
Dysfunctional and Dysregulated Nitric Oxide Synthases in Cardiovascular Disease: Mechanisms and Therapeutic Potential

{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Nitric Oxide Signalling
3. Structure and Function of Nitric Oxide Synthases
4. Physiological Role of Nitric Oxide Synthases in the Cardiovascular System
4.1. Endothelial NOS
4.2. Neuronal NOS
4.3. Inducible NOS
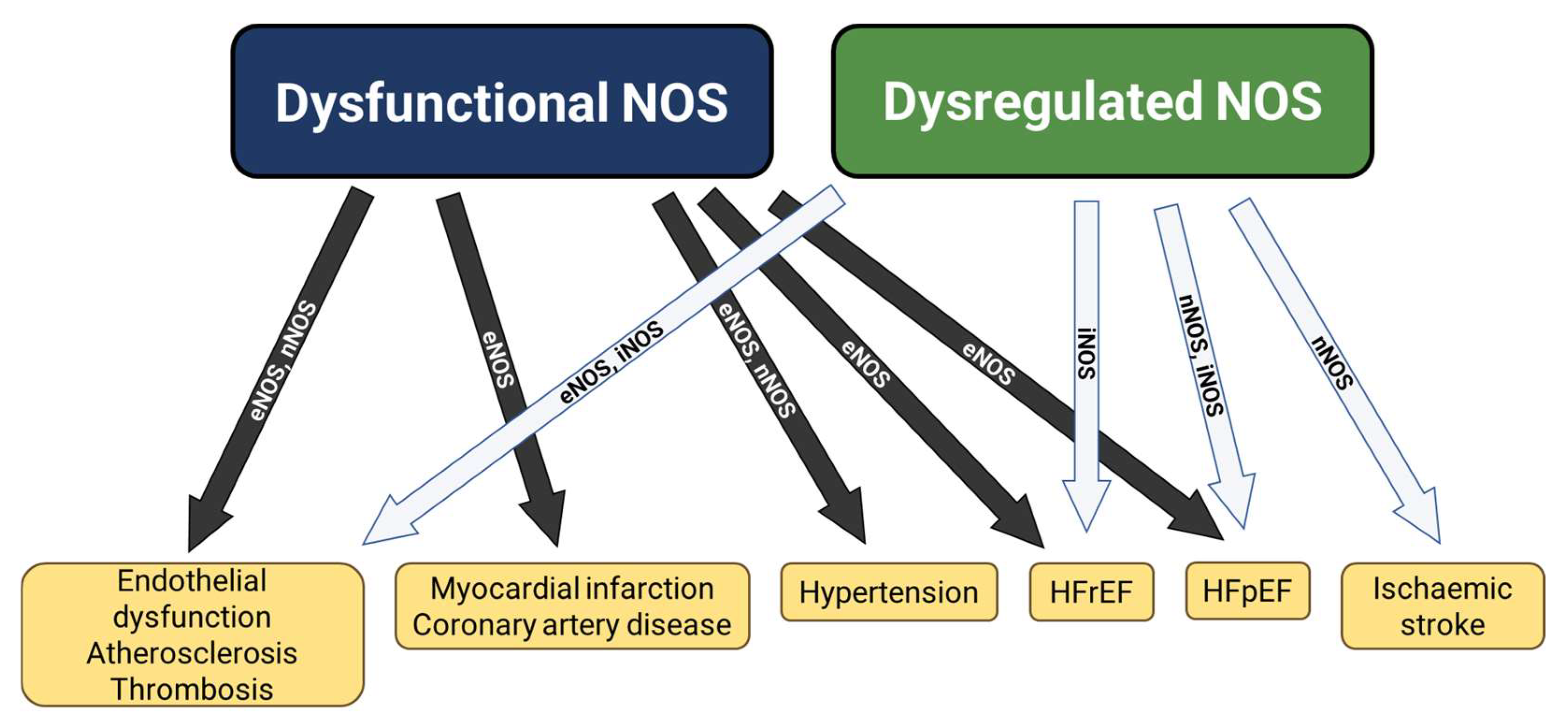
5. Dysfunctional NOS in Cardiovascular Disease
5.1. Hypertension
5.2. Endothelial Dysfunction
5.3. Myocardial Infarction and Ischaemia/Reperfusion Injury
5.4. Heart Failure
5.5. Therapeutic Potential
5.6. L-Arginine
5.7. BH4
5.8. NO Donors
5.9. NOS Transcriptional Regulators/Enhancers
5.10. Gene Therapy
6. Dysregulated NOS in Cardiovascular Disease
6.1. Endothelial Dysfunction, Inflammation, and Oxidative Stress
6.2. NO and Mitochondria
6.3. Heart Failure with Preserved Ejection Fraction (HFpEF)
6.4. Coronary Microvascular Disease (CMD)
6.5. Ischaemia-Reperfusion Injury in the Heart
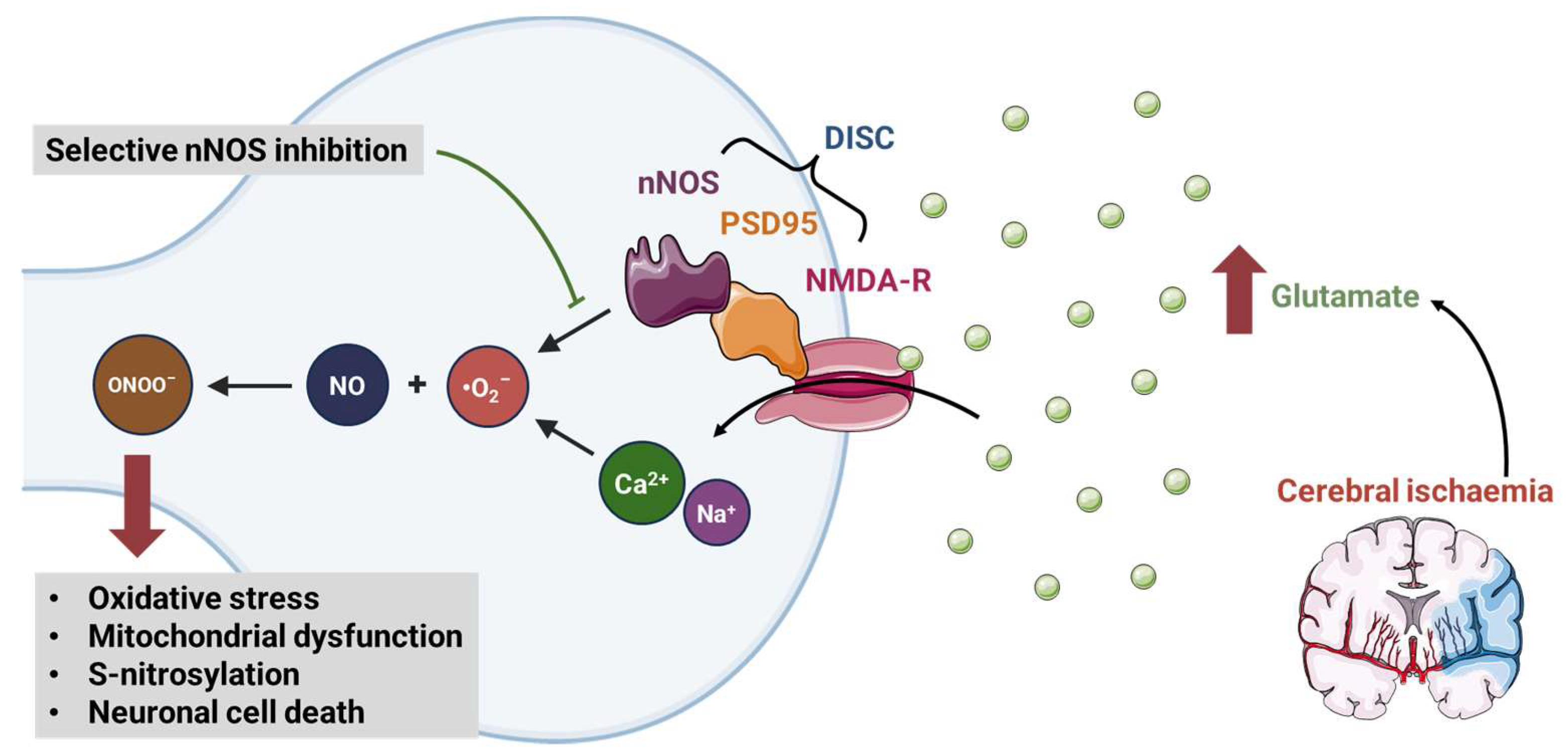
6.6. Post-Stroke Reperfusion Injury
6.7. Therapeutic Potential of NOS Inhibitors
6.8. Safety Concerns with NOS Inhibitors
7. Conclusions, Limitations, and Future Directions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Barouch, L.A.; Harrison, R.W.; Skaf, M.W.; Rosas, G.O.; Cappola, T.P.; Kobeissi, Z.A.; Hobai, I.A.; Lemmon, C.A.; Burnett, A.L.; O’Rourke, B.; et al. Nitric Oxide Regulates the Heart by Spatial Confinement of Nitric Oxide Synthase Isoforms. Nature 2002, 416, 337–339. [Google Scholar] [CrossRef]
- Lundberg, J.O.; Weitzberg, E. Nitric Oxide Signaling in Health and Disease. Cell 2022, 185, 2853–2878. [Google Scholar] [CrossRef]
- O’Gallagher, K.; Puledda, F.; O’Daly, O.; Ryan, M.; Dancy, L.; Chowienczyk, P.J.; Zelaya, F.; Goadsby, P.J.; Shah, A.M. Neuronal Nitric Oxide Synthase Regulates Regional Brain Perfusion in Healthy Humans. Cardiovasc. Res. 2022, 118, 1321–1329. [Google Scholar] [CrossRef]
- Cinelli, M.A.; Do, H.T.; Miley, G.P.; Silverman, R.B. Inducible Nitric Oxide Synthase: Regulation, Structure, and Inhibition. Med. Res. Rev. 2020, 40, 158–189. [Google Scholar] [CrossRef]
- Ahmad, A.; Dempsey, S.; Daneva, Z.; Azam, M.; Li, N.; Li, P.-L.; Ritter, J. Role of Nitric Oxide in the Cardiovascular and Renal Systems. Int. J. Mol. Sci. 2018, 19, 2605. [Google Scholar] [CrossRef]
- Lincoln, T.M.; Dey, N.; Sellak, H. Invited Review: CGMP-Dependent Protein Kinase Signaling Mechanisms in Smooth Muscle: From the Regulation of Tone to Gene Expression. J. Appl. Physiol. 2001, 91, 1421–1430. [Google Scholar] [CrossRef]
- Fernando, V.; Zheng, X.; Walia, Y.; Sharma, V.; Letson, J.; Furuta, S. S-Nitrosylation: An Emerging Paradigm of Redox Signaling. Antioxidants 2019, 8, 404. [Google Scholar] [CrossRef]
- Nakamura, T.; Oh, C.; Zhang, X.; Lipton, S.A. Protein S-Nitrosylation and Oxidation Contribute to Protein Misfolding in Neurodegeneration. Free Radic. Biol. Med. 2021, 172, 562–577. [Google Scholar] [CrossRef]
- Zhang, Y.; Deng, Y.; Yang, X.; Xue, H.; Lang, Y. The Relationship Between Protein S-Nitrosylation and Human Diseases: A Review. Neurochem. Res. 2020, 45, 2815–2827. [Google Scholar] [CrossRef]
- Gantner, B.N.; LaFond, K.M.; Bonini, M.G. Nitric Oxide in Cellular Adaptation and Disease. Redox Biol. 2020, 34, 101550. [Google Scholar] [CrossRef]
- Förstermann, U.; Closs, E.I.; Pollock, J.S.; Nakane, M.; Schwarz, P.; Gath, I.; Kleinert, H. Nitric Oxide Synthase Isozymes. Characterization, Purification, Molecular Cloning, and Functions. Hypertension 1994, 23, 1121–1131. [Google Scholar] [CrossRef]
- Spiller, F.; Oliveira Formiga, R.; Fernandes da Silva Coimbra, J.; Alves-Filho, J.C.; Cunha, T.M.; Cunha, F.Q. Targeting Nitric Oxide as a Key Modulator of Sepsis, Arthritis and Pain. Nitric Oxide 2019, 89, 32–40. [Google Scholar] [CrossRef]
- Abu-Soud, H.M.; Rousseau, D.L.; Stuehr, D.J. Nitric Oxide Binding to the Heme of Neuronal Nitric-Oxide Synthase Links Its Activity to Changes in Oxygen Tension. J. Biol. Chem. 1996, 271, 32515–32518. [Google Scholar] [CrossRef] [PubMed]
- Dweik, R.A.; Laskowski, D.; Abu-Soud, H.M.; Kaneko, F.T.; Hutte, R.; Stuehr, D.J.; Erzurum, S.C. Nitric Oxide Synthesis in the Lung. Regulation by Oxygen through a Kinetic Mechanism. J. Clin. Investig. 1998, 101, 660–666. [Google Scholar] [CrossRef]
- Haque, M.M.; Panda, K.; Tejero, J.; Aulak, K.S.; Fadlalla, M.A.; Mustovich, A.T.; Stuehr, D.J. A Connecting Hinge Represses the Activity of Endothelial Nitric Oxide Synthase. Proc. Natl. Acad. Sci. USA 2007, 104, 9254–9259. [Google Scholar] [CrossRef] [PubMed]
- Searles, C.D. Transcriptional and Posttranscriptional Regulation of Endothelial Nitric Oxide Synthase Expression. Am. J. Physiol.-Cell Physiol. 2006, 291, C803–C816. [Google Scholar] [CrossRef] [PubMed]
- Farah, C.; Michel, L.Y.M.; Balligand, J.-L. Nitric Oxide Signalling in Cardiovascular Health and Disease. Nat. Rev. Cardiol. 2018, 15, 292–316. [Google Scholar] [CrossRef]
- Seddon, M.; Melikian, N.; Dworakowski, R.; Shabeeh, H.; Jiang, B.; Byrne, J.; Casadei, B.; Chowienczyk, P.; Shah, A.M. Effects of Neuronal Nitric Oxide Synthase on Human Coronary Artery Diameter and Blood Flow In Vivo. Circulation 2009, 119, 2656–2662. [Google Scholar] [CrossRef]
- Seddon, M.D.; Chowienczyk, P.J.; Brett, S.E.; Casadei, B.; Shah, A.M. Neuronal Nitric Oxide Synthase Regulates Basal Microvascular Tone in Humans In Vivo. Circulation 2008, 117, 1991–1996. [Google Scholar] [CrossRef]
- Park, S.-K.; La Salle, D.T.; Cerbie, J.; Cho, J.M.; Bledsoe, A.; Nelson, A.; Morgan, D.E.; Richardson, R.S.; Shiu, Y.-T.; Boudina, S.; et al. Elevated Arterial Shear Rate Increases Indexes of Endothelial Cell Autophagy and Nitric Oxide Synthase Activation in Humans. Am. J. Physiol.-Heart Circ. Physiol. 2019, 316, H106–H112. [Google Scholar] [CrossRef]
- Cyr, A.R.; Huckaby, L.V.; Shiva, S.S.; Zuckerbraun, B.S. Nitric Oxide and Endothelial Dysfunction. Crit. Care Clin. 2020, 36, 307–321. [Google Scholar] [CrossRef]
