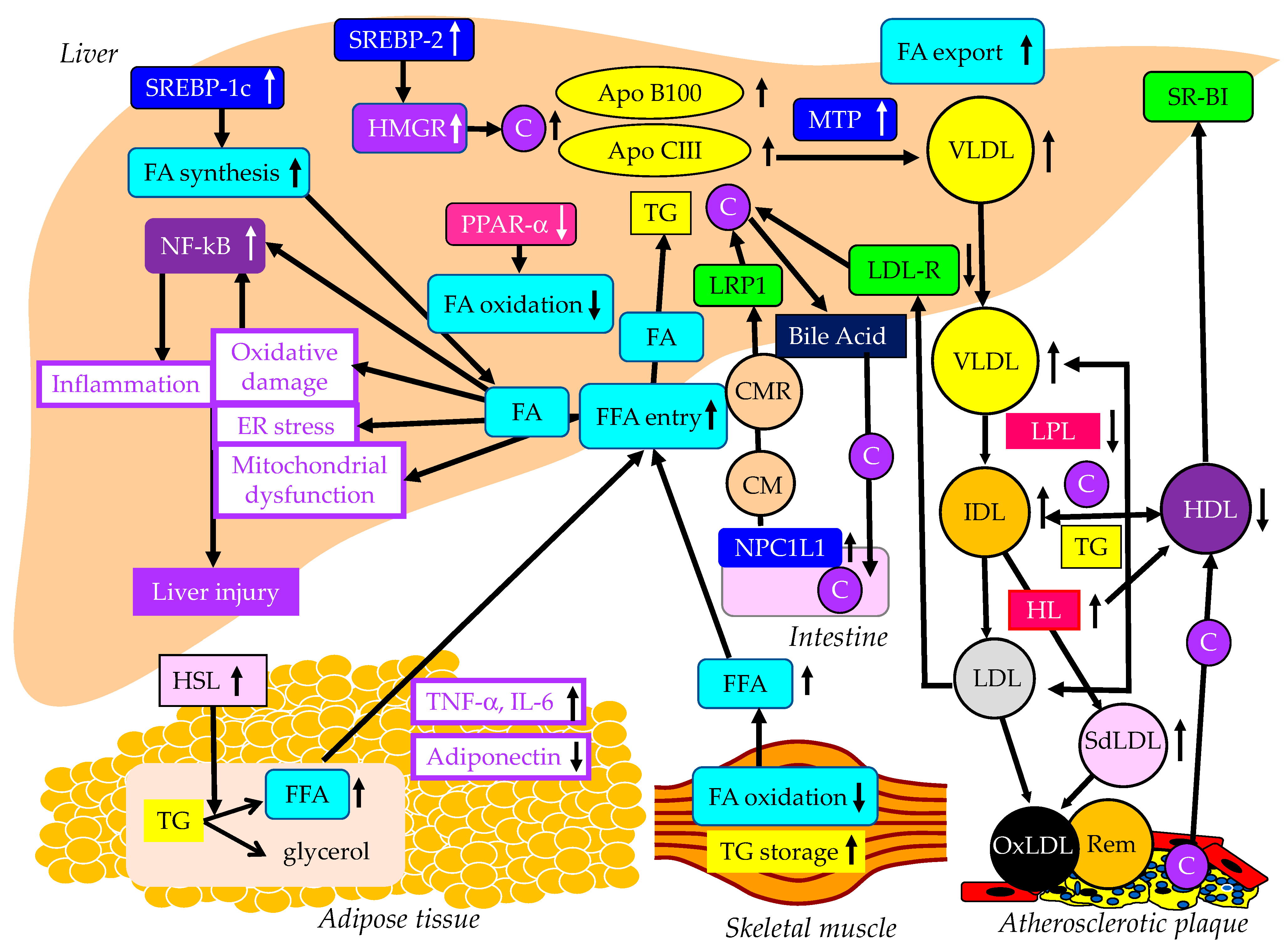
2. The Metabolic Abnormalities That Induce MASLD
3. The Association of MASLD with ASCVD
4. The Therapeutic Approaches for MASLD—Lifestyle Modification and Surgical Interventions
4.1. Lifestyle Modification
4.1.1. Diet
4.1.2. Exercise
4.1.3. Diet and Exercise
4.2. Surgical Interventions
4.2.1. Bariatric Surgery
4.2.2. Intragastric Balloons
5. Pharmacological Interventions for MASLD
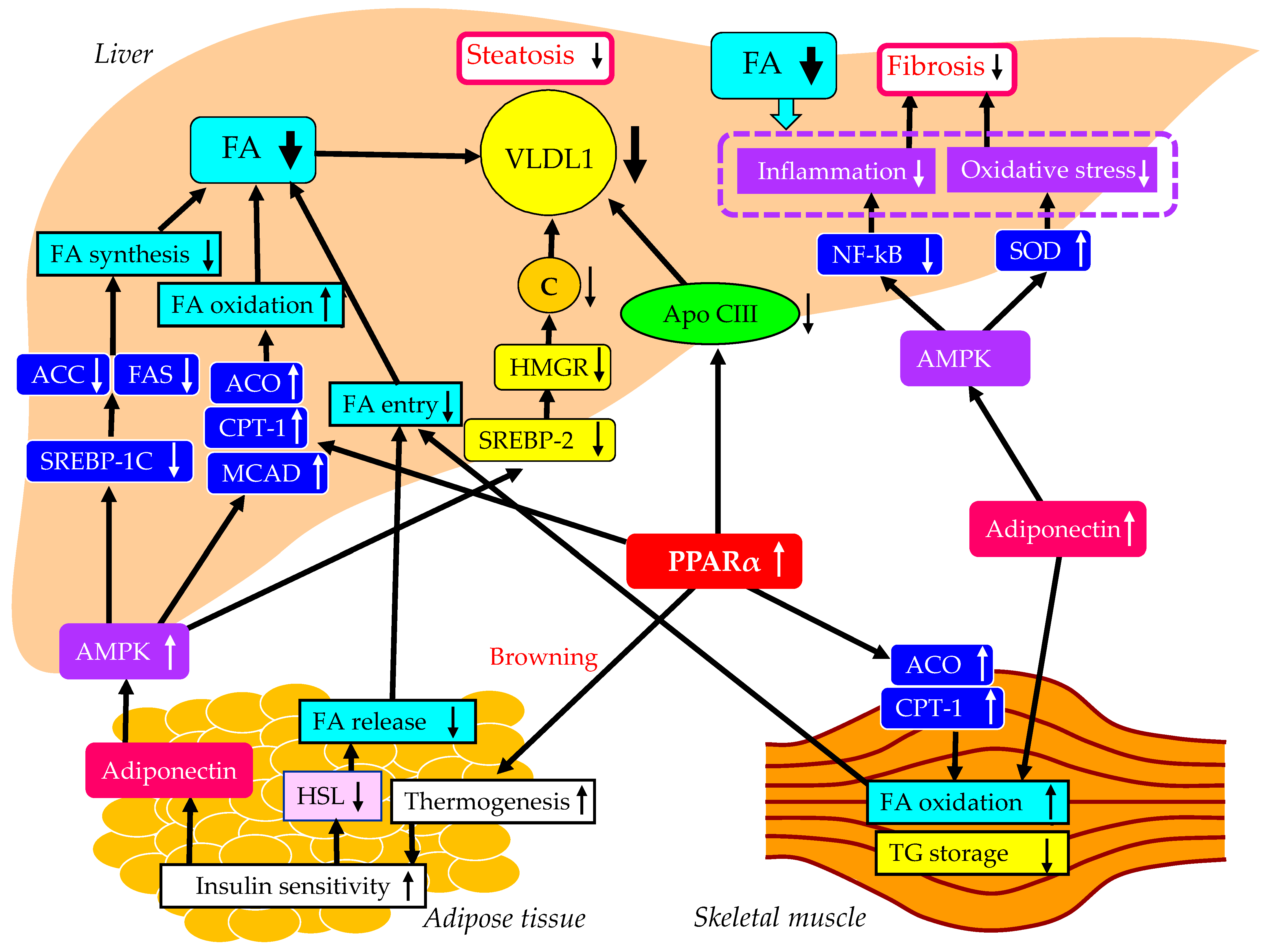
5.1. Pemafibrate
5.1.1. Effects of Pemafibrate on Liver Enzymes, Hepatic Steatosis and Fibrosis
5.1.2. The Underlying Mechanisms for the Improvement of MASLD Using Pemafibrate
5.1.3. The Vasculoprotective Effects of Pemafibrate
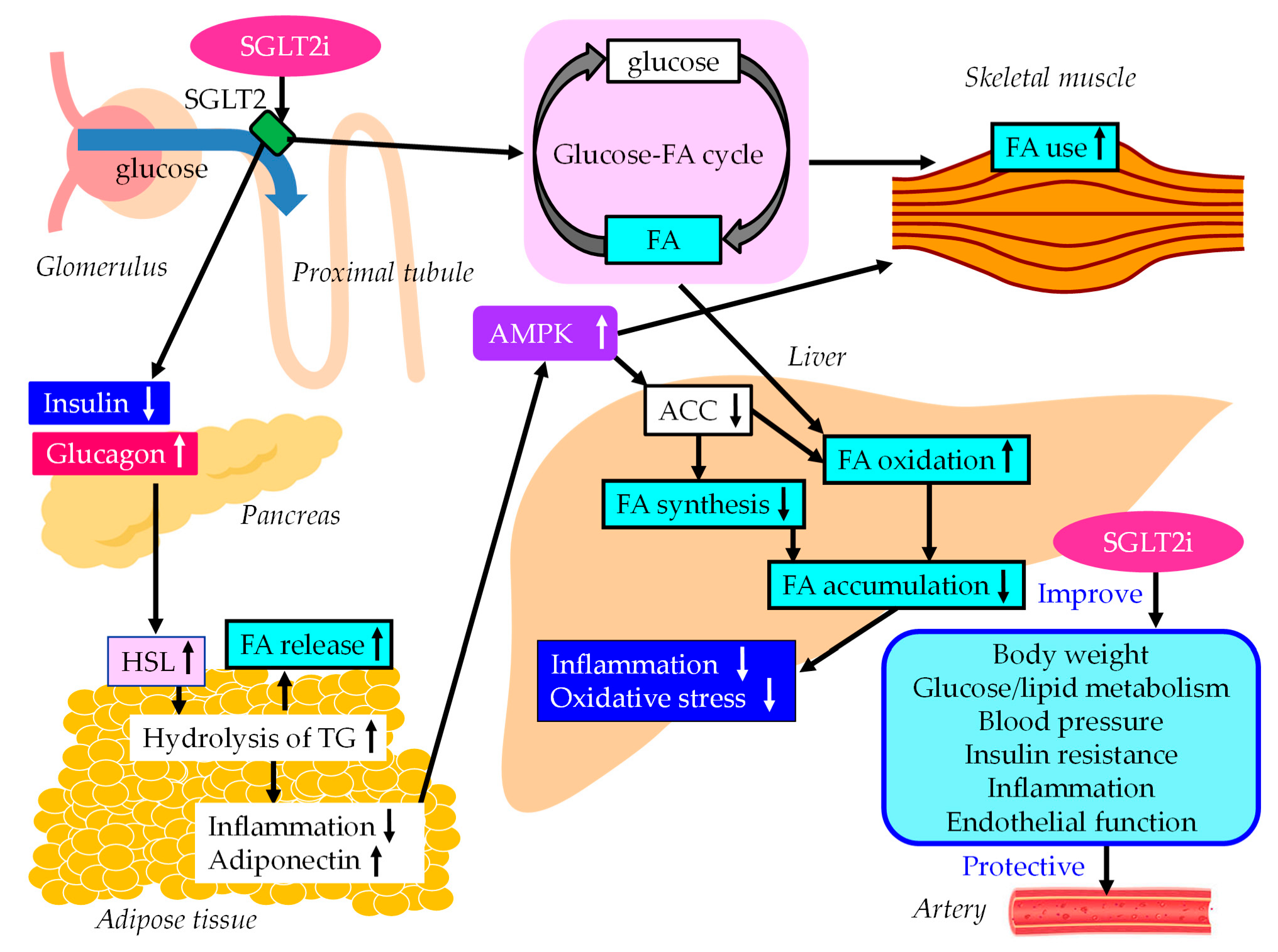
5.2. SGLT2i
5.2.1. Effects of SGLT2i on Liver Enzymes, Hepatic Steatosis and Fibrosis
5.2.2. The Underlying Mechanisms for the Improvement of MASLD Using SGLT2is
5.2.3. The Vasculoprotective Effects of SGLT2i
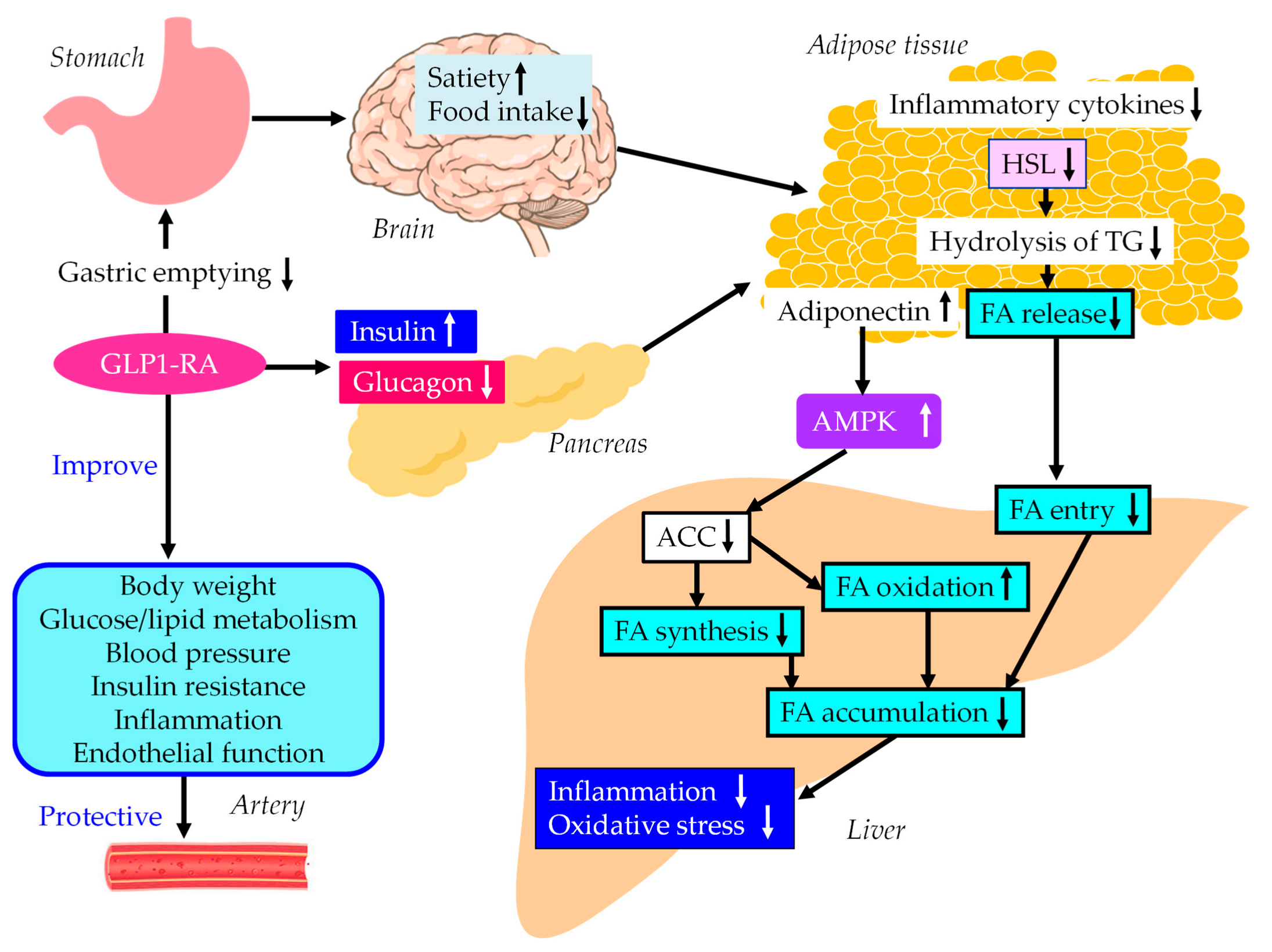
5.3. GLP-1RAs
5.3.1. Effects of GLP-1RAs on Liver Enzymes, Hepatic Steatosis and Fibrosis
5.3.2. The Underlying Mechanisms for MASLD Treatment using GLP-1RAs
5.3.3. The Vasculoprotective Effects of GLP-1RAs
5.4. Comparison of the Efficacy of SGLT2i/GLP-1RA/PPAR Agonists in MASLD
5.5. Effects of Combination Therapy Using SGLT2is and GLP-1RAs in MASLD
5.6. Effects of the Combination Therapy of SGLT2is and Pemafibrate on MASLD
6. The Limitations of Our Review
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| ACC | acetyl-CoA carboxylase |
| ACO | acyl-CoA oxidase |
| ALT | alanine aminotransferase |
| APRI | aminotransferase-to-platelet ratio index |
| AST | aspartate aminotransferase |
| ASCVD | atherosclerotic cardiovascular disease |
| AMPK | adenosine monophosphate-activated protein kinase |
| BMI | body mass index |
| C | cholesterol |
| CETP | cholesterol ester transfer protein |
| CI | confidence interval |
| CM | chylomicron |
| CMR | chylomicron remnant |
| CPT-1 | carnitine palmitoyl-transferase 1 |
| CRP | C-reactive protein |
| CVD | cardiovascular diseases |
| GGT | gamma-glutamyl transferase |
| FA | fatty acid |
| FAS | fatty acid synthase |
| FFA | free fatty acid |
| FIB-4 | fibrosis-4 |
| GLP-1RA | glucagon-like peptide-1 receptor agonists |
| HDL-C | high-density lipoprotein cholesterol |
| HMGR | 3-hydroxy-3-methyl-glutaryl-CoA reductase |
| HL | hepatic lipase |
| HOMA-IR | homeostasis model assessment of insulin resistance |
| HR | hazard ratio |
| HSL | hormone-sensitive lipase |
| IDL | intermediate-density lipoprotein |
| IL | interleukin |
| IRS-2 | insulin receptor substrate 2 |
| LDL | low-density lipoprotein |
| LPL | lipoprotein lipase |
| LRP1 | LDL receptor-related protein 1 |
| MACE | major adverse cardiovascular events |
| MASH | metabolic-dysfunction-associated steatohepatitis |
| MASLD | metabolic-dysfunction-associated steatotic liver disease |
| MCAD | medium-chain acyl-CoA dehydrogenase |
| MD | mean difference |
| MRI | magnetic resonance imaging |
| MTP | microsomal TG transfer protein |
| NAFLD | nonalcoholic fatty liver disease |
| NASH | nonalcoholic steatohepatitis |
| NF-κB | nuclear factor-κB |
| NPC1L1 | Niemann-Pick C1-like 1 |
| OR | odds ratio |
| OxLDL | oxidized LDL |
| PPAR | peroxisome proliferator-activated receptor |
| RCTs | randomized controlled trials |
| Rem | remnant lipoproteins |
| ROS | reactive oxygen species |
| SdLDL | small dense LDL |
| SGLT2i | sodium-glucose cotransporter 2 inhibitors |
| SOD | superoxide dismutase |
| SR-BI | scavenger receptor class B type I |
| SREBP | sterol regulatory element binding protein |
| TNF-α | tumor necrosis factor-alpha |
| TG | triglyceride |
| VLDL | very low-density lipoprotein |
| WC | waist circumference |
References
- Rinella, M.E.; Lazarus, J.V.; Ratziu, V.; Francque, S.M.; Sanyal, A.J.; Kanwal, F.; Romero, D.; Abdelmalek, M.F.; Anstee, Q.M.; Arab, J.P.; et al. A multi-society Delphi consensus statement on new fatty liver disease nomenclature. Ann. Hepatol. 2023, 101133. [Google Scholar] [CrossRef]
- Younossi, Z.; Anstee, Q.M.; Marietti, M.; Hardy, T.; Henry, L.; Eslam, M.; George, J.; Bugianesi, E. Global burden of NAFLD and NASH: Trends, predictions, risk factors and prevention. Nat. Rev. Gastroenterol. Hepatol. 2018, 15, 11–20. [Google Scholar] [CrossRef]
- Le, M.H.; Yeo, Y.H.; Zou, B.; Barnet, S.; Henry, L.; Cheung, R.; Nguyen, M.H. Forecasted 2040 global prevalence of nonalcoholic fatty liver disease using hierarchical bayesian approach. Clin. Mol. Hepatol. 2022, 28, 841–850. [Google Scholar] [CrossRef]
- Godoy-Matos, A.F.; Silva Júnior, W.S.; Valerio, C.M. NAFLD as a continuum: From obesity to metabolic syndrome and diabetes. Diabetol. Metab. Syndr. 2020, 12, 60. [Google Scholar] [CrossRef] [PubMed]
- Chalasani, N.; Younossi, Z.; Lavine, J.E.; Charlton, M.; Cusi, K.; Rinella, M.; Harrison, S.A.; Brunt, E.M.; Sanyal, A.J. The diagnosis and management of nonalcoholic fatty liver disease: Practice guidance from the American Association for the Study of Liver Diseases. Hepatology 2018, 67, 328–357. [Google Scholar] [CrossRef] [PubMed]
- Yoneda, M.; Yamamoto, T.; Honda, Y.; Imajo, K.; Ogawa, Y.; Kessoku, T.; Kobayashi, T.; Nogami, A.; Higurashi, T.; Kato, S.; et al. Risk of cardiovascular disease in patients with fatty liver disease as defined from the metabolic dysfunction associated fatty liver disease or nonalcoholic fatty liver disease point of view: A retrospective nationwide claims database study in Japan. J. Gastroenterol. 2021, 56, 1022–1032. [Google Scholar] [CrossRef]
- Pang, Q.; Zhang, J.Y.; Song, S.D.; Qu, K.; Xu, X.S.; Liu, S.S.; Liu, C. Central obesity and nonalcoholic fatty liver disease risk after adjusting for body mass index. World. J. Gastroenterol. 2015, 21, 1650–1662. [Google Scholar] [CrossRef] [PubMed]
