
Experimental and Bioinformatic Insights into the Effects of Epileptogenic Variants on the Function and Trafficking of the GABA Transporter GAT-1
 , , , ,
, , , ,
Abstract
:1. Introduction
2. Results
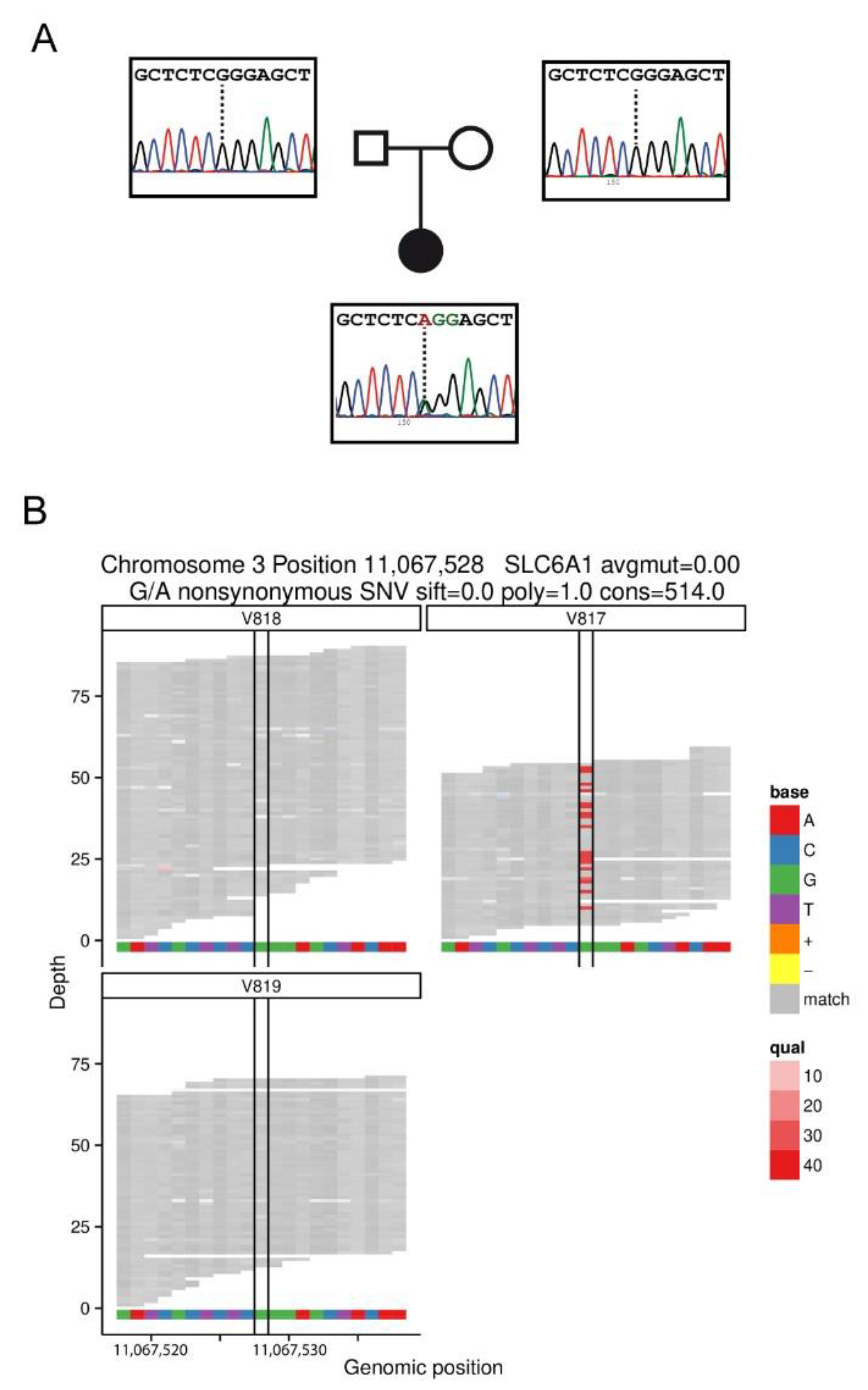
2.1. Patients with the Variant G307R of the SLC6A1 Gene