- Costa, D.; Benincasa, G.; Lucchese, R.; Infante, T.; Nicoletti, G.F.; Napoli, C. Effect of Nitric Oxide Reduction on Arterial Thrombosis. Scand. Cardiovasc. J. 2019, 53, 1–8. [Google Scholar] [CrossRef]
- Theofilis, P.; Sagris, M.; Oikonomou, E.; Antonopoulos, A.S.; Siasos, G.; Tsioufis, C.; Tousoulis, D. Inflammatory Mechanisms Contributing to Endothelial Dysfunction. Biomedicines 2021, 9, 781. [Google Scholar] [CrossRef]
- Tran, N.; Garcia, T.; Aniqa, M.; Ali, S.; Ally, A.; Nauli, S.M. Endothelial Nitric Oxide Synthase (ENOS) and the Cardiovascular System: In Physiology and in Disease States. Am. J. Biomed. Sci. Res. 2022, 15, 153–177. [Google Scholar] [PubMed]
- Tejero, J.; Shiva, S.; Gladwin, M.T. Sources of Vascular Nitric Oxide and Reactive Oxygen Species and Their Regulation. Physiol. Rev. 2019, 99, 311–379. [Google Scholar] [CrossRef]
- Gkaliagkousi, E.; Ritter, J.; Ferro, A. Platelet-Derived Nitric Oxide Signaling and Regulation. Circ. Res. 2007, 101, 654–662. [Google Scholar] [CrossRef]
- Gresele, P.; Momi, S.; Guglielmini, G. Nitric Oxide-Enhancing or -Releasing Agents as Antithrombotic Drugs. Biochem. Pharmacol. 2019, 166, 300–312. [Google Scholar] [CrossRef]
- Morrell, C.N.; Matsushita, K.; Chiles, K.; Scharpf, R.B.; Yamakuchi, M.; Mason, R.J.A.; Bergmeier, W.; Mankowski, J.L.; Baldwin, W.M.; Faraday, N.; et al. Regulation of Platelet Granule Exocytosis by S-Nitrosylation. Proc. Natl. Acad. Sci. USA 2005, 102, 3782–3787. [Google Scholar] [CrossRef] [PubMed]
- Corban, M.T.; Lerman, L.O.; Lerman, A. Endothelial Dysfunction. Arterioscler. Thromb. Vasc. Biol. 2019, 39, 1272–1274. [Google Scholar] [CrossRef]
- Gallo, G.; Volpe, M.; Savoia, C. Endothelial Dysfunction in Hypertension: Current Concepts and Clinical Implications. Front. Med. 2022, 8, 798958. [Google Scholar] [CrossRef] [PubMed]
- Zuchi, C.; Tritto, I.; Carluccio, E.; Mattei, C.; Cattadori, G.; Ambrosio, G. Role of Endothelial Dysfunction in Heart Failure. Heart Fail. Rev. 2020, 25, 21–30. [Google Scholar] [CrossRef]
- Kuhlencordt, P.J.; Gyurko, R.; Han, F.; Scherrer-Crosbie, M.; Aretz, T.H.; Hajjar, R.; Picard, M.H.; Huang, P.L. Accelerated Atherosclerosis, Aortic Aneurysm Formation, and Ischemic Heart Disease in Apolipoprotein E/Endothelial Nitric Oxide Synthase Double-Knockout Mice. Circulation 2001, 104, 448–454. [Google Scholar] [CrossRef]
- Hong, F.; Liang, X.; Liu, W.; Lv, S.; He, S.; Kuang, H.; Yang, S. Roles of ENOS in Atherosclerosis Treatment. Inflamm. Res. 2019, 68, 429–441. [Google Scholar] [CrossRef]
- Man, A.W.C.; Zhou, Y.; Xia, N.; Li, H. Endothelial Nitric Oxide Synthase in the Perivascular Adipose Tissue. Biomedicines 2022, 10, 1754. [Google Scholar] [CrossRef] [PubMed]
- Balligand, J.-L.; Feron, O.; Dessy, C. ENOS Activation by Physical Forces: From Short-Term Regulation of Contraction to Chronic Remodeling of Cardiovascular Tissues. Physiol. Rev. 2009, 89, 481–534. [Google Scholar] [CrossRef] [PubMed]
- Massion, P.B.; Feron, O.; Dessy, C.; Balligand, J.-L. Nitric Oxide and Cardiac Function. Circ. Res. 2003, 93, 388–398. [Google Scholar] [CrossRef] [PubMed]
- Seddon, M.; Shah, A.; Casadei, B. Cardiomyocytes as Effectors of Nitric Oxide Signalling. Cardiovasc. Res. 2007, 75, 315–326. [Google Scholar] [CrossRef] [PubMed]
- Umar, S.; van der Laarse, A. Nitric Oxide and Nitric Oxide Synthase Isoforms in the Normal, Hypertrophic, and Failing Heart. Mol. Cell Biochem. 2010, 333, 191–201. [Google Scholar] [CrossRef] [PubMed]
- Huang, A.; Sun, D.; Shesely, E.G.; Levee, E.M.; Koller, A.; Kaley, G. Neuronal NOS-Dependent Dilation to Flow in Coronary Arteries of Male ENOS-KO Mice. Am. J. Physiol.-Heart Circ. Physiol. 2002, 282, H429–H436. [Google Scholar] [CrossRef] [PubMed]
- Buchwalow, I.B.; Podzuweit, T.; Böcker, W.; Samoilova, V.E.; Thomas, S.; Wellner, M.; Baba, H.A.; Robenek, H.; Schnekenburger, J.; Lerch, M.M. Vascular Smooth Muscle and Nitric Oxide Synthase. FASEB J. 2002, 16, 500–508. [Google Scholar] [CrossRef]
- Cotter, M.A.; Cameron, N.E.; Nangle, M.R. An in Vitro Investigation of Aorta and Corpus Cavernosum from ENOS and NNOS Gene-Deficient Mice. Pflug. Arch. Eur. J. Physiol. 2004, 448, 139–145. [Google Scholar] [CrossRef]
- Toda, N.; Okamura, T. Modulation of Renal Blood Flow and Vascular Tone by Neuronal Nitric Oxide Synthase-Derived Nitric Oxide. J. Vasc. Res. 2011, 48, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Melikian, N.; Seddon, M.D.; Casadei, B.; Chowienczyk, P.J.; Shah, A.M. Neuronal Nitric Oxide Synthase and Human Vascular Regulation. Trends Cardiovasc. Med. 2009, 19, 256–262. [Google Scholar] [CrossRef]
- Shabeeh, H.; Khan, S.; Jiang, B.; Brett, S.; Melikian, N.; Casadei, B.; Chowienczyk, P.J.; Shah, A.M. Blood Pressure in Healthy Humans Is Regulated by Neuronal NO Synthase. Hypertension 2017, 69, 970–976. [Google Scholar] [CrossRef] [PubMed]
- Ally, A.; Powell, I.; Ally, M.M.; Chaitoff, K.; Nauli, S.M. Role of Neuronal Nitric Oxide Synthase on Cardiovascular Functions in Physiological and Pathophysiological States. Nitric Oxide 2020, 102, 52–73. [Google Scholar] [CrossRef] [PubMed]
- Danson, E.; Choate, J.; Paterson, D. Cardiac Nitric Oxide: Emerging Role for NNOS in Regulating Physiological Function. Pharmacol. Ther. 2005, 106, 57–74. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.H.; Jin, C.Z.; Jang, J.H.; Wang, Y. Molecular Mechanisms of Neuronal Nitric Oxide Synthase in Cardiac Function and Pathophysiology. J. Physiol. 2014, 592, 3189–3200. [Google Scholar] [CrossRef] [PubMed]
- Seddon, M.; Looi, Y.H.; Shah, A.M. Oxidative Stress and Redox Signalling in Cardiac Hypertrophy and Heart Failure. Heart 2007, 93, 903–907. [Google Scholar] [CrossRef]
- O’Gallagher, K.; Rosentreter, R.E.; Elaine Soriano, J.; Roomi, A.; Saleem, S.; Lam, T.; Roy, R.; Gordon, G.R.; Raj, S.R.; Chowienczyk, P.J.; et al. The Effect of a Neuronal Nitric Oxide Synthase Inhibitor on Neurovascular Regulation in Humans. Circ. Res. 2022, 131, 952–961. [Google Scholar] [CrossRef]