- Younossi, Z.M.; Golabi, P.; de Avila, L.; Paik, J.M.; Srishord, M.; Fukui, N.; Qiu, Y.; Burns, L.; Afendy, A.; Nader, F. The global epidemiology of NAFLD and NASH in patients with type 2 diabetes: A systematic review and meta-analysis. J. Hepatol. 2019, 71, 793–801. [Google Scholar] [CrossRef] [PubMed]
- Li, G.; Peng, Y.; Chen, Z.; Li, H.; Liu, D.; Ye, X. Bidirectional Association between Hypertension and NAFLD: A Systematic Review and Meta-Analysis of Observational Studies. Int. J. Endocrinol. 2022, 2022, 8463640. [Google Scholar] [CrossRef]
- Assy, N.; Kaita, K.; Mymin, D.; Levy, C.; Rosser, B.; Minuk, G. Fatty infiltration of liver in hyperlipidemic patients. Dig. Dis. Sci. 2000, 45, 1929–1934. [Google Scholar] [CrossRef]
- Sanyal, A.J.; Campbell-Sargent, C.; Mirshahi, F.; Rizzo, W.B.; Contos, M.J.; Sterling, R.K.; Luketic, V.A.; Shiffman, M.L.; Clore, J.N. Nonalcoholic steatohepatitis: Association of insulin resistance and mitochondrial abnormalities. Gastroenterology 2001, 120, 1183–1192. [Google Scholar] [CrossRef] [PubMed]
- Vollenweider, P.; Ménard, B.; Nicod, P. Insulin resistance, defective insulin receptor substrate 2-associated phosphatidylinositol-3’ kinase activation, and impaired atypical protein kinase C (zeta/lambda) activation in myotubes from obese patients with impaired glucose tolerance. Diabetes 2002, 51, 1052–1059. [Google Scholar] [CrossRef] [PubMed]
- Xing, L.J.; Zhang, L.; Liu, T.; Hua, Y.Q.; Zheng, P.Y.; Ji, G. Berberine reducing insulin resistance by up-regulating IRS-2 mRNA expression in nonalcoholic fatty liver disease (NAFLD) rat liver. Eur. J. Pharmacol. 2011, 668, 467–471. [Google Scholar] [CrossRef] [PubMed]
- Berg, A.H.; Scherer, P.E. Adipose tissue, inflammation, and cardiovascular disease. Circ. Res. 2005, 96, 939–949. [Google Scholar] [CrossRef] [PubMed]
- Yanai, H.; Hirowatari, Y.; Ito, K.; Kurosawa, H.; Tada, N.; Yoshida, H. Understanding of Diabetic Dyslipidemia by Using the Anion-Exchange High Performance Liquid Chromatography Data. J. Clin. Med. Res. 2016, 8, 424–426. [Google Scholar] [CrossRef]
- Kelley, D.E.; Goodpaster, B.H. Skeletal muscle triglyceride. An aspect of regional adiposity and insulin resistance. Diabetes. Care 2001, 24, 933–941. [Google Scholar] [CrossRef]
- Fisher, E.A. The degradation of apolipoprotein B100: Multiple opportunities to regulate VLDL triglyceride production by different proteolytic pathways. Biochim. Biophys. Acta 2012, 1821, 778–781. [Google Scholar] [CrossRef]
- Taghibiglou, C.; Carpentier, A.; Van Iderstine, S.C.; Chen, B.; Rudy, D.; Aiton, A.; Lewis, G.F.; Adeli, K. Mechanisms of hepatic very low density lipoprotein overproduction in insulin resistance. Evidence for enhanced lipoprotein assembly, reduced intracellular ApoB degradation, and increased microsomal triglyceride transfer protein in a fructose-fed hamster model. J. Biol. Chem. 2000, 275, 8416–8425. [Google Scholar]
- Chen, M.; Breslow, J.L.; Li, W.; Leff, T. Transcriptional regulation of the apoC-III gene by insulin in diabetic mice: Correlation with changes in plasma triglyceride levels. J. Lipid. Res. 1994, 35, 1918–1924. [Google Scholar] [CrossRef]
- Avramoglu, R.K.; Basciano, H.; Adeli, K. Lipid and lipoprotein dysregulation in insulin resistant states. Clin. Chim. Acta 2006, 368, 1–19. [Google Scholar] [CrossRef]
- Duran, E.K.; Pradhan, A.D. Triglyceride-Rich Lipoprotein Remnants and Cardiovascular Disease. Clin. Chem. 2021, 67, 183–196. [Google Scholar] [CrossRef] [PubMed]
- Barrows, B.R.; Parks, E.J. Contributions of different fatty acid sources to very low-density lipoprotein-triacylglycerol in the fasted and fed states. J. Clin. Endocrinol. Metab. 2006, 91, 1446–1452. [Google Scholar] [CrossRef] [PubMed]
- Taskinen, M.R.; Adiels, M.; Westerbacka, J.; Söderlund, S.; Kahri, J.; Lundbom, N.; Lundbom, J.; Hakkarainen, A.; Olofsson, S.O.; Orho-Melander, M.; et al. Dual metabolic defects are required to produce hypertriglyceridemia in obese subjects. Arterioscler. Thromb. Vasc. Biol. 2011, 31, 2144–2150. [Google Scholar] [CrossRef] [PubMed]
- Musso, G.; Gambino, R.; De Michieli, F.; Biroli, G.; Fagà, E.; Pagano, G.; Cassader, M. Association of liver disease with postprandial large intestinal triglyceride-rich lipoprotein accumulation and pro/antioxidant imbalance in normolipidemic non-alcoholic steatohepatitis. Ann. Med. 2008, 40, 383–394. [Google Scholar] [CrossRef]
- Qin, B.; Anderson, R.A.; Kuzuya, T.; Kitaura, Y.; Shimomura, Y. Multiple factors and pathways involved in hepatic very low density lipoprotein-apoB100 overproduction in Otsuka Long-Evans Tokushima Fatty rats. Atherosclerosis 2012, 222, 409–416. [Google Scholar] [CrossRef]
- Grove, J.I.; Lo, P.C.K.; Shrine, N.; Barwell, J.; Wain, L.V.; Tobin, M.D.; Salter, A.M.; Borkar, A.N.; Cuevas-Ocaña, S.; Bennett, N.; et al. Identification and characterisation of a rare MTTP variant underlying hereditary non-alcoholic fatty liver disease. JHEP Rep. 2023, 5, 100764. [Google Scholar] [CrossRef]
- Hong, S.; Gordon, D.; Stec, D.E.; Hinds, T.D. Bilirubin: A Ligand of the PPARα Nuclear Receptor. In Nuclear Receptors: The Art and Science of Modulator Design and Discovery; Badr, M.Z., Ed.; Springer International Publishing: Cham, Switzerland, 2021; pp. 463–482. [Google Scholar]
- Moreno-Fernandez, M.E.; Giles, D.A.; Stankiewicz, T.E.; Sheridan, R.; Karns, R.; Cappelletti, M.; Lampe, K.; Mukherjee, R.; Sina, C.; Sallese, A.; et al. Peroxisomal β-oxidation regulates whole body metabolism, inflammatory vigor, and pathogenesis of nonalcoholic fatty liver disease. JCI Insight 2018, 3, e93626. [Google Scholar] [CrossRef]
- Hinds, T.D., Jr.; Hosick, P.A.; Chen, S.; Tukey, R.H.; Hankins, M.W.; Nestor-Kalinoski, A.; Stec, D.E. Mice with hyperbilirubinemia due to Gilbert’s syndrome polymorphism are resistant to hepatic steatosis by decreased serine 73 phosphorylation of PPARα. Am. J. Physiol. Endocrinol. Metab. 2017, 312, E244–E252. [Google Scholar] [CrossRef]