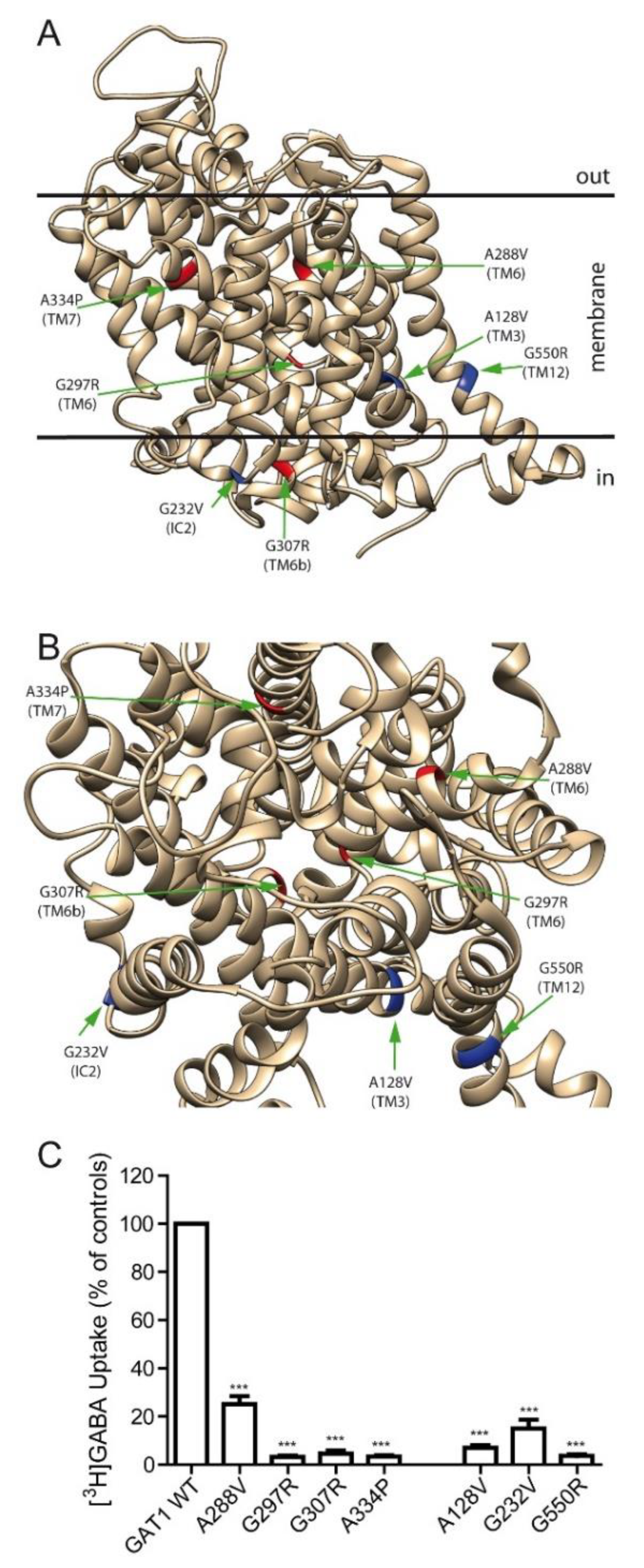
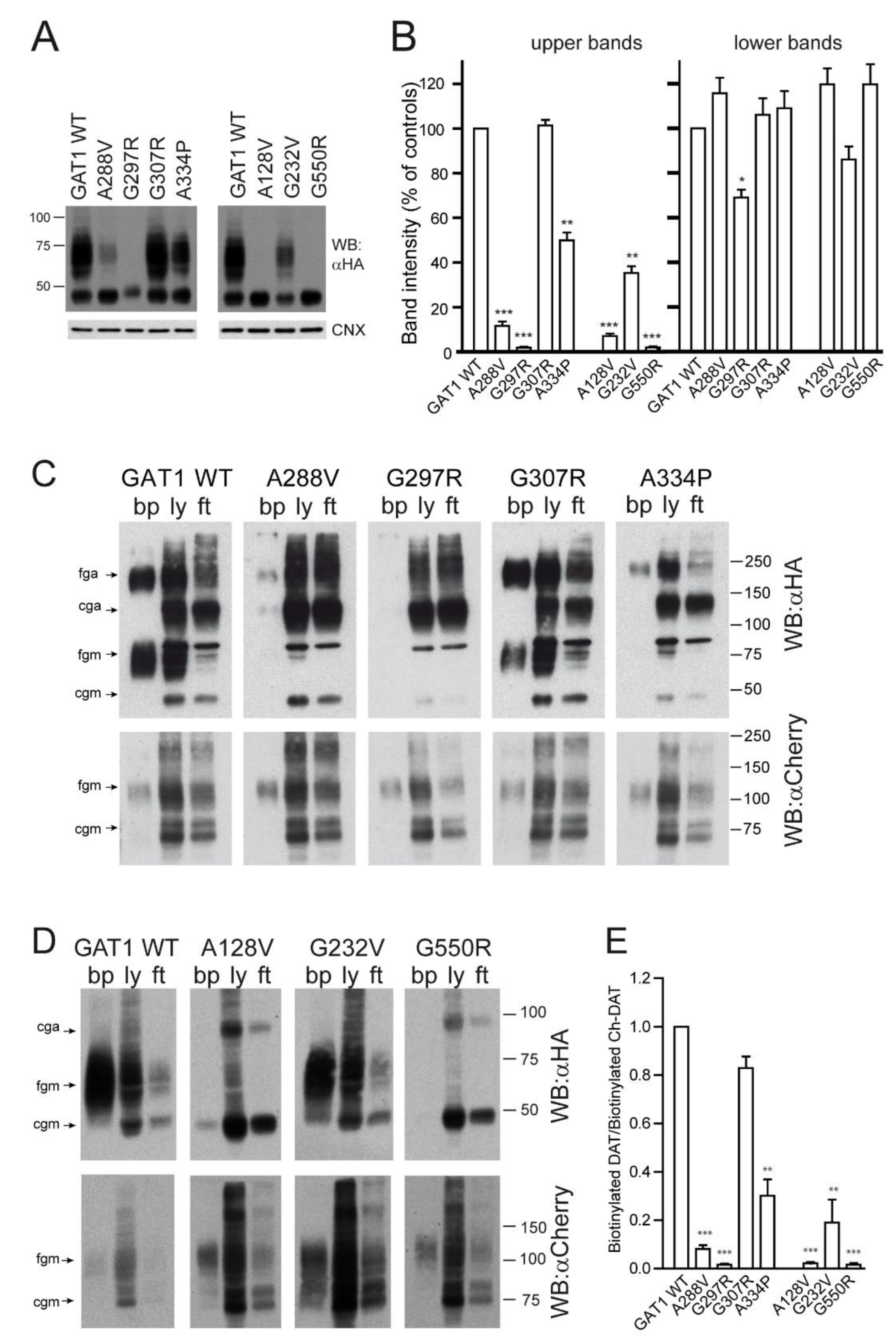
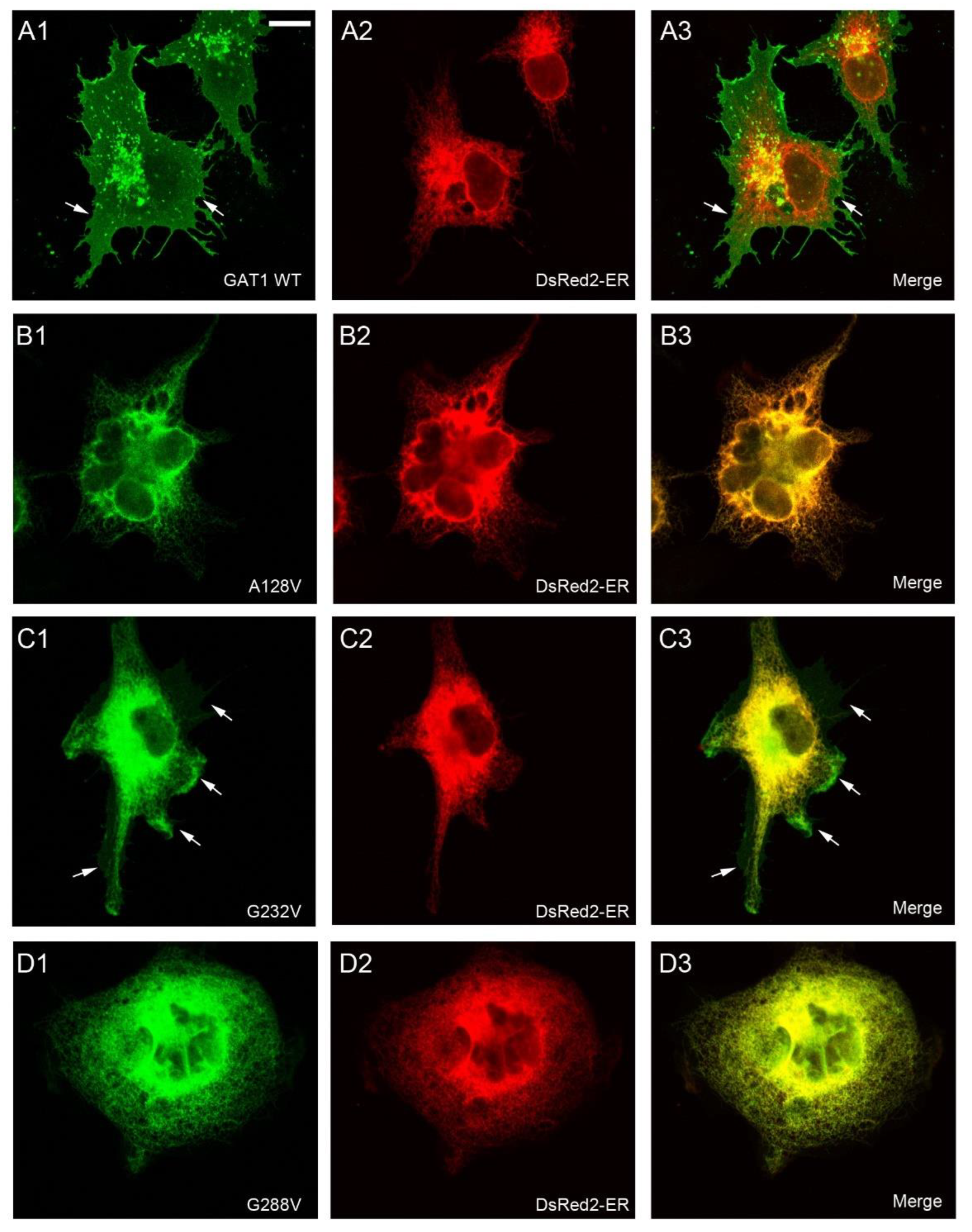