- Claassen, J.A.H.R.; Thijssen, D.H.J.; Panerai, R.B.; Faraci, F.M. Regulation of Cerebral Blood Flow in Humans: Physiology and Clinical Implications of Autoregulation. Physiol. Rev. 2021, 101, 1487–1559. [Google Scholar] [CrossRef]
- Phillips, A.A.; Chan, F.H.; Zheng, M.M.Z.; Krassioukov, A.V.; Ainslie, P.N. Neurovascular Coupling in Humans: Physiology, Methodological Advances and Clinical Implications. J. Cereb. Blood Flow. Metab. 2016, 36, 647–664. [Google Scholar] [CrossRef]
- Okamoto, H.; Hudetz, A.G.; Roman, R.J.; Bosnjak, Z.J.; Kampine, J.P. Neuronal NOS-Derived NO Plays Permissive Role in Cerebral Blood Flow Response to Hypercapnia. Am. J. Physiol.-Heart Circ. Physiol. 1997, 272, H559–H566. [Google Scholar] [CrossRef]
- Hudetz, A.G.; Shen, H.; Kampine, J.P. Nitric Oxide from Neuronal NOS Plays Critical Role in Cerebral Capillary Flow Response to Hypoxia. Am. J. Physiol.-Heart Circ. Physiol. 1998, 274, H982–H989. [Google Scholar] [CrossRef]
- Kourosh-Arami, M.; Hosseini, N.; Mohsenzadegan, M.; Komaki, A.; Joghataei, M.T. Neurophysiologic Implications of Neuronal Nitric Oxide Synthase. Rev. Neurosci. 2020, 31, 617–636. [Google Scholar] [CrossRef]
- Khan, S.G.; Melikian, N.; Shabeeh, H.; Cabaco, A.R.; Martin, K.; Khan, F.; O’Gallagher, K.; Chowienczyk, P.J.; Shah, A.M. The Human Coronary Vasodilatory Response to Acute Mental Stress Is Mediated by Neuronal Nitric Oxide Synthase. Am. J. Physiol.-Heart Circ. Physiol. 2017, 313, H578–H583. [Google Scholar] [CrossRef]
- Khan, S.G.; Geer, A.; Fok, H.W.; Shabeeh, H.; Brett, S.E.; Shah, A.M.; Chowienczyk, P.J. Impaired Neuronal Nitric Oxide Synthase–Mediated Vasodilator Responses to Mental Stress in Essential Hypertension. Hypertension 2015, 65, 903–909. [Google Scholar] [CrossRef]
- Hemmens, B.; Mayer, B. Enzymology of Nitric Oxide Synthases. In Nitric Oxide Protocols; Humana Press: Totowa, NJ, USA, 1998; pp. 1–32. [Google Scholar]
- Hevel, J.M.; White, K.A.; Marletta, M.A. Purification of the Inducible Murine Macrophage Nitric Oxide Synthase. Identification as a Flavoprotein. J. Biol. Chem. 1991, 266, 22789–22791. [Google Scholar] [CrossRef]
- Venema, R.C.; Sayegh, H.S.; Kent, J.D.; Harrison, D.G. Identification, Characterization, and Comparison of the Calmodulin-Binding Domains of the Endothelial and Inducible Nitric Oxide Synthases. J. Biol. Chem. 1996, 271, 6435–6440. [Google Scholar] [CrossRef]
- Bath, P.M.; Coleman, C.M.; Gordon, A.L.; Lim, W.S.; Webb, A.J. Nitric Oxide for the Prevention and Treatment of Viral, Bacterial, Protozoal and Fungal Infections. F1000Res 2021, 10, 536. [Google Scholar] [CrossRef]
- Chakravortty, D.; Hensel, M. Inducible Nitric Oxide Synthase and Control of Intracellular Bacterial Pathogens. Microbes Infect. 2003, 5, 621–627. [Google Scholar] [CrossRef]
- Jayaraj, R.L.; Azimullah, S.; Beiram, R.; Jalal, F.Y.; Rosenberg, G.A. Neuroinflammation: Friend and Foe for Ischemic Stroke. J. Neuroinflammation 2019, 16, 142. [Google Scholar] [CrossRef]
- Justo, A.F.O.; Suemoto, C.K. The Modulation of Neuroinflammation by Inducible Nitric Oxide Synthase. J. Cell Commun. Signal 2022, 16, 155–158. [Google Scholar] [CrossRef]
- Król, M.; Kepinska, M. Human Nitric Oxide Synthase—Its Functions, Polymorphisms, and Inhibitors in the Context of Inflammation, Diabetes and Cardiovascular Diseases. Int. J. Mol. Sci. 2020, 22, 56. [Google Scholar] [CrossRef]
- Lind, M.; Hayes, A.; Caprnda, M.; Petrovic, D.; Rodrigo, L.; Kruzliak, P.; Zulli, A. Inducible Nitric Oxide Synthase: Good or Bad? Biomed. Pharmacother. 2017, 93, 370–375. [Google Scholar] [CrossRef]
- Chen, J.; Ye, Z.; Wang, X.; Chang, J.; Yang, M.; Zhong, H.; Hong, F.; Yang, S. Nitric Oxide Bioavailability Dysfunction Involves in Atherosclerosis. Biomed. Pharmacother. 2018, 97, 423–428. [Google Scholar] [CrossRef]
- López, A.; Lorente, J.A.; Steingrub, J.; Bakker, J.; McLuckie, A.; Willatts, S.; Brockway, M.; Anzueto, A.; Holzapfel, L.; Breen, D.; et al. Multiple-Center, Randomized, Placebo-Controlled, Double-Blind Study of the Nitric Oxide Synthase Inhibitor 546C88: Effect on Survival in Patients with Septic Shock*. Crit. Care Med. 2004, 32, 21–30. [Google Scholar] [CrossRef]
- Chung, A.W.; Anand, K.; Anselme, A.C.; Chan, A.A.; Gupta, N.; Venta, L.A.; Schwartz, M.R.; Qian, W.; Xu, Y.; Zhang, L.; et al. A Phase 1/2 Clinical Trial of the Nitric Oxide Synthase Inhibitor L-NMMA and Taxane for Treating Chemoresistant Triple-Negative Breast Cancer. Sci. Transl. Med. 2021, 13, eabj5070. [Google Scholar] [CrossRef]
- Zou, M.-H.; Cohen, R.A.; Ullrich, V. Peroxynitrite and Vascular Endothelial Dysfunction in Diabetes Mellitus. Endothelium 2004, 11, 89–97. [Google Scholar] [CrossRef]
- Yang, Y.-M.; Huang, A.; Kaley, G.; Sun, D. ENOS Uncoupling and Endothelial Dysfunction in Aged Vessels. Am. J. Physiol. Heart Circ. Physiol. 2009, 297, H1829–H1836. [Google Scholar] [CrossRef]
- Gebhart, V.; Reiß, K.; Kollau, A.; Mayer, B.; Gorren, A.C.F. Site and Mechanism of Uncoupling of Nitric-Oxide Synthase: Uncoupling by Monomerization and Other Misconceptions. Nitric Oxide 2019, 89, 14–21. [Google Scholar] [CrossRef]
- Griendling, K.K.; Camargo, L.L.; Rios, F.J.; Alves-Lopes, R.; Montezano, A.C.; Touyz, R.M. Oxidative Stress and Hypertension. Circ. Res. 2021, 128, 993–1020. [Google Scholar] [CrossRef]
- Wu, Y.; Ding, Y.; Ramprasath, T.; Zou, M.-H. Oxidative Stress, GTPCH1, and Endothelial Nitric Oxide Synthase Uncoupling in Hypertension. Antioxid. Redox Signal 2021, 34, 750–764. [Google Scholar] [CrossRef]
- Zhang, Y.H. Neuronal Nitric Oxide Synthase in Hypertension—An Update. Clin. Hypertens. 2016, 22, 20. [Google Scholar] [CrossRef]
- Gliozzi, M.; Scicchitano, M.; Bosco, F.; Musolino, V.; Carresi, C.; Scarano, F.; Maiuolo, J.; Nucera, S.; Maretta, A.; Paone, S.; et al. Modulation of Nitric Oxide Synthases by Oxidized LDLs: Role in Vascular Inflammation and Atherosclerosis Development. Int. J. Mol. Sci. 2019, 20, 3294. [Google Scholar] [CrossRef] [PubMed]