- Stec, D.E.; John, K.; Trabbic, C.J.; Luniwal, A.; Hankins, M.W.; Baum, J.; Hinds, T.D., Jr. Bilirubin Binding to PPARα Inhibits Lipid Accumulation. PLoS ONE 2016, 11, e0153427. [Google Scholar] [CrossRef]
- Hinds, T.D., Jr.; Adeosun, S.O.; Alamodi, A.A.; Stec, D.E. Does bilirubin prevent hepatic steatosis through activation of the PPARα nuclear receptor? Med. Hypotheses 2016, 95, 54–57. [Google Scholar] [CrossRef]
- Hinds, T.D., Jr.; Burns, K.A.; Hosick, P.A.; McBeth, L.; Nestor-Kalinoski, A.; Drummond, H.A.; AlAmodi, A.A.; Hankins, M.W.; Heuvel, J.P.V.; Stec, D.E. Biliverdin Reductase A Attenuates Hepatic Steatosis by Inhibition of Glycogen Synthase Kinase (GSK) 3β Phosphorylation of Serine 73 of Peroxisome Proliferator-activated Receptor (PPAR) α. J. Biol. Chem. 2016, 291, 25179–25191. [Google Scholar] [CrossRef]
- Francque, S.; Verrijken, A.; Caron, S.; Prawitt, J.; Paumelle, R.; Derudas, B.; Lefebvre, P.; Taskinen, M.R.; Van Hul, W.; Mertens, I.; et al. PPARα gene expression correlates with severity and histological treatment response in patients with non-alcoholic steatohepatitis. J. Hepatol. 2015, 63, 164–173. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Nakajima, T.; Gonzalez, F.J.; Tanaka, N. PPARs as Metabolic Regulators in the Liver: Lessons from Liver-Specific PPAR-Null Mice. Int. J. Mol. Sci. 2020, 21, 2061. [Google Scholar] [CrossRef] [PubMed]
- Paiva, A.A.; Raposo, H.F.; Wanschel, A.C.; Nardelli, T.R.; Oliveira, H.C. Apolipoprotein CIII Overexpression-Induced Hypertriglyceridemia Increases Nonalcoholic Fatty Liver Disease in Association with Inflammation and Cell Death. Oxid. Med. Cell. Longev. 2017, 2017, 1838679. [Google Scholar] [CrossRef] [PubMed]
- Roden, M.; Stingl, H.; Chandramouli, V.; Schumann, W.C.; Hofer, A.; Landau, B.R.; Nowotny, P.; Waldhäusl, W.; Shulman, G.I. Effects of free fatty acid elevation on postabsorptive endogenous glucose production and gluconeogenesis in humans. Diabetes 2000, 49, 701–707. [Google Scholar] [CrossRef] [PubMed]
- Wen, H.; Gris, D.; Lei, Y.; Jha, S.; Zhang, L.; Huang, M.T.; Brickey, W.J.; Ting, J.P. Fatty acid-induced NLRP3-ASC inflammasome activation interferes with insulin signaling. Nat. Immunol. 2011, 12, 408–415. [Google Scholar] [CrossRef]
- Neuschwander-Tetri, B.A. Hepatic lipotoxicity and the pathogenesis of nonalcoholic steatohepatitis: The central role of nontriglyceride fatty acid metabolites. Hepatology 2010, 52, 774–788. [Google Scholar] [CrossRef]
- Fuchs, M.; Sanyal, A.J. Lipotoxicity in NASH. J. Hepatol. 2012, 56, 291–293. [Google Scholar] [CrossRef]
- Sinha, R.A. Autophagy: A Cellular Guardian against Hepatic Lipotoxicity. Genes 2023, 14, 553. [Google Scholar] [CrossRef]
- Yamaguchi, K.; Yang, L.; McCall, S.; Huang, J.; Yu, X.X.; Pandey, S.K.; Bhanot, S.; Monia, B.P.; Li, Y.X.; Diehl, A.M. Inhibiting triglyceride synthesis improves hepatic steatosis but exacerbates liver damage and fibrosis in obese mice with nonalcoholic steatohepatitis. Hepatology 2007, 45, 1366–1374. [Google Scholar] [CrossRef]
- Neuschwander-Tetri, B.A. Nontriglyceride hepatic lipotoxicity: The new paradigm for the pathogenesis of NASH. Curr. Gastroenterol. Rep. 2010, 12, 49–56. [Google Scholar] [CrossRef] [PubMed]
- Van Rooyen, D.M.; Farrell, G.C. SREBP-2: A link between insulin resistance, hepatic cholesterol, and inflammation in NASH. J. Gastroenterol. Hepatol. 2011, 26, 789–792. [Google Scholar] [CrossRef]
- Angulo, P.; Kleiner, D.E.; Dam-Larsen, S.; Adams, L.A.; Bjornsson, E.S.; Charatcharoenwitthaya, P.; Mills, P.R.; Keach, J.C.; Lafferty, H.D.; Stahler, A.; et al. Liver Fibrosis, but No Other Histologic Features, Is Associated With Long-term Outcomes of Patients With Nonalcoholic Fatty Liver Disease. Gastroenterology 2015, 149, 389–397. [Google Scholar] [CrossRef] [PubMed]
- Musso, G.; Gambino, R.; Cassader, M.; Pagano, G. Meta-analysis: Natural history of non-alcoholic fatty liver disease (NAFLD) and diagnostic accuracy of non-invasive tests for liver disease severity. Ann. Med. 2011, 43, 617–649. [Google Scholar] [CrossRef] [PubMed]
- Eguchi, Y.; Hyogo, H.; Ono, M.; Mizuta, T.; Ono, N.; Fujimoto, K.; Chayama, K.; Saibara, T.; JSG-NAFLD. Prevalence and associated metabolic factors of nonalcoholic fatty liver disease in the general population from 2009 to 2010 in Japan: A multicenter large retrospective study. J. Gastroenterol. 2012, 47, 586–595. [Google Scholar] [CrossRef] [PubMed]
- Yanai, H.; Adachi, H.; Hakoshima, M.; Katsuyama, H. Atherogenic Lipoproteins for the Statin Residual Cardiovascular Disease Risk. Int. J. Mol. Sci. 2022, 23, 13499. [Google Scholar] [CrossRef]
- Nikkila, E.A.; Huttunen, J.K.; Ehnholm, C. Postheparin plasma lipoprotein lipase and hepatic lipase in diabetes mellitus. Relationship to plasma triglyceride metabolism. Diabetes 1977, 26, 11–21. [Google Scholar]
- Nikkila, E.A.; Taskinen, M.R.; Kekki, M. Relation of plasma high-density lipoprotein cholesterol to lipoprotein-lipase activity in adipose tissue and skeletal muscle of man. Atherosclerosis 1978, 29, 497–501. [Google Scholar] [CrossRef]
- Kasim, S.E.; Tseng, K.; Jen, K.L.; Khilnani, S. Significance of hepatic triglyceride lipase activity in the regulation of serum high density lipoproteins in type II diabetes mellitus. J. Clin. Endocrinol. Metab. 1987, 65, 183–187. [Google Scholar] [CrossRef]
- Vega, G.L.; Grundy, S.M. Effect of statins on metabolism of apo-B-containing lipoproteins in hypertriglyceridemic men. Am. J. Cardiol. 1998, 81, 36B–42B. [Google Scholar] [CrossRef]
- Zambon, A.; Austin, M.A.; Brown, B.G.; Hokanson, J.E.; Brunzell, J.D. Effect of hepatic lipase on LDL in normal men and those with coronary artery disease. Arterioscler. Thromb. 1993, 13, 147–153. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, E.A.; Reaven, P.D. Lipolysis of triglyceride-rich lipoproteins, vascular inflammation, and atherosclerosis. Biochim. Biophys. Acta 2012, 1821, 858–866. [Google Scholar] [CrossRef]