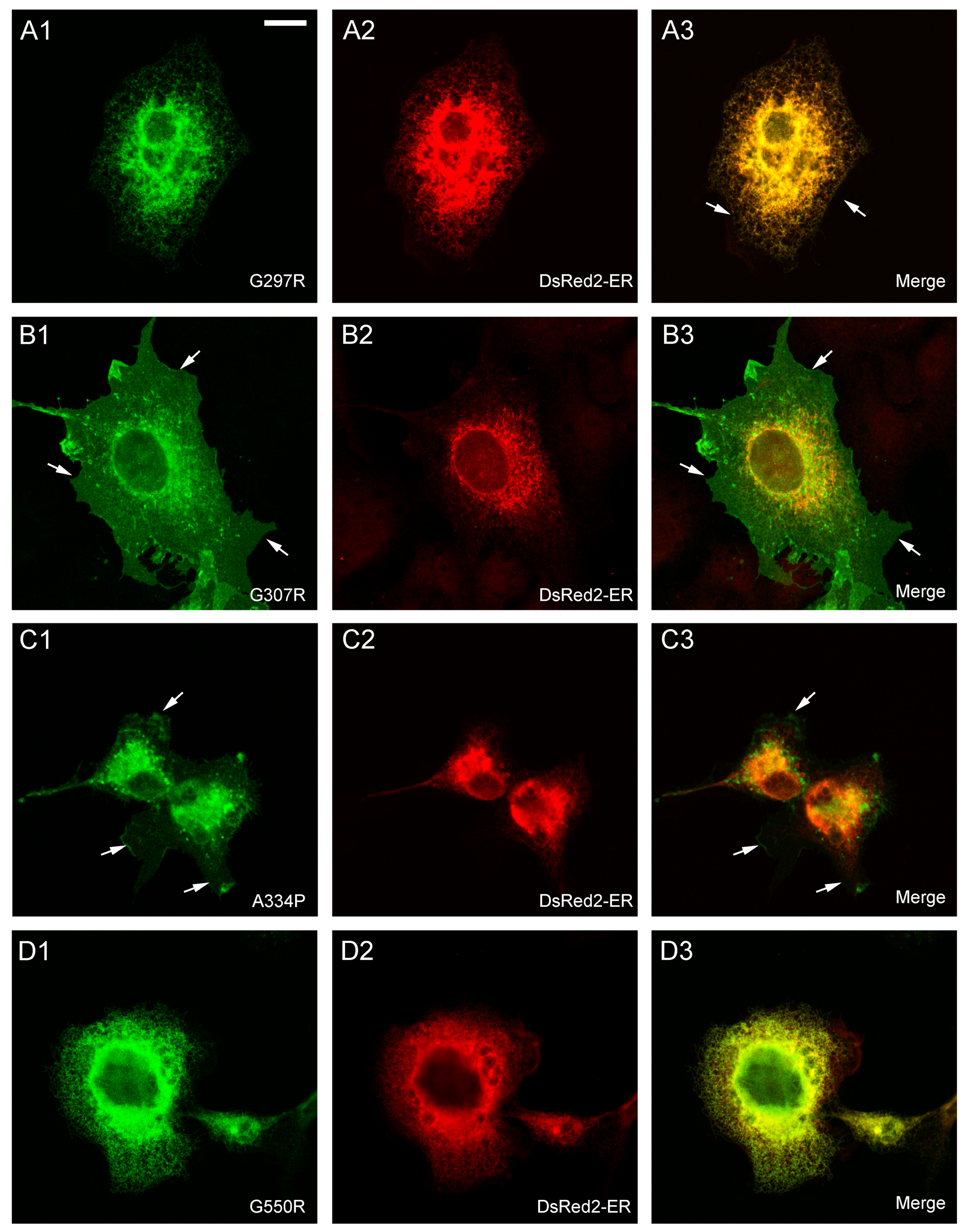
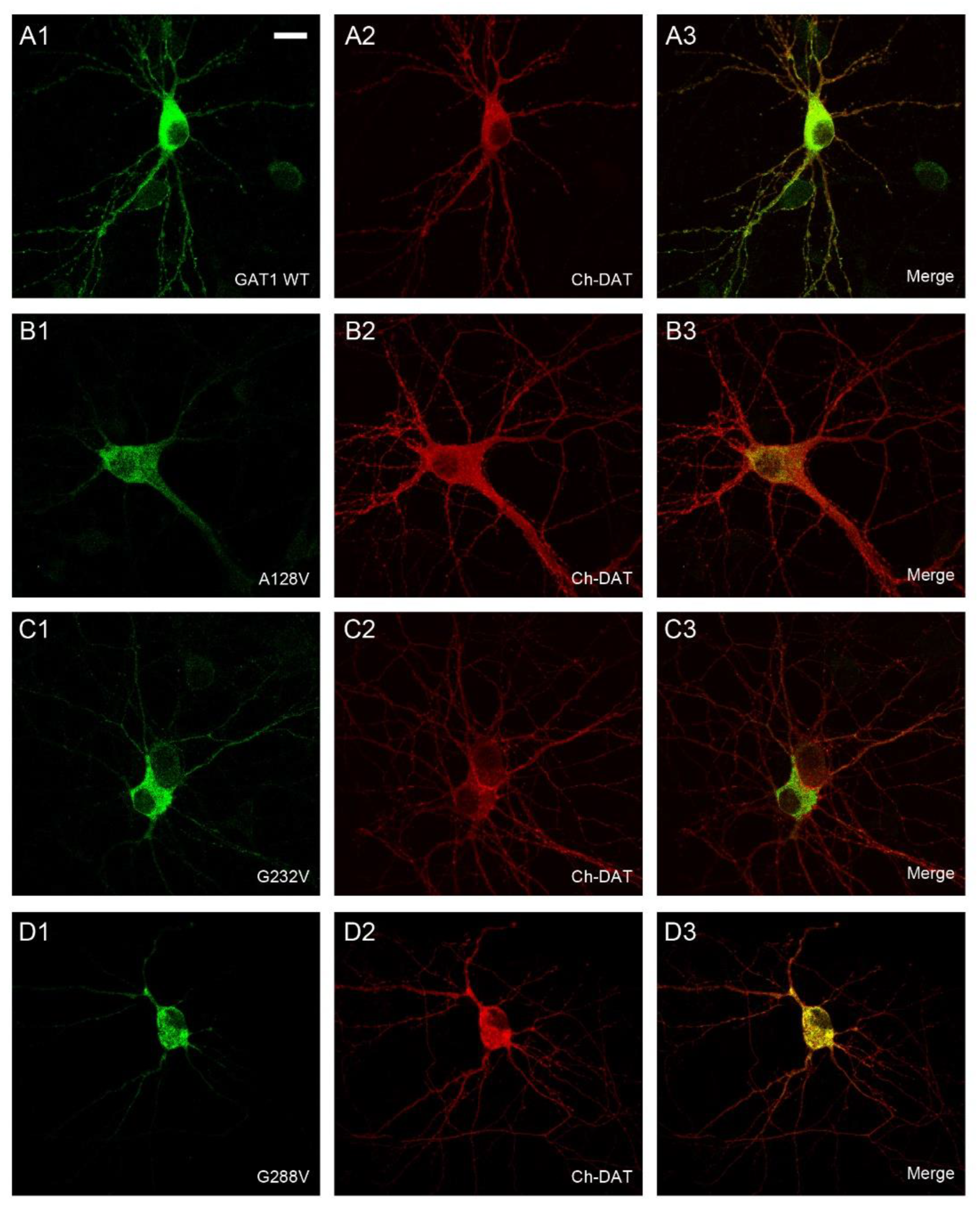