- Liu, P.; Zhu, W.; Chen, C.; Yan, B.; Zhu, L.; Chen, X.; Peng, C. The Mechanisms of Lysophosphatidylcholine in the Development of Diseases. Life Sci. 2020, 247, 117443. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, J.A.; Kirkby, N.S. Eicosanoids, Prostacyclin and Cyclooxygenase in the Cardiovascular System. Br. J. Pharmacol. 2019, 176, 1038–1050. [Google Scholar] [CrossRef]
- Pong, T.; Huang, P.L. Effects of Nitric Oxide on Atherosclerosis. In Atherosclerosis; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2015; pp. 353–364. [Google Scholar]
- Kuhlencordt, P.J.; Hötten, S.; Schödel, J.; Rützel, S.; Hu, K.; Widder, J.; Marx, A.; Huang, P.L.; Ertl, G. Atheroprotective Effects of Neuronal Nitric Oxide Synthase in Apolipoprotein E Knockout Mice. Arterioscler. Thromb. Vasc. Biol. 2006, 26, 1539–1544. [Google Scholar] [CrossRef]
- Costa, E.D.; Rezende, B.A.; Cortes, S.F.; Lemos, V.S. Neuronal Nitric Oxide Synthase in Vascular Physiology and Diseases. Front. Physiol. 2016, 7, 206. [Google Scholar] [CrossRef] [PubMed]
- Mahdi, A.; Kövamees, O.; Pernow, J. Improvement in Endothelial Function in Cardiovascular Disease—Is Arginase the Target? Int. J. Cardiol. 2020, 301, 207–214. [Google Scholar] [CrossRef]
- Janaszak-Jasiecka, A.; Płoska, A.; Wierońska, J.M.; Dobrucki, L.W.; Kalinowski, L. Endothelial Dysfunction Due to ENOS Uncoupling: Molecular Mechanisms as Potential Therapeutic Targets. Cell Mol. Biol. Lett. 2023, 28, 21. [Google Scholar] [CrossRef]
- Siragusa, M.; Fleming, I. The ENOS Signalosome and Its Link to Endothelial Dysfunction. Pflug. Arch. 2016, 468, 1125–1137. [Google Scholar] [CrossRef] [PubMed]
- Scherrer-Crosbie, M.; Ullrich, R.; Bloch, K.D.; Nakajima, H.; Nasseri, B.; Aretz, H.T.; Lindsey, M.L.; Vançon, A.-C.; Huang, P.L.; Lee, R.T.; et al. Endothelial Nitric Oxide Synthase Limits Left Ventricular Remodeling After Myocardial Infarction in Mice. Circulation 2001, 104, 1286–1291. [Google Scholar] [CrossRef] [PubMed]
- Saraiva, R.M.; Minhas, K.M.; Raju, S.V.Y.; Barouch, L.A.; Pitz, E.; Schuleri, K.H.; Vandegaer, K.; Li, D.; Hare, J.M. Deficiency of Neuronal Nitric Oxide Synthase Increases Mortality and Cardiac Remodeling After Myocardial Infarction. Circulation 2005, 112, 3415–3422. [Google Scholar] [CrossRef]
- Dawson, D.; Lygate, C.A.; Zhang, M.-H.; Hulbert, K.; Neubauer, S.; Casadei, B. NNOS Gene Deletion Exacerbates Pathological Left Ventricular Remodeling and Functional Deterioration After Myocardial Infarction. Circulation 2005, 112, 3729–3737. [Google Scholar] [CrossRef]
- Bell, R.M.; Yellon, D.M. The Contribution of Endothelial Nitric Oxide Synthase to Early Ischaemic Preconditioning: The Lowering of the Preconditioning Threshold. An Investigation in ENOS Knockout Mice. Cardiovasc. Res. 2001, 52, 274–280. [Google Scholar] [CrossRef] [PubMed]
- Takano, H.; Manchikalapudi, S.; Tang, X.L.; Qiu, Y.; Rizvi, A.; Jadoon, A.K.; Zhang, Q.; Bolli, R. Nitric Oxide Synthase Is the Mediator of Late Preconditioning against Myocardial Infarction in Conscious Rabbits. Circulation 1998, 98, 441–449. [Google Scholar] [CrossRef]
- Bolli, R. Cardioprotective Function of Inducible Nitric Oxide Synthase and Role of Nitric Oxide in Myocardial Ischemia and Preconditioning: An Overview of a Decade of Research. J. Mol. Cell Cardiol. 2001, 33, 1897–1918. [Google Scholar] [CrossRef]
- Lee, H.-M.; Choi, J.W.; Choi, M.S. Role of Nitric Oxide and Protein S-Nitrosylation in Ischemia-Reperfusion Injury. Antioxidants 2021, 11, 57. [Google Scholar] [CrossRef]
- Griffiths, K.; Lee, J.J.; Frenneaux, M.P.; Feelisch, M.; Madhani, M. Nitrite and Myocardial Ischaemia Reperfusion Injury. Where Are We Now? Pharmacol. Ther. 2021, 223, 107819. [Google Scholar] [CrossRef]
- Jones, S.P.; Girod, W.G.; Palazzo, A.J.; Granger, D.N.; Grisham, M.B.; Jourd’Heuil, D.; Huang, P.L.; Lefer, D.J. Myocardial Ischemia-Reperfusion Injury Is Exacerbated in Absence of Endothelial Cell Nitric Oxide Synthase. Am. J. Physiol. Heart Circ. Physiol. 1999, 276, H1567–H1573. [Google Scholar] [CrossRef] [PubMed]
- Jones, S.P.; Greer, J.J.M.; Kakkar, A.K.; Ware, P.D.; Turnage, R.H.; Hicks, M.; van Haperen, R.; de Crom, R.; Kawashima, S.; Yokoyama, M.; et al. Endothelial Nitric Oxide Synthase Overexpression Attenuates Myocardial Reperfusion Injury. Am. J. Physiol. Heart Circ. Physiol. 2004, 286, H276–H282. [Google Scholar] [CrossRef]
- Schiattarella, G.G.; Altamirano, F.; Tong, D.; French, K.M.; Villalobos, E.; Kim, S.Y.; Luo, X.; Jiang, N.; May, H.I.; Wang, Z.V.; et al. Nitrosative Stress Drives Heart Failure with Preserved Ejection Fraction. Nature 2019, 568, 351–356. [Google Scholar] [CrossRef]
- Piech, A.; Massart, P.E.; Dessy, C.; Feron, O.; Havaux, X.; Morel, N.; Vanoverschelde, J.-L.; Donckier, J.; Balligand, J.-L. Decreased Expression of Myocardial ENOS and Caveolin in Dogs with Hypertrophic Cardiomyopathy. Am. J. Physiol. Heart Circ. Physiol. 2002, 282, H219–H231. [Google Scholar] [CrossRef] [PubMed]
- Drexler, H.; Kästner, S.; Strobel, A.; Studer, R.; Brodde, O.E.; Hasenfuß, G. Expression, Activity and Functional Significance of Inducible Nitric Oxide Synthase in the Failing Human Heart. J. Am. Coll. Cardiol. 1998, 32, 955–963. [Google Scholar] [CrossRef]
- Damy, T.; Ratajczak, P.; Shah, A.M.; Camors, E.; Marty, I.; Hasenfuss, G.; Marotte, F.; Samuel, J.-L.; Heymes, C. Increased Neuronal Nitric Oxide Synthase-Derived NO Production in the Failing Human Heart. Lancet 2004, 363, 1365–1367. [Google Scholar] [CrossRef]
- Nagareddy, P.R.; Xia, Z.; McNeill, J.H.; MacLeod, K.M. Increased Expression of INOS Is Associated with Endothelial Dysfunction and Impaired Pressor Responsiveness in Streptozotocin-Induced Diabetes. Am. J. Physiol. Heart Circ. Physiol. 2005, 289, H2144–H2152. [Google Scholar] [CrossRef]
- Szelid, Z.; Pokreisz, P.; Liu, X.; Vermeersch, P.; Marsboom, G.; Gillijns, H.; Pellens, M.; Verbeken, E.; Werf, F.; Collen, D.; et al. Cardioselective Nitric Oxide Synthase 3 Gene Transfer Protects against Myocardial Reperfusion Injury. Basic. Res. Cardiol. 2010, 105, 169–179. [Google Scholar] [CrossRef]