- Yanai, H.; Adachi, H.; Hakoshima, M.; Katsuyama, H. Molecular Biological and Clinical Understanding of the Statin Residual Cardiovascular Disease Risk and Peroxisome Proliferator-Activated Receptor Alpha Agonists and Ezetimibe for Its Treatment. Int. J. Mol. Sci. 2022, 23, 3418. [Google Scholar] [CrossRef] [PubMed]
- Chandra, N.C. A comprehensive account of insulin and LDL receptor activity over the years: A highlight on their signaling and functional role. J. Biochem. Mol. Toxicol. 2021, 35, e22840. [Google Scholar] [CrossRef] [PubMed]
- Lally, S.; Owens, D.; Tomkin, G.H. Genes that affect cholesterol synthesis, cholesterol absorption, and chylomicron assembly: The relationship between the liver and intestine in control and streptozotosin diabetic rats. Metabolism 2007, 56, 430–438. [Google Scholar] [CrossRef]
- Actis Dato, V.; Chiabrando, G.A. The Role of Low-Density Lipoprotein Receptor-Related Protein 1 in Lipid Metabolism, Glucose Homeostasis and Inflammation. Int. J. Mol. Sci. 2018, 19, 1780. [Google Scholar] [CrossRef]
- Borén, J.; Williams, K.J. The central role of arterial retention of cholesterol-rich apolipoprotein-B-containing lipoproteins in the pathogenesis of atherosclerosis: A triumph of simplicity. Curr. Opin. Lipidol. 2016, 27, 473–483. [Google Scholar] [CrossRef] [PubMed]
- Ahn, J.; Jun, D.W.; Lee, H.Y.; Moon, J.H. Critical appraisal for low-carbohydrate diet in nonalcoholic fatty liver disease: Review and meta-analyses. Clin. Nutr. 2019, 38, 2023–2030. [Google Scholar] [CrossRef]
- Houttu, V.; Csader, S.; Nieuwdorp, M.; Holleboom, A.G.; Schwab, U. Dietary Interventions in Patients With Non-alcoholic Fatty Liver Disease: A Systematic Review and Meta-Analysis. Front. Nutr. 2021, 8, 716783. [Google Scholar] [CrossRef]
- Haigh, L.; Kirk, C.; El Gendy, K.; Gallacher, J.; Errington, L.; Mathers, J.C.; Anstee, Q.M. The effectiveness and acceptability of Mediterranean diet and calorie restriction in non-alcoholic fatty liver disease (NAFLD): A systematic review and meta-analysis. Clin. Nutr. 2022, 41, 1913–1931. [Google Scholar] [CrossRef]
- Cho, Y.; Hong, N.; Kim, K.W.; Cho, S.J.; Lee, M.; Lee, Y.H.; Lee, Y.H.; Kang, E.S.; Cha, B.S.; Lee, B.W. The Effectiveness of Intermittent Fasting to Reduce Body Mass Index and Glucose Metabolism: A Systematic Review and Meta-Analysis. J. Clin. Med. 2019, 8, 1645. [Google Scholar] [CrossRef] [PubMed]
- Harris, L.; Hamilton, S.; Azevedo, L.B.; Olajide, J.; De Brún, C.; Waller, G.; Whittaker, V.; Sharp, T.; Lean, M.; Hankey, C.; et al. Intermittent fasting interventions for treatment of overweight and obesity in adults: A systematic review and meta-analysis. JBI Database Syst. Rev. Implement. Rep. 2018, 16, 507–547. [Google Scholar] [CrossRef] [PubMed]
- Rizza, W.; Veronese, N.; Fontana, L. What are the roles of calorie restriction and diet quality in promoting healthy longevity? Ageing. Res. Rev. 2014, 13, 38–45. [Google Scholar] [CrossRef] [PubMed]
- Yin, C.; Li, Z.; Xiang, Y.; Peng, H.; Yang, P.; Yuan, S.; Zhang, X.; Wu, Y.; Huang, M.; Li, J. Effect of Intermittent Fasting on Non-Alcoholic Fatty Liver Disease: Systematic Review and Meta-Analysis. Front. Nutr. 2021, 8, 709683. [Google Scholar] [CrossRef] [PubMed]
- Lange, M.; Nadkarni, D.; Martin, L.; Newberry, C.; Kumar, S.; Kushner, T. Intermittent fasting improves hepatic end points in nonalcoholic fatty liver disease: A systematic review and meta-analysis. Hepatol. Commun. 2023, 7, e0212. [Google Scholar] [CrossRef]
- Orci, L.A.; Gariani, K.; Oldani, G.; Delaune, V.; Morel, P.; Toso, C. Exercise-based Interventions for Nonalcoholic Fatty Liver Disease: A Meta-analysis and Meta-regression. Clin. Gastroenterol. Hepatol. 2016, 14, 1398–1411. [Google Scholar] [CrossRef]
- Katsagoni, C.N.; Georgoulis, M.; Papatheodoridis, G.V.; Panagiotakos, D.B.; Kontogianni, M.D. Effects of lifestyle interventions on clinical characteristics of patients with non-alcoholic fatty liver disease: A meta-analysis. Metabolism 2017, 68, 119–132. [Google Scholar] [CrossRef]
- González-Ruiz, K.; Ramírez-Vélez, R.; Correa-Bautista, J.E.; Peterson, M.D.; García-Hermoso, A. The Effects of Exercise on Abdominal Fat and Liver Enzymes in Pediatric Obesity: A Systematic Review and Meta-Analysis. Child. Obes. 2017, 13, 272–282. [Google Scholar] [CrossRef]
- Medrano, M.; Cadenas-Sanchez, C.; Álvarez-Bueno, C.; Cavero-Redondo, I.; Ruiz, J.R.; Ortega, F.B.; Labayen, I. Evidence-Based Exercise Recommendations to Reduce Hepatic Fat Content in Youth- a Systematic Review and Meta-Analysis. Prog. Cardiovasc. Dis. 2018, 61, 222–231. [Google Scholar] [CrossRef]
- Sargeant, J.A.; Gray, L.J.; Bodicoat, D.H.; Willis, S.A.; Stensel, D.J.; Nimmo, M.A.; Aithal, G.P.; King, J.A. The effect of exercise training on intrahepatic triglyceride and hepatic insulin sensitivity: A systematic review and meta-analysis. Obes. Rev. 2018, 19, 1446–1459. [Google Scholar] [CrossRef]
- Musso, G.; Cassader, M.; Rosina, F.; Gambino, R. Impact of current treatments on liver disease, glucose metabolism and cardiovascular risk in non-alcoholic fatty liver disease (NAFLD): A systematic review and meta-analysis of randomised trials. Diabetologia 2012, 55, 885–904. [Google Scholar] [CrossRef]
- Fernández, T.; Viñuela, M.; Vidal, C.; Barrera, F. Lifestyle changes in patients with non-alcoholic fatty liver disease: A systematic review and meta-analysis. PLoS ONE 2022, 17, e0263931. [Google Scholar] [CrossRef] [PubMed]
- Buchwald, H.; Avidor, Y.; Braunwald, E.; Jensen, M.D.; Pories, W.; Fahrbach, K.; Schoelles, K. Bariatric surgery: A systematic review and meta-analysis. JAMA 2004, 292, 1724–1737. [Google Scholar] [CrossRef] [PubMed]
- Adams, T.D.; Gress, R.E.; Smith, S.C.; Halverson, R.C.; Simper, S.C.; Rosamond, W.D.; Lamonte, M.J.; Stroup, A.M.; Hunt, S.C. Long-term mortality after gastric bypass surgery. N. Engl. J. Med. 2007, 357, 753–761. [Google Scholar] [CrossRef]
- Pontiroli, A.E.; Morabito, A. Long-term prevention of mortality in morbid obesity through bariatric surgery. A systematic review and meta-analysis of trials performed with gastric banding and gastric bypass. Ann. Surg. 2011, 253, 484–487. [Google Scholar] [CrossRef]
- Bower, G.; Toma, T.; Harling, L.; Jiao, L.R.; Efthimiou, E.; Darzi, A.; Athanasiou, T.; Ashrafian, H. Bariatric Surgery and Non-Alcoholic Fatty Liver Disease: A Systematic Review of Liver Biochemistry and Histology. Obes. Surg. 2015, 25, 2280–2289. [Google Scholar] [CrossRef] [PubMed]
- Popov, V.B.; Thompson, C.C.; Kumar, N.; Ciarleglio, M.M.; Deng, Y.; Laine, L. Effect of Intragastric Balloons on Liver Enzymes: A Systematic Review and Meta-Analysis. Dig. Dis. Sci. 2016, 61, 2477–2487. [Google Scholar] [CrossRef]
- Chandan, S.; Mohan, B.P.; Khan, S.R.; Facciorusso, A.; Ramai, D.; Kassab, L.L.; Bhogal, N.; Asokkumar, R.; Lopez-Nava, G.; McDonough, S.; et al. Efficacy and Safety of Intragastric Balloon (IGB) in Non-alcoholic Fatty Liver Disease (NAFLD): A Comprehensive Review and Meta-analysis. Obes. Surg. 2021, 31, 1271–1279. [Google Scholar] [CrossRef]
- Kamata, S.; Honda, A.; Ishii, I. Current Clinical Trial Status and Future Prospects of PPAR-Targeted Drugs for Treating Nonalcoholic Fatty Liver Disease. Biomolecules 2023, 13, 1264. [Google Scholar] [CrossRef]
- Lee, M.; Saver, J.L.; Towfighi, A.; Chow, J.; Ovbiagele, B. Efficacy of fibrates for cardiovascular risk reduction in persons with atherogenic dyslipidemia: A meta-analysis. Atherosclerosis 2011, 217, 492–498. [Google Scholar] [CrossRef]
- Yanai, H.; Katsuyama, H.; Hakoshima, M. Effects of a Novel Selective Peroxisome Proliferator-Activated Receptor α Modulator, Pemafibrate, on Metabolic Parameters: A Retrospective Longitudinal Study. Biomedicines 2022, 10, 401. [Google Scholar] [CrossRef] [PubMed]
- Yanai, H.; Hakoshima, M.; Adachi, H.; Katsuyama, H. Multi-Organ Protective Effects of Sodium Glucose Cotransporter 2 Inhibitors. Int. J. Mol. Sci. 2021, 22, 4416. [Google Scholar] [CrossRef] [PubMed]
- Yanai, H.; Adachi, H.; Hakoshima, M.; Katsuyama, H. Glucagon-Like Peptide 1 Receptor Agonists Versus Sodium-Glucose Cotransporter 2 Inhibitors for Atherosclerotic Cardiovascular Disease in Patients With Type 2 Diabetes. Cardiol. Res. 2023, 14, 12–21. [Google Scholar] [CrossRef] [PubMed]
- Yamazaki, Y.; Abe, K.; Toma, T.; Nishikawa, M.; Ozawa, H.; Okuda, A.; Araki, T.; Oda, S.; Inoue, K.; Shibuya, K.; et al. Design and synthesis of highly potent and selective human peroxisome proliferator-activated receptor alpha agonists. Bioorg. Med. Chem. Lett. 2007, 17, 4689–4693. [Google Scholar] [CrossRef]
- Katsuyama, H.; Yanai, H.; Adachi, H.; Hakoshima, M. A Significant Effect of Pemafibrate on Hepatic Steatosis and Fibrosis Indexes in Patients With Hypertriglyceridemia. Gastroenterol. Res. 2023, 16, 240–243. [Google Scholar] [CrossRef]
- Nakajima, A.; Eguchi, Y.; Yoneda, M.; Imajo, K.; Tamaki, N.; Suganami, H.; Nojima, T.; Tanigawa, R.; Iizuka, M.; Iida, Y.; et al. Randomised clinical trial: Pemafibrate, a novel selective peroxisome proliferator-activated receptor α modulator (SPPARMα), versus placebo in patients with non-alcoholic fatty liver disease. Aliment. Pharmacol. Ther. 2021, 54, 1263–1277. [Google Scholar] [CrossRef]
- Morishita, A.; Oura, K.; Takuma, K.; Nakahara, M.; Tadokoro, T.; Fujita, K.; Tani, J.; Shi, T.; Himoto, T.; Tatsuta, M.; et al. Pemafibrate improves liver dysfunction and non-invasive surrogates for liver fibrosis in patients with non-alcoholic fatty liver disease with hypertriglyceridemia: A multicenter study. Hepatol. Int. 2023, 17, 606–614. [Google Scholar] [CrossRef]
- Ahima, R.S. Adipose tissue as an endocrine organ. Obesity 2006, 14, 242S–249S. [Google Scholar] [CrossRef]
- Fonseca-Alaniz, M.H.; Takada, J.; Alonso-Vale, M.I.; Lima, F.B. Adipose tissue as an endocrine organ: From theory to practice. J. Pediatr. 2007, 83, S192–S203. [Google Scholar] [CrossRef]
- Kim, S.H.; Plutzky, J. Brown Fat and Browning for the Treatment of Obesity and Related Metabolic Disorders. Diabetes. Metab. J. 2016, 40, 12–21. [Google Scholar] [CrossRef]
- Rachid, T.L.; Silva-Veiga, F.M.; Graus-Nunes, F.; Bringhenti, I.; Mandarim-de-Lacerda, C.A.; Souza-Mello, V. Differential actions of PPAR-alpha and PPAR-beta/delta on beige adipocyte formation: A study in the subcutaneous white adipose tissue of obese male mice. PLoS ONE 2018, 13, e0191365. [Google Scholar] [CrossRef] [PubMed]
- Maia-Fernandes, T.; Roncon-Albuquerque, R., Jr.; Leite-Moreira, A.F. Cardiovascular actions of adiponectin: Pathophysiologic implications. Rev. Port. Cardiol. 2008, 27, 1431–1449. [Google Scholar] [PubMed]
- Yanai, H.; Yoshida, H. Beneficial Effects of Adiponectin on Glucose and Lipid Metabolism and Atherosclerotic Progression: Mechanisms and Perspectives. Int. J. Mol. Sci. 2019, 20, 1190. [Google Scholar] [CrossRef] [PubMed]
- Haluzík, M.M.; Haluzík, M. PPAR-alpha and insulin sensitivity. Physiol. Res. 2006, 55, 115–122. [Google Scholar] [CrossRef] [PubMed]
- Staels, B.; Dallongeville, J.; Auwerx, J.; Schoonjans, K.; Leitersdorf, E.; Fruchart, J.C. Mechanism of action of fibrates on lipid and lipoprotein metabolism. Circulation 1998, 98, 2088–2093. [Google Scholar] [CrossRef]
- Kim, H.; Haluzik, M.; Asghar, Z.; Yau, D.; Joseph, J.W.; Fernandez, A.M.; Reitman, M.L.; Yakar, S.; Stannard, B.; Heron-Milhavet, L.; et al. Peroxisome proliferator-activated receptor-alpha agonist treatment in a transgenic model of type 2 diabetes reverses the lipotoxic state and improves glucose homeostasis. Diabetes 2003, 52, 1770–1778. [Google Scholar] [CrossRef]