- Janssens, S.; Pokreisz, P.; Schoonjans, L.; Pellens, M.; Vermeersch, P.; Tjwa, M.; Jans, P.; Scherrer-Crosbie, M.; Picard, M.H.; Szelid, Z.; et al. Cardiomyocyte-Specific Overexpression of Nitric Oxide Synthase 3 Improves Left Ventricular Performance and Reduces Compensatory Hypertrophy After Myocardial Infarction. Circ. Res. 2004, 94, 1256–1262. [Google Scholar] [CrossRef] [PubMed]
- Jones, S.P.; Greer, J.J.M.; van Haperen, R.; Duncker, D.J.; de Crom, R.; Lefer, D.J. Endothelial Nitric Oxide Synthase Overexpression Attenuates Congestive Heart Failure in Mice. Proc. Natl. Acad. Sci. USA 2003, 100, 4891–4896. [Google Scholar] [CrossRef] [PubMed]
- Smith, R.S.; Agata, J.; Xia, C.-F.; Chao, L.; Chao, J. Human Endothelial Nitric Oxide Synthase Gene Delivery Protects against Cardiac Remodeling and Reduces Oxidative Stress after Myocardial Infarction. Life Sci. 2005, 76, 2457–2471. [Google Scholar] [CrossRef]
- Khanna, S.; Singh, G.B.; Khullar, M. Nitric Oxide Synthases and Diabetic Cardiomyopathy. Nitric Oxide 2014, 43, 29–34. [Google Scholar] [CrossRef] [PubMed]
- Sorop, O.; Heinonen, I.; van Kranenburg, M.; van de Wouw, J.; de Beer, V.J.; Nguyen, I.T.N.; Octavia, Y.; van Duin, R.W.B.; Stam, K.; van Geuns, R.-J.; et al. Multiple Common Comorbidities Produce Left Ventricular Diastolic Dysfunction Associated with Coronary Microvascular Dysfunction, Oxidative Stress, and Myocardial Stiffening. Cardiovasc. Res. 2018, 114, 954–964. [Google Scholar] [CrossRef]
- O’Gallagher, K.; Shah, A.M. Modelling the Complexity of Heart Failure with Preserved Ejection Fraction. Cardiovasc. Res. 2018, 114, 919–921. [Google Scholar] [CrossRef] [PubMed]
- Paulus, W.J.; Tschöpe, C. A Novel Paradigm for Heart Failure With Preserved Ejection Fraction. J. Am. Coll. Cardiol. 2013, 62, 263–271. [Google Scholar] [CrossRef] [PubMed]
- Suvorava, T.; Metry, S.; Pick, S.; Kojda, G. Alterations in Endothelial Nitric Oxide Synthase Activity and Their Relevance to Blood Pressure. Biochem. Pharmacol. 2022, 205, 115256. [Google Scholar] [CrossRef] [PubMed]
- Gorabi, A.M.; Kiaie, N.; Hajighasemi, S.; Banach, M.; Penson, P.E.; Jamialahmadi, T.; Sahebkar, A. Statin-Induced Nitric Oxide Signaling: Mechanisms and Therapeutic Implications. J. Clin. Med. 2019, 8, 2051. [Google Scholar] [CrossRef]
- Ding, Y.; Vaziri, N.D. Nifedipine and Diltiazem but Not Verapamil Up-Regulate Endothelial Nitric-Oxide Synthase Expression. J. Pharmacol. Exp. Ther. 2000, 292, 606–609. [Google Scholar]
- Daiber, A.; Xia, N.; Steven, S.; Oelze, M.; Hanf, A.; Kröller-Schön, S.; Münzel, T.; Li, H. New Therapeutic Implications of Endothelial Nitric Oxide Synthase (ENOS) Function/Dysfunction in Cardiovascular Disease. Int. J. Mol. Sci. 2019, 20, 187. [Google Scholar] [CrossRef] [PubMed]
- Rodrigues-Krause, J.; Krause, M.; Rocha, I.; Umpierre, D.; Fayh, A. Association of L-Arginine Supplementation with Markers of Endothelial Function in Patients with Cardiovascular or Metabolic Disorders: A Systematic Review and Meta-Analysis. Nutrients 2018, 11, 15. [Google Scholar] [CrossRef] [PubMed]
- Sun, T.; Zhou, W.; Luo, X.; Tang, Y.; Shi, H. Oral L-Arginine Supplementation in Acute Myocardial Infarction Therapy: A Meta-Analysis of Randomized Controlled Trials. Clin. Cardiol. 2009, 32, 649–652. [Google Scholar] [CrossRef]
- Schulman, S.P.; Becker, L.C.; Kass, D.A.; Champion, H.C.; Terrin, M.L.; Forman, S.; Ernst, K.V.; Kelemen, M.D.; Townsend, S.N.; Capriotti, A.; et al. L-Arginine Therapy in Acute Myocardial Infarction. JAMA 2006, 295, 58. [Google Scholar] [CrossRef]
- Liu, X.; Hou, L.; Xu, D.; Chen, A.; Yang, L.; Zhuang, Y.; Xu, Y.; Fassett, J.T.; Chen, Y. Effect of Asymmetric Dimethylarginine (ADMA) on Heart Failure Development. Nitric Oxide 2016, 54, 73–81. [Google Scholar] [CrossRef]
- Gawrys, J.; Gajecki, D.; Szahidewicz-Krupska, E.; Doroszko, A. Intraplatelet L-Arginine-Nitric Oxide Metabolic Pathway: From Discovery to Clinical Implications in Prevention and Treatment of Cardiovascular Disorders. Oxid. Med. Cell Longev. 2020, 2020, 1015908. [Google Scholar] [CrossRef]
- Chin-Dusting, J.P.F.; Willems, L.; Kaye, D.M. L-Arginine Transporters in Cardiovascular Disease: A Novel Therapeutic Target. Pharmacol. Ther. 2007, 116, 428–436. [Google Scholar] [CrossRef]
- Hattori, Y.; Hattori, S.; Wang, X.; Satoh, H.; Nakanishi, N.; Kasai, K. Oral Administration of Tetrahydrobiopterin Slows the Progression of Atherosclerosis in Apolipoprotein E-Knockout Mice. Arterioscler. Thromb. Vasc. Biol. 2007, 27, 865–870. [Google Scholar] [CrossRef]
- Li, L.; Chen, W.; Rezvan, A.; Jo, H.; Harrison, D.G. Tetrahydrobiopterin Deficiency and Nitric Oxide Synthase Uncoupling Contribute to Atherosclerosis Induced by Disturbed Flow. Arterioscler. Thromb. Vasc. Biol. 2011, 31, 1547–1554. [Google Scholar] [CrossRef]
- Moens, A.L.; Takimoto, E.; Tocchetti, C.G.; Chakir, K.; Bedja, D.; Cormaci, G.; Ketner, E.A.; Majmudar, M.; Gabrielson, K.; Halushka, M.K.; et al. Reversal of Cardiac Hypertrophy and Fibrosis From Pressure Overload by Tetrahydrobiopterin. Circulation 2008, 117, 2626–2636. [Google Scholar] [CrossRef]
- Tratsiakovich, Y.; Gonon, A.T.; Kiss, A.; Yang, J.; Böhm, F.; Tornvall, P.; Settergren, M.; Channon, K.M.; Sjöquist, P.-O.; Pernow, J. Myocardial Protection by Co-Administration of l-Arginine and Tetrahydrobiopterin during Ischemia and Reperfusion. Int. J. Cardiol. 2013, 169, 83–88. [Google Scholar] [CrossRef]
- Tiefenbacher, C.P.; Bleeke, T.; Vahl, C.; Amann, K.; Vogt, A.; Kübler, W. Endothelial Dysfunction of Coronary Resistance Arteries Is Improved by Tetrahydrobiopterin in Atherosclerosis. Circulation 2000, 102, 2172–2179. [Google Scholar] [CrossRef]
- Cunnington, C.; Van Assche, T.; Shirodaria, C.; Kylintireas, I.; Lindsay, A.C.; Lee, J.M.; Antoniades, C.; Margaritis, M.; Lee, R.; Cerrato, R.; et al. Systemic and Vascular Oxidation Limits the Efficacy of Oral Tetrahydrobiopterin Treatment in Patients With Coronary Artery Disease. Circulation 2012, 125, 1356–1366. [Google Scholar] [CrossRef]