- Qiang, X.; Xu, L.; Zhang, M.; Zhang, P.; Wang, Y.; Wang, Y.; Zhao, Z.; Chen, H.; Liu, X.; Zhang, Y. Demethyleneberberine attenuates non-alcoholic fatty liver disease with activation of AMPK and inhibition of oxidative stress. Biochem. Biophys. Res. Commun. 2016, 472, 603–609. [Google Scholar] [CrossRef]
- Liu, S.; Jing, F.; Yu, C.; Gao, L.; Qin, Y.; Zhao, J. AICAR-Induced Activation of AMPK Inhibits TSH/SREBP-2/HMGCR Pathway in Liver. PLoS ONE 2015, 10, e0124951. [Google Scholar] [CrossRef]
- Day, E.A.; Ford, R.J.; Steinberg, G.R. AMPK as a Therapeutic Target for Treating Metabolic Diseases. Trends Endocrinol. Metab. 2017, 28, 545–560. [Google Scholar] [CrossRef]
- Yun, H.; Park, S.; Kim, M.J.; Yang, W.K.; Im, D.U.; Yang, K.R.; Hong, J.; Choe, W.; Kang, I.; Kim, S.S.; et al. AMP-activated protein kinase mediates the antioxidant effects of resveratrol through regulation of the transcription factor FoxO1. FEBS J. 2014, 281, 4421–4438. [Google Scholar] [CrossRef]
- Yanai, H.; Adachi, H.; Hakoshima, M.; Katsuyama, H. Postprandial Hyperlipidemia: Its Pathophysiology, Diagnosis, Atherogenesis, and Treatments. Int. J. Mol. Sci. 2023, 24, 13942. [Google Scholar] [CrossRef]
- Chinetti, G.; Lestavel, S.; Bocher, V.; Remaley, A.T.; Neve, B.; Torra, I.P.; Teissier, E.; Minnich, A.; Jaye, M.; Duverger, N.; et al. PPAR-alpha and PPAR-gamma activators induce cholesterol removal from human macrophage foam cells through stimulation of the ABCA1 pathway. Nat. Med. 2001, 7, 53–58. [Google Scholar] [CrossRef]
- Das Pradhan, A.; Glynn, R.J.; Fruchart, J.C.; MacFadyen, J.G.; Zaharris, E.S.; Everett, B.M.; Campbell, S.E.; Oshima, R.; Amarenco, P.; Blom, D.J.; et al. Triglyceride Lowering with Pemafibrate to Reduce Cardiovascular Risk. N. Engl. J. Med. 2022, 387, 1923–1934. [Google Scholar] [CrossRef]
- Vallon, V.; Platt, K.A.; Cunard, R.; Schroth, J.; Whaley, J.; Thomson, S.C.; Koepsell, H.; Rieg, T. SGLT2 mediates glucose reabsorption in the early proximal tubule. J. Am. Soc. Nephrol. 2011, 22, 104–112. [Google Scholar] [CrossRef]
- Jabbour, S.A.; Goldstein, B.J. Sodium glucose co-transporter 2 inhibitors: Blocking renal tubular reabsorption of glucose to improve glycaemic control in patients with diabetes. Int. J. Clin. Pract. 2008, 62, 1279–1284. [Google Scholar] [CrossRef]
- Yanai, H.; Katsuyama, H.; Hamasaki, H.; Adachi, H.; Moriyama, S.; Yoshikawa, R.; Sako, A. Sodium-Glucose Cotransporter 2 Inhibitors: Possible Anti-Atherosclerotic Effects Beyond Glucose Lowering. J. Clin. Med. Res. 2016, 8, 10–14. [Google Scholar] [CrossRef]
- Katsuyama, H.; Hamasaki, H.; Adachi, H.; Moriyama, S.; Kawaguchi, A.; Sako, A.; Mishima, S.; Yanai, H. Effects of Sodium-Glucose Cotransporter 2 Inhibitors on Metabolic Parameters in Patients With Type 2 Diabetes: A Chart-Based Analysis. J. Clin. Med. Res. 2016, 8, 237–243. [Google Scholar] [CrossRef]
- Yanai, H.; Hakoshima, M.; Adachi, H.; Kawaguchi, A.; Waragai, Y.; Harigae, T.; Masui, Y.; Kakuta, K.; Hamasaki, H.; Katsuyama, H.; et al. Effects of Six Kinds of Sodium-Glucose Cotransporter 2 Inhibitors on Metabolic Parameters, and Summarized Effect and Its Correlations with Baseline Data. J. Clin. Med. Res. 2017, 9, 605–612. [Google Scholar] [CrossRef]
- Shah, A.G.; Lydecker, A.; Murray, K.; Tetri, B.N.; Contos, M.J.; Sanyal, A.J.; Nash Clinical Research, N. Comparison of noninvasive markers of fibrosis in patients with nonalcoholic fatty liver disease. Clin. Gastroenterol. Hepatol. 2009, 7, 1104–1112. [Google Scholar] [CrossRef]
- Katsuyama, H.; Hakoshima, M.; Iijima, T.; Adachi, H.; Hidekatsu Yanai, H. Effects of Sodium-Glucose Cotransporter 2 Inhibitors on Hepatic Fibrosis in Patients With Type 2 Diabetes: A Chart-Based Analysis. J. Endocrinol. Metab. 2020, 10, 1–7. [Google Scholar] [CrossRef]
- Mino, M.; Kakazu, E.; Sano, A.; Katsuyama, H.; Hakoshima, M.; Yanai, H.; Aoki, Y.; Imamura, M.; Yamazoe, T.; Mori, T.; et al. Effects of sodium glucose cotransporter 2 inhibitors and pioglitazone on FIB-4 index in metabolic-associated fatty liver disease. Hepatol. Res. 2023, 53, 618–628. [Google Scholar] [CrossRef]
- Coelho, F.D.S.; Borges-Canha, M.; von Hafe, M.; Neves, J.S.; Vale, C.; Leite, A.R.; Carvalho, D.; Leite-Moreira, A. Effects of sodium-glucose co-transporter 2 inhibitors on liver parameters and steatosis: A meta-analysis of randomized clinical trials. Diabetes. Metab. Res. Rev. 2021, 37, e3413. [Google Scholar] [CrossRef]
- Mantovani, A.; Petracca, G.; Csermely, A.; Beatrice, G.; Targher, G. Sodium-Glucose Cotransporter-2 Inhibitors for Treatment of Nonalcoholic Fatty Liver Disease: A Meta-Analysis of Randomized Controlled Trials. Metabolites 2020, 11, 22. [Google Scholar] [CrossRef]
- Sinha, B.; Datta, D.; Ghosal, S. Meta-analysis of the effects of sodium glucose cotransporter 2 inhibitors in non-alcoholic fatty liver disease patients with type 2 diabetes. JGH Open 2020, 5, 219–227. [Google Scholar] [CrossRef]
- Wong, C.; Yaow, C.Y.L.; Ng, C.H.; Chin, Y.H.; Low, Y.F.; Lim, A.Y.L.; Muthiah, M.D.; Khoo, C.M. Sodium-Glucose Co-Transporter 2 Inhibitors for Non-Alcoholic Fatty Liver Disease in Asian Patients With Type 2 Diabetes: A Meta-Analysis. Front. Endocrinol. 2021, 11, 609135. [Google Scholar] [CrossRef]
- Wei, Q.; Xu, X.; Guo, L.; Li, J.; Li, L. Effect of SGLT2 Inhibitors on Type 2 Diabetes Mellitus With Non-Alcoholic Fatty Liver Disease: A Meta-Analysis of Randomized Controlled Trials. Front. Endocrinol. 2021, 12, 635556. [Google Scholar] [CrossRef]
- Murawaki, Y.; Ikuta, K.; Koda, M.; Kawasaki, H. Serum type III procollagen peptide, type IV collagen 7S domain, central triple-helix of type IV collagen and tissue inhibitor of metalloproteinases in patients with chronic viral liver disease: Relationship to liver histology. Hepatology 1994, 20, 780–787. [Google Scholar] [CrossRef]
- Manousou, P.; Kalambokis, G.; Grillo, F.; Watkins, J.; Xirouchakis, E.; Pleguezuelo, M.; Leandro, G.; Arvaniti, V.; Germani, G.; Patch, D.; et al. Serum ferritin is a discriminant marker for both fibrosis and inflammation in histologically proven non-alcoholic fatty liver disease patients. Liver Int. 2011, 31, 730–739. [Google Scholar] [CrossRef]
- Kowdley, K.V.; Belt, P.; Wilson, L.A.; Yeh, M.M.; Neuschwander-Tetri, B.A.; Chalasani, N.; Sanyal, A.J.; Nelson, J.E.; NASH Clinical Research Network. Serum ferritin is an independent predictor of histologic severity and advanced fibrosis in patients with nonalcoholic fatty liver disease. Hepatology 2012, 55, 77–85. [Google Scholar] [CrossRef]
- Jung, J.Y.; Shim, J.J.; Park, S.K.; Ryoo, J.H.; Choi, J.M.; Oh, I.H.; Jung, K.W.; Cho, H.; Ki, M.; Won, Y.J.; et al. Serum ferritin level is associated with liver steatosis and fibrosis in Korean general population. Hepatol. Int. 2019, 13, 222–233. [Google Scholar] [CrossRef]
- Song, T.; Chen, S.; Zhao, H.; Wang, F.; Song, H.; Tian, D.; Yang, Q.; Qi, L. Meta-analysis of the effect of sodium-glucose cotransporter 2 inhibitors on hepatic fibrosis in patients with type 2 diabetes mellitus complicated with non-alcoholic fatty liver disease. Hepatol. Res. 2021, 51, 641–651. [Google Scholar] [CrossRef]
- Sasso, M.; Beaugrand, M.; de Ledinghen, V.; Douvin, C.; Marcellin, P.; Poupon, R.; Sandrin, L.; Miette, V. Controlled attenuation parameter (CAP): A novel VCTE™ guided ultrasonic attenuation measurement for the evaluation of hepatic steatosis: Preliminary study and validation in a cohort of patients with chronic liver disease from various causes. Ultrasound Med. Biol. 2010, 36, 1825–1835. [Google Scholar] [CrossRef]
- Piazzolla, V.A.; Mangia, A. Noninvasive Diagnosis of NAFLD and NASH. Cells 2020, 9, 1005. [Google Scholar] [CrossRef]
- Vuppalanchi, R.; Siddiqui, M.S.; Van Natta, M.L.; Hallinan, E.; Brandman, D.; Kowdley, K.; Neuschwander-Tetri, B.A.; Loomba, R.; Dasarathy, S.; Abdelmalek, M.; et al. Performance characteristics of vibration-controlled transient elastography for evaluation of nonalcoholic fatty liver disease. Hepatology 2018, 67, 134–144. [Google Scholar] [CrossRef]
- Jin, Z.; Yuan, Y.; Zheng, C.; Liu, S.; Weng, H. Effects of sodium-glucose co-transporter 2 inhibitors on liver fibrosis in non-alcoholic fatty liver disease patients with type 2 diabetes mellitus: An updated meta-analysis of randomized controlled trials. J. Diabetes Complicat. 2023, 37, 108558. [Google Scholar] [CrossRef]
- Obata, A.; Kubota, N.; Kubota, T.; Iwamoto, M.; Sato, H.; Sakurai, Y.; Takamoto, I.; Katsuyama, H.; Suzuki, Y.; Fukazawa, M.; et al. Tofogliflozin Improves Insulin Resistance in Skeletal Muscle and Accelerates Lipolysis in Adipose Tissue in Male Mice. Endocrinology 2016, 157, 1029–1042. [Google Scholar] [CrossRef]
- Wang, D.; Liu, J.; Zhong, L.; Li, S.; Zhou, L.; Zhang, Q.; Li, M.; Xiao, X. The effect of sodium-glucose cotransporter 2 inhibitors on biomarkers of inflammation: A systematic review and meta-analysis of randomized controlled trials. Front. Pharmacol. 2022, 13, 1045235. [Google Scholar] [CrossRef]
- Xu, L.; Nagata, N.; Nagashimada, M.; Zhuge, F.; Ni, Y.; Chen, G.; Mayoux, E.; Kaneko, S.; Ota, T. SGLT2 inhibition by empagliflozin promotes fat utilization and browning and attenuates inflammation and insulin resistance by polarizing M2 macrophages in diet-induced obese mice. EBioMedicine 2017, 20, 137–149. [Google Scholar] [CrossRef]
- Randle, P.J.; Garland, P.B.; Hales, C.N.; Newsholme, E.A. The glucose fatty-acid cycle. Its role in insulin sensitivity and the metabolic disturbances of diabetes mellitus. Lancet 1963, 1, 785–789. [Google Scholar] [CrossRef]
- Kolb, H.; Kempf, K.; Röhling, M.; Lenzen-Schulte, M.; Schloot, N.C.; Martin, S. Ketone bodies: From enemy to friend and guardian angel. BMC Med. 2021, 19, 313. [Google Scholar] [CrossRef]
- Vasilakou, D.; Karagiannis, T.; Athanasiadou, E.; Mainou, M.; Liakos, A.; Bekiari, E.; Sarigianni, M.; Matthews, D.R.; Tsapas, A. Sodium-glucose cotransporter 2 inhibitors for type 2 diabetes: A systematic review and meta-analysis. Ann. Intern. Med. 2013, 159, 262–274. [Google Scholar] [CrossRef]
- Sanchez-Garcia, A.; Simental-Mendia, M.; Millan-Alanis, J.M.; Simental-Mendia, L.E. Effect of sodium-glucose co-transporter 2 inhibitors on lipid profile: A systematic review and meta-analysis of 48 randomized controlled trials. Pharmacol. Res. 2020, 160, 105068. [Google Scholar] [CrossRef]
- Li, D.; Wu, T.; Wang, T.; Wei, H.; Wang, A.; Tang, H.; Song, Y. Effects of sodium glucose cotransporter 2 inhibitors on risk of dyslipidemia among patients with type 2 diabetes: A systematic review and meta-analysis of randomized controlled trials. Pharmacoepidemiol. Drug Saf. 2020, 29, 582–590. [Google Scholar] [CrossRef]
- Yanai, H.; Adachi, H.; Hakoshima, M.; Katsuyama, H. Significance of Endothelial Dysfunction Amelioration for Sodium-Glucose Cotransporter 2 Inhibitor-Induced Improvements in Heart Failure and Chronic Kidney Disease in Diabetic Patients. Metabolites 2023, 13, 736. [Google Scholar] [CrossRef]
- Rahman, H.; Khan, S.U.; Lone, A.N.; Ghosh, P.; Kunduru, M.; Sharma, S.; Sattur, S.; Kaluski, E. Sodium-Glucose Cotransporter-2 Inhibitors and Primary Prevention of Atherosclerotic Cardiovascular Disease: A Meta-Analysis of Randomized Trials and Systematic Review. J. Am. Heart Assoc. 2023, 12, e030578. [Google Scholar] [CrossRef]
- Katsuyama, H.; Hakoshima, M.; Umeyama, S.; Iida, S.; Adachi, H.; Yanai, H. Real-World Efficacy of Glucagon-like Peptide-1 (GLP-1) Receptor Agonist, Dulaglutide, on Metabolic Parameters in Japanese Patients with Type 2 Diabetes: A Retrospective Longitudinal Study. Biomedicines 2023, 11, 869. [Google Scholar] [CrossRef]
- Carbone, L.J.; Angus, P.W.; Yeomans, N.D. Incretin-based therapies for the treatment of non-alcoholic fatty liver disease: A systematic review and meta-analysis. J. Gastroenterol. Hepatol. 2016, 31, 23–31. [Google Scholar] [CrossRef]