- De Maria, R.; Campolo, J.; Frontali, M.; Taroni, F.; Federico, A.; Inzitari, D.; Tavani, A.; Romano, S.; Puca, E.; Orzi, F.; et al. Effects of Sapropterin on Endothelium-Dependent Vasodilation in Patients With CADASIL. Stroke 2014, 45, 2959–2966. [Google Scholar] [CrossRef]
- Webb, A.J.; Patel, N.; Loukogeorgakis, S.; Okorie, M.; Aboud, Z.; Misra, S.; Rashid, R.; Miall, P.; Deanfield, J.; Benjamin, N.; et al. Acute Blood Pressure Lowering, Vasoprotective, and Antiplatelet Properties of Dietary Nitrate via Bioconversion to Nitrite. Hypertension 2008, 51, 784–790. [Google Scholar] [CrossRef]
- Banez, M.J.; Geluz, M.I.; Chandra, A.; Hamdan, T.; Biswas, O.S.; Bryan, N.S.; Von Schwarz, E.R. A Systemic Review on the Antioxidant and Anti-Inflammatory Effects of Resveratrol, Curcumin, and Dietary Nitric Oxide Supplementation on Human Cardiovascular Health. Nutr. Res. 2020, 78, 11–26. [Google Scholar] [CrossRef]
- Liu, Y.; Croft, K.D.; Hodgson, J.M.; Mori, T.; Ward, N.C. Mechanisms of the Protective Effects of Nitrate and Nitrite in Cardiovascular and Metabolic Diseases. Nitric Oxide 2020, 96, 35–43. [Google Scholar] [CrossRef]
- Petraina, A.; Nogales, C.; Krahn, T.; Mucke, H.; Lüscher, T.F.; Fischmeister, R.; Kass, D.A.; Burnett, J.C.; Hobbs, A.J.; Schmidt, H.H.H.W. Cyclic GMP Modulating Drugs in Cardiovascular Diseases: Mechanism-Based Network Pharmacology. Cardiovasc. Res. 2021, 118, 2085–2102. [Google Scholar] [CrossRef] [PubMed]
- Münzel, T.; Daiber, A. Inorganic Nitrite and Nitrate in Cardiovascular Therapy: A Better Alternative to Organic Nitrates as Nitric Oxide Donors? Vasc. Pharmacol. 2018, 102, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Redfield, M.M.; Anstrom, K.J.; Levine, J.A.; Koepp, G.A.; Borlaug, B.A.; Chen, H.H.; LeWinter, M.M.; Joseph, S.M.; Shah, S.J.; Semigran, M.J.; et al. Isosorbide Mononitrate in Heart Failure with Preserved Ejection Fraction. N. Engl. J. Med. 2015, 373, 2314–2324. [Google Scholar] [CrossRef]
- Fraccarollo, D.; Widder, J.D.; Galuppo, P.; Thum, T.; Tsikas, D.; Hoffmann, M.; Ruetten, H.; Ertl, G.; Bauersachs, J. Improvement in Left Ventricular Remodeling by the Endothelial Nitric Oxide Synthase Enhancer AVE9488 After Experimental Myocardial Infarction. Circulation 2008, 118, 818–827. [Google Scholar] [CrossRef]
- Westermann, D.; Riad, A.; Richter, U.; Jäger, S.; Savvatis, K.; Schuchardt, M.; Bergmann, N.; Tölle, M.; Nagorsen, D.; Gotthardt, M.; et al. Enhancement of the Endothelial NO Synthase Attenuates Experimental Diastolic Heart Failure. Basic. Res. Cardiol. 2009, 104, 499–509. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Chen, C.; Feng, C.; Tang, A.; Ma, Y.; He, X.; Li, Y.; He, J.; Dong, Y. AVE 3085, a Novel Endothelial Nitric Oxide Synthase Enhancer, Attenuates Cardiac Remodeling in Mice through the Smad Signaling Pathway. Arch. Biochem. Biophys. 2015, 570, 8–13. [Google Scholar] [CrossRef]
- Schäfer, A.; Fraccarollo, D.; Widder, J.; Eigenthaler, M.; Ertl, G.; Bauersachs, J. Inhibition of Platelet Activation in Rats with Severe Congestive Heart Failure by a Novel Endothelial Nitric Oxide Synthase Transcription Enhancer. Eur. J. Heart Fail. 2009, 11, 336–341. [Google Scholar] [CrossRef]
- Yang, Q.; Xue, H.-M.; Wong, W.-T.; Tian, X.-Y.; Huang, Y.; Tsui, S.K.; Ng, P.K.; Wohlfart, P.; Li, H.; Xia, N.; et al. AVE3085, an Enhancer of Endothelial Nitric Oxide Synthase, Restores Endothelial Function and Reduces Blood Pressure in Spontaneously Hypertensive Rats. Br. J. Pharmacol. 2011, 163, 1078–1085. [Google Scholar] [CrossRef] [PubMed]
- Cheang, W.S.; Wong, W.T.; Tian, X.Y.; Yang, Q.; Lee, H.K.; He, G.-W.; Yao, X.; Huang, Y. Endothelial Nitric Oxide Synthase Enhancer Reduces Oxidative Stress and Restores Endothelial Function in Db/Db Mice. Cardiovasc. Res. 2011, 92, 267–275. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.-L.; Yin, H.; Huang, J. Inhibition of TGF-Β1 Signaling by ENOS Gene Transfer Improves Ventricular Remodeling after Myocardial Infarction through Angiogenesis and Reduction of Apoptosis. Cardiovasc. Pathol. 2007, 16, 221–230. [Google Scholar] [CrossRef]
- Torondel, B.; Nandi, M.; Kelly, P.; Wojciak-Stothard, B.; Fleming, I.; Leiper, J. Adenoviral-Mediated Overexpression of DDAH Improves Vascular Tone Regulation. Vasc. Med. 2010, 15, 205–213. [Google Scholar] [CrossRef] [PubMed]
- Chauhan, S.D.; Seggara, G.; Vo, P.A.; Macallister, R.J.; Hobbs, A.J.; Ahluwalia, A. Protection against Lipopolysaccharide-induced Endothelial Dysfunction in Resistance and Conduit Vasculature of INOS Knockout Mice. FASEB J. 2003, 17, 773–775. [Google Scholar] [CrossRef]
- Wildhirt, S.M.; Dudek, R.R.; Suzuki, H.; Bing, R.J. Involvement of Inducible Nitric Oxide Synthase in the Inflammatory Process of Myocardial Infarction. Int. J. Cardiol. 1995, 50, 253–261. [Google Scholar] [CrossRef]
- Arstall, M.A.; Sawyer, D.B.; Fukazawa, R.; Kelly, R.A. Cytokine-Mediated Apoptosis in Cardiac Myocytes. Circ. Res. 1999, 85, 829–840. [Google Scholar] [CrossRef]
- D’Oria, R.; Schipani, R.; Leonardini, A.; Natalicchio, A.; Perrini, S.; Cignarelli, A.; Laviola, L.; Giorgino, F. The Role of Oxidative Stress in Cardiac Disease: From Physiological Response to Injury Factor. Oxid. Med. Cell Longev. 2020, 2020, 5732956. [Google Scholar] [CrossRef] [PubMed]
- Dubois-deruy, E.; Peugnet, V.; Turkieh, A.; Pinet, F. Oxidative Stress in Cardiovascular Diseases. Antioxidants 2020, 9, 864. [Google Scholar] [CrossRef]
- Haynes, V.; Elfering, S.L.; Squires, R.J.; Traaseth, N.; Solien, J.; Ettl, A.; Giulivi, C. Mitochondrial Nitric-Oxide Synthase: Role in Pathophysiology. IUBMB Life 2003, 55, 599–603. [Google Scholar] [CrossRef] [PubMed]
- Poderoso, J.J.; Helfenberger, K.; Poderoso, C. The Effect of Nitric Oxide on Mitochondrial Respiration. Nitric Oxide 2019, 88, 61–72. [Google Scholar] [CrossRef] [PubMed]
- Bailey, J.D.; Diotallevi, M.; Nicol, T.; McNeill, E.; Shaw, A.; Chuaiphichai, S.; Hale, A.; Starr, A.; Nandi, M.; Stylianou, E.; et al. Nitric Oxide Modulates Metabolic Remodeling in Inflammatory Macrophages through TCA Cycle Regulation and Itaconate Accumulation. Cell Rep. 2019, 28, 218–230.e7. [Google Scholar] [CrossRef]
- Ow, Y.L.P.; Green, D.R.; Hao, Z.; Mak, T.W. Cytochrome c: Functions beyond Respiration. Nat. Rev. Mol. Cell Biol. 2008, 9, 532–542. [Google Scholar] [CrossRef]
- Yoon, S.; Kim, M.; Lee, H.; Kang, G.; Bedi, K.; Margulies, K.B.; Jain, R.; Nam, K.-I.; Kook, H.; Eom, G.H. S-Nitrosylation of Histone Deacetylase 2 by Neuronal Nitric Oxide Synthase as a Mechanism of Diastolic Dysfunction. Circulation 2021, 143, 1912–1925. [Google Scholar] [CrossRef]
- Solomon, S.D.; McMurray, J.J.V.; Claggett, B.; de Boer, R.A.; DeMets, D.; Hernandez, A.F.; Inzucchi, S.E.; Kosiborod, M.N.; Lam, C.S.P.; Martinez, F.; et al. Dapagliflozin in Heart Failure with Mildly Reduced or Preserved Ejection Fraction. N. Engl. J. Med. 2022, 387, 1089–1098. [Google Scholar] [CrossRef] [PubMed]
- Kondo, H.; Akoumianakis, I.; Badi, I.; Akawi, N.; Kotanidis, C.P.; Polkinghorne, M.; Stadiotti, I.; Sommariva, E.; Antonopoulos, A.S.; Carena, M.C.; et al. Effects of Canagliflozin on Human Myocardial Redox Signalling: Clinical Implications. Eur. Heart J. 2021, 42, 4947–4960. [Google Scholar] [CrossRef]
- Rahman, H.; Demir, O.M.; Khan, F.; Ryan, M.; Ellis, H.; Mills, M.T.; Chiribiri, A.; Webb, A.; Perera, D. Physiological Stratification of Patients With Angina Due to Coronary Microvascular Dysfunction. J. Am. Coll. Cardiol. 2020, 75, 2538–2549. [Google Scholar] [CrossRef] [PubMed]
- Sinha, A.; Rahman, H.; Webb, A.; Shah, A.M.; Perera, D. Untangling the Pathophysiologic Link between Coronary Microvascular Dysfunction and Heart Failure with Preserved Ejection Fraction. Eur. Heart J. 2021, 42, 4431–4441. [Google Scholar] [CrossRef]
- He, J.; Liu, D.; Zhao, L.; Zhou, D.; Rong, J.; Zhang, L.; Xia, Z. Myocardial Ischemia/Reperfusion Injury: Mechanisms of Injury and Implications for Management (Review). Exp. Ther. Med. 2022, 23, 430. [Google Scholar] [CrossRef]
- Heusch, G. Myocardial Ischaemia–Reperfusion Injury and Cardioprotection in Perspective. Nat. Rev. Cardiol. 2020, 17, 773–789. [Google Scholar] [CrossRef]
- Damy, T.; Ratajczak, P.; Robidel, E.; Bendall, J.K.; Oliviéro, P.; Boczkowski, J.; Ebrahimian, T.; Marotte, F.; Samuel, J.-L.; Heymes, C. Up-regulation of Cardiac Nitric Oxide Synthase 1-derived Nitric Oxide after Myocardial Infarction in Senescent Rats. FASEB J. 2003, 17, 1–22. [Google Scholar] [CrossRef]
- Yu, X.; Ge, L.; Niu, L.; Lian, X.; Ma, H.; Pang, L. The Dual Role of Inducible Nitric Oxide Synthase in Myocardial Ischemia/Reperfusion Injury: Friend or Foe? Oxid. Med. Cell Longev. 2018, 2018, 8364848. [Google Scholar] [CrossRef] [PubMed]
- Wilmes, V.; Scheiper, S.; Roehr, W.; Niess, C.; Kippenberger, S.; Steinhorst, K.; Verhoff, M.A.; Kauferstein, S. Increased Inducible Nitric Oxide Synthase (INOS) Expression in Human Myocardial Infarction. Int. J. Legal Med. 2020, 134, 575–581. [Google Scholar] [CrossRef] [PubMed]
- Schulz, R.; Wambolt, R. Inhibition of Nitric Oxide Synthesis Protects the Isolated Working Rabbit Heart from Ischaemia-Reperfusion Injury. Cardiovasc. Res. 1995, 30, 432–439. [Google Scholar] [CrossRef] [PubMed]
- Yasmin, W. Generation of Peroxynitrite Contributes to Ischemia-Reperfusion Injury in Isolated Rat Hearts. Cardiovasc. Res. 1997, 33, 422–432. [Google Scholar] [CrossRef] [PubMed]
- Patel, V.C.; Yellon, D.M.; Singh, K.J.; Neild, G.H.; Woolfson, R.G. Inhibition of Nitric Oxide Limits Infarct Size in the in Situ Rabbit Heart. Biochem. Biophys. Res. Commun. 1993, 194, 234–238. [Google Scholar] [CrossRef]
- Ramasamy, R.; Hwang, Y.C.; Liu, Y.; Son, N.H.; Ma, N.; Parkinson, J.; Sciacca, R.; Albala, A.; Edwards, N.; Szabolcs, M.J.; et al. Metabolic and Functional Protection by Selective Inhibition of Nitric Oxide Synthase 2 During Ischemia-Reperfusion in Isolated Perfused Hearts. Circulation 2004, 109, 1668–1673. [Google Scholar] [CrossRef]
- Hu, A.; Jiao, X.; Gao, E.; Koch, W.J.; Sharifi-Azad, S.; Grunwald, Z.; Ma, X.L.; Sun, J.-Z. Chronic β-Adrenergic Receptor Stimulation Induces Cardiac Apoptosis and Aggravates Myocardial Ischemia/Reperfusion Injury by Provoking Inducible Nitric-Oxide Synthase-Mediated Nitrative Stress. J. Pharmacol. Exp. Ther. 2006, 318, 469–475. [Google Scholar] [CrossRef]
- Cotter, G.; Kaluski, E.; Blatt, A.; Milovanov, O.; Moshkovitz, Y.; Zaidenstein, R.; Salah, A.; Alon, D.; Michovitz, Y.; Metzger, M.; et al. L-NMMA (a Nitric Oxide Synthase Inhibitor) Is Effective in the Treatment of Cardiogenic Shock. Circulation 2000, 101, 1358–1361. [Google Scholar] [CrossRef]
- Cotter, G.; Kaluski, E.; Milo, O.; Blatt, A.; Salah, A.; Hendler, A.; Krakover, R.; Golick, A.; Vered, Z. LINCS: L-NAME (a NO Synthase Inhibitor) In the Treatment of Refractory Cardiogenic Shock A Prospective Randomized Study. Eur. Heart J. 2003, 24, 1287–1295. [Google Scholar] [CrossRef] [PubMed]
- Dzavik, V.; Cotter, G.; Reynolds, H.R.; Alexander, J.H.; Ramanathan, K.; Stebbins, A.L.; Hathaway, D.; Farkouh, M.E.; Ohman, E.M.; Baran, D.A.; et al. Effect of Nitric Oxide Synthase Inhibition on Haemodynamics and Outcome of Patients with Persistent Cardiogenic Shock Complicating Acute Myocardial Infarction: A Phase II Dose-Ranging Study. Eur. Heart J. 2007, 28, 1109–1116. [Google Scholar] [CrossRef]
- The TRIUMPH Investigators. Effect of Tilarginine Acetate in Patients With Acute Myocardial Infarction and Cardiogenic Shock. JAMA 2007, 297, 1657. [Google Scholar] [CrossRef]
- Shen, Z.; Xiang, M.; Chen, C.; Ding, F.; Wang, Y.; Shang, C.; Xin, L.; Zhang, Y.; Cui, X. Glutamate Excitotoxicity: Potential Therapeutic Target for Ischemic Stroke. Biomed. Pharmacother. 2022, 151, 113125. [Google Scholar] [CrossRef]
- Yang, Q.; Huang, Q.; Hu, Z.; Tang, X. Potential Neuroprotective Treatment of Stroke: Targeting Excitotoxicity, Oxidative Stress, and Inflammation. Front. Neurosci. 2019, 13, 1036. [Google Scholar] [CrossRef]
- Choi, D.W. Excitotoxicity: Still Hammering the Ischemic Brain in 2020. Front. Neurosci. 2020, 14, 579953. [Google Scholar] [CrossRef]