- Dong, Y.; Lv, Q.; Li, S.; Wu, Y.; Li, L.; Li, J.; Zhang, F.; Sun, X.; Tong, N. Efficacy and safety of glucagon-like peptide-1 receptor agonists in non-alcoholic fatty liver disease: A systematic review and meta-analysis. Clin. Res. Hepatol. Gastroenterol. 2017, 41, 284–295. [Google Scholar] [CrossRef]
- Mantovani, A.; Petracca, G.; Beatrice, G.; Csermely, A.; Lonardo, A.; Targher, G. Glucagon-Like Peptide-1 Receptor Agonists for Treatment of Nonalcoholic Fatty Liver Disease and Nonalcoholic Steatohepatitis: An Updated Meta-Analysis of Randomized Controlled Trials. Metabolites 2021, 11, 73. [Google Scholar] [CrossRef]
- Davies, M.J.; D’Alessio, D.A.; Fradkin, J.; Kernan, W.N.; Mathieu, C.; Mingrone, G.; Rossing, P.; Tsapas, A.; Wexler, D.J.; Buse, J.B. Management of hyperglycaemia in type 2 diabetes, 2018. A consensus report by the American Diabetes Association (ADA) and the European Association for the Study of Diabetes (EASD). Diabetologia 2018, 61, 2461–2498. [Google Scholar] [CrossRef]
- Buse, J.B.; Wexler, D.J.; Tsapas, A.; Rossing, P.; Mingrone, G.; Mathieu, C.; D’Alessio, D.A.; Davies, M.J. 2019 update to: Management of hyperglycaemia in type 2 diabetes, 2018. A consensus report by the American Diabetes Association (ADA) and the European Association for the Study of Diabetes (EASD). Diabetologia 2020, 63, 221–228. [Google Scholar] [CrossRef] [PubMed]
- Nauck, M.A.; Quast, D.R.; Wefers, J.; Meier, J.J. GLP-1 receptor agonists in the treatment of type 2 diabetes—State-of-the-art. Mol. Metab. 2021, 46, 101102. [Google Scholar] [CrossRef] [PubMed]
- Krieger, J.P. Intestinal glucagon-like peptide-1 effects on food intake: Physiological relevance and emerging mechanisms. Peptides 2020, 131, 170342. [Google Scholar] [CrossRef] [PubMed]
- Bray, J.J.H.; Foster-Davies, H.; Salem, A.; Hoole, A.L.; Obaid, D.R.; Halcox, J.P.J.; Stephens, J.W. Glucagon-like peptide-1 receptor agonists improve biomarkers of inflammation and oxidative stress: A systematic review and meta-analysis of randomised controlled trials. Diabetes Obes. Metab. 2021, 23, 1806–1822. [Google Scholar] [CrossRef] [PubMed]
- Katout, M.; Zhu, H.; Rutsky, J.; Shah, P.; Brook, R.D.; Zhong, J.; Rajagopalan, S. Effect of GLP-1 mimetics on blood pressure and relationship to weight loss and glycemia lowering: Results of a systematic meta-analysis and meta-regression. Am. J. Hypertens. 2014, 27, 130–139. [Google Scholar] [CrossRef]
- Berg, G.; Barchuk, M.; Lobo, M.; Nogueira, J.P. Effect of glucagon-like peptide-1 (GLP-1) analogues on epicardial adipose tissue: A meta-analysis. Diabetes. Metab. Syndr. 2022, 16, 102562. [Google Scholar] [CrossRef] [PubMed]
- Rahman, A.; Alqaisi, S.; Saith, S.E.; Alzakhari, R.; Levy, R. The Impact of Glucagon-Like Peptide-1 Receptor Agonist on the Cardiovascular Outcomes in Patients With Type 2 Diabetes Mellitus: A Meta-Analysis and Systematic Review. Cardiol. Res. 2023, 14, 250–260. [Google Scholar] [CrossRef]
- Gu, Y.; Sun, L.; Zhang, W.; Kong, T.; Zhou, R.; He, Y.; Deng, C.; Yang, L.; Kong, J.; Chen, Y.; et al. Comparative efficacy of 5 sodium-glucose cotransporter protein-2 (SGLT-2) inhibitor and 4 glucagon-like peptide-1 (GLP-1) receptor agonist drugs in non-alcoholic fatty liver disease: A GRADE-assessed systematic review and network meta-analysis of randomized controlled trials. Front. Pharmacol. 2023, 14, 1102792. [Google Scholar]
- Yan, H.; Huang, C.; Shen, X.; Li, J.; Zhou, S.; Li, W. GLP-1 RAs and SGLT-2 Inhibitors for Insulin Resistance in Nonalcoholic Fatty Liver Disease: Systematic Review and Network Meta-Analysis. Front. Endocrinol. 2022, 13, 923606. [Google Scholar] [CrossRef]
- Mantovani, A.; Byrne, C.D.; Targher, G. Efficacy of peroxisome proliferator-activated receptor agonists, glucagon-like peptide-1 receptor agonists, or sodium-glucose cotransporter-2 inhibitors for treatment of non-alcoholic fatty liver disease: A systematic review. Lancet. Gastroenterol. Hepatol. 2022, 7, 367–378. [Google Scholar] [CrossRef]
- Shinozaki, S.; Tahara, T.; Miura, K.; Lefor, A.K.; Yamamoto, H. Effectiveness of One-Year Pemafibrate Therapy on Non-Alcoholic Fatty Liver Disease Refractory to Long-Term Sodium Glucose Cotransporter-2 Inhibitor Therapy: A Pilot Study. Life 2023, 13, 1327. [Google Scholar] [CrossRef] [PubMed]
- Angulo, P.; Keach, J.C.; Batts, K.P.; Lindor, K.D. Independent predictors of liver fibrosis in patients with non-alcoholic steatohepatitis. Hepatology 1999, 30, 1356–1362. [Google Scholar] [CrossRef] [PubMed]
- Lin, Z.H.; Xin, Y.N.; Dong, Q.J.; Wang, Q.; Jiang, X.J.; Zhan, S.H.; Sun, Y.; Xuan, S.Y. Performance of the aspartate aminotransferase-to-platelet ratio index for the staging of hepatitis C-related fibrosis: An updated meta-analysis. Hepatology 2011, 53, 726–736. [Google Scholar] [CrossRef] [PubMed]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yanai, H.; Adachi, H.; Hakoshima, M.; Iida, S.; Katsuyama, H. Metabolic-Dysfunction-Associated Steatotic Liver Disease—Its Pathophysiology, Association with Atherosclerosis and Cardiovascular Disease, and Treatments. Int. J. Mol. Sci. 2023, 24, 15473. https://doi.org/10.3390/ijms242015473
Yanai H, Adachi H, Hakoshima M, Iida S, Katsuyama H. Metabolic-Dysfunction-Associated Steatotic Liver Disease—Its Pathophysiology, Association with Atherosclerosis and Cardiovascular Disease, and Treatments. International Journal of Molecular Sciences. 2023; 24(20):15473. https://doi.org/10.3390/ijms242015473
Chicago/Turabian StyleYanai, Hidekatsu, Hiroki Adachi, Mariko Hakoshima, Sakura Iida, and Hisayuki Katsuyama. 2023. "Metabolic-Dysfunction-Associated Steatotic Liver Disease—Its Pathophysiology, Association with Atherosclerosis and Cardiovascular Disease, and Treatments" International Journal of Molecular Sciences 24, no. 20: 15473. https://doi.org/10.3390/ijms242015473