- Zhu, L.-J.; Li, F.; Zhu, D.-Y. NNOS and Neurological, Neuropsychiatric Disorders: A 20-Year Story. Neurosci. Bull. 2023, 39, 1439–1453. [Google Scholar] [CrossRef]
- Lai, T.W.; Zhang, S.; Wang, Y.T. Excitotoxicity and Stroke: Identifying Novel Targets for Neuroprotection. Prog. Neurobiol. 2014, 115, 157–188. [Google Scholar] [CrossRef] [PubMed]
- Dawson, V.; Dawson, T.; Bartley, D.; Uhl, G.; Snyder, S. Mechanisms of Nitric Oxide-Mediated Neurotoxicity in Primary Brain Cultures. J. Neurosci. 1993, 13, 2651–2661. [Google Scholar] [CrossRef] [PubMed]
- Dawson, V.L.; Dawson, T.M.; London, E.D.; Bredt, D.S.; Snyder, S.H. Nitric Oxide Mediates Glutamate Neurotoxicity in Primary Cortical Cultures. Proc. Natl. Acad. Sci. USA 1991, 88, 6368–6371. [Google Scholar] [CrossRef]
- Dawson, V.; Kizushi, V.; Huang, P.; Snyder, S.; Dawson, T. Resistance to Neurotoxicity in Cortical Cultures from Neuronal Nitric Oxide Synthase-Deficient Mice. J. Neurosci. 1996, 16, 2479–2487. [Google Scholar] [CrossRef] [PubMed]
- Huang, Z.; Huang, P.L.; Panahian, N.; Dalkara, T.; Fishman, M.C.; Moskowitz, M.A. Effects of Cerebral Ischemia in Mice Deficient in Neuronal Nitric Oxide Synthase. Science 1994, 265, 1883–1885. [Google Scholar] [CrossRef] [PubMed]
- Hill, M.D.; Martin, R.H.; Mikulis, D.; Wong, J.H.; Silver, F.L.; terBrugge, K.G.; Milot, G.; Clark, W.M.; MacDonald, R.L.; Kelly, M.E.; et al. Safety and Efficacy of NA-1 in Patients with Iatrogenic Stroke after Endovascular Aneurysm Repair (ENACT): A Phase 2, Randomised, Double-Blind, Placebo-Controlled Trial. Lancet Neurol. 2012, 11, 942–950. [Google Scholar] [CrossRef] [PubMed]
- Hill, M.D.; Goyal, M.; Menon, B.K.; Nogueira, R.G.; McTaggart, R.A.; Demchuk, A.M.; Poppe, A.Y.; Buck, B.H.; Field, T.S.; Dowlatshahi, D.; et al. Efficacy and Safety of Nerinetide for the Treatment of Acute Ischaemic Stroke (ESCAPE-NA1): A Multicentre, Double-Blind, Randomised Controlled Trial. Lancet 2020, 395, 878–887. [Google Scholar] [CrossRef]
- NIWA, M.; INAO, S.; TAKAYASU, M.; KAWAI, T.; KAJITA, Y.; NIHASHI, T.; KABEYA, R.; SUGIMOTO, T.; YOSHIDA, J. Time Course of Expression of Three Nitric Oxide Synthase Isoforms After Transient Middle Cerebral Artery Occlusion in Rats. Neurol. Med. Chir. 2001, 41, 63–73. [Google Scholar] [CrossRef]
- Nelson, R.J.; Demas, G.E.; Huang, P.L.; Fishman, M.C.; Dawson, V.L.; Dawson, T.M.; Snyder, S.H. Behavioural Abnormalities in Male Mice Lacking Neuronal Nitric Oxide Synthase. Nature 1995, 378, 383–386. [Google Scholar] [CrossRef]
- Furfine, E.S.; Harmon, M.F.; Paith, J.E.; Knowles, R.G.; Salter, M.; Kiff, R.J.; Duffy, C.; Hazelwood, R.; Oplinger, J.A.; Garvey, E.P. Potent and Selective Inhibition of Human Nitric Oxide Synthases. Selective Inhibition of Neuronal Nitric Oxide Synthase by S-Methyl-L-Thiocitrulline and S-Ethyl-L-Thiocitrulline. J. Biol. Chem. 1994, 269, 26677–26683. [Google Scholar] [CrossRef]
- Zhang, J.; Xu, M.; Dence, C.S.; Sherman, E.L.; McCarthy, T.J.; Welch, M.J. Synthesis, in Vivo Evaluation and PET Study of a Carbon-11-Labeled Neuronal Nitric Oxide Synthase (NNOS) Inhibitor S-Methyl-L-Thiocitrulline. J. Nucl. Med. 1997, 38, 1273–1278. [Google Scholar]
- Kim, J.H.; Yenari, M.A.; Giffard, R.G.; Cho, S.W.; Park, K.A.; Lee, J.E. Agmatine Reduces Infarct Area in a Mouse Model of Transient Focal Cerebral Ischemia and Protects Cultured Neurons from Ischemia-like Injury. Exp. Neurol. 2004, 189, 122–130. [Google Scholar] [CrossRef]
- Kotagale, N.R.; Taksande, B.G.; Inamdar, N.N. Neuroprotective Offerings by Agmatine. Neurotoxicology 2019, 73, 228–245. [Google Scholar] [CrossRef]
- Yang, M.Z.; Mun, C.H.; Choi, Y.J.; Baik, J.H.; Park, K.A.; Lee, W.T.; Lee, J.E. Agmatine Inhibits Matrix Metalloproteinase-9 via Endothelial Nitric Oxide Synthase in Cerebral Endothelial Cells. Neurol. Res. 2007, 29, 749–754. [Google Scholar] [CrossRef]
- Kim, D.J.; Kim, D.I.; Lee, S.K.; Suh, S.H.; Lee, Y.J.; Kim, J.; Chung, T.S.; Lee, J.E. Protective Effect of Agmatine on a Reperfusion Model after Transient Cerebral Ischemia: Temporal Evolution on Perfusion MR Imaging and Histopathologic Findings. AJNR Am. J. Neuroradiol. 2006, 27, 780–785. [Google Scholar] [PubMed]
- Feng, Y.; Piletz, J.E.; Leblanc, M.H. Agmatine Suppresses Nitric Oxide Production and Attenuates Hypoxic-Ischemic Brain Injury in Neonatal Rats. Pediatr. Res. 2002, 52, 606–611. [Google Scholar] [CrossRef] [PubMed]
- Tegtmeier, F.; Schinzel, R.; Beer, R.; Bulters, D.; LeFrant, J.-Y.; Sahuquillo, J.; Unterberg, A.; Andrews, P.; Belli, A.; Ibanez, J.; et al. Efficacy of Ronopterin (VAS203) in Patients with Moderate and Severe Traumatic Brain Injury (NOSTRA Phase III Trial): Study Protocol of a Confirmatory, Placebo-Controlled, Randomised, Double Blind, Multi-Centre Study. Trials 2020, 21, 80. [Google Scholar] [CrossRef] [PubMed]
- Ott, C.; Bosch, A.; Winzer, N.; Friedrich, S.; Schinzel, R.; Tegtmeier, F.; Schmieder, R.E. Effects of the Nitric Oxide Synthase Inhibitor Ronopterin (VAS203) on Renal Function in Healthy Volunteers. Br. J. Clin. Pharmacol. 2019, 85, 900–907. [Google Scholar] [CrossRef] [PubMed]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Roy, R.; Wilcox, J.; Webb, A.J.; O’Gallagher, K. Dysfunctional and Dysregulated Nitric Oxide Synthases in Cardiovascular Disease: Mechanisms and Therapeutic Potential. Int. J. Mol. Sci. 2023, 24, 15200. https://doi.org/10.3390/ijms242015200
Roy R, Wilcox J, Webb AJ, O’Gallagher K. Dysfunctional and Dysregulated Nitric Oxide Synthases in Cardiovascular Disease: Mechanisms and Therapeutic Potential. International Journal of Molecular Sciences. 2023; 24(20):15200. https://doi.org/10.3390/ijms242015200
Chicago/Turabian StyleRoy, Roman, Joshua Wilcox, Andrew J. Webb, and Kevin O’Gallagher. 2023. "Dysfunctional and Dysregulated Nitric Oxide Synthases in Cardiovascular Disease: Mechanisms and Therapeutic Potential" International Journal of Molecular Sciences 24, no. 20: 15200. https://doi.org/10.3390/ijms242015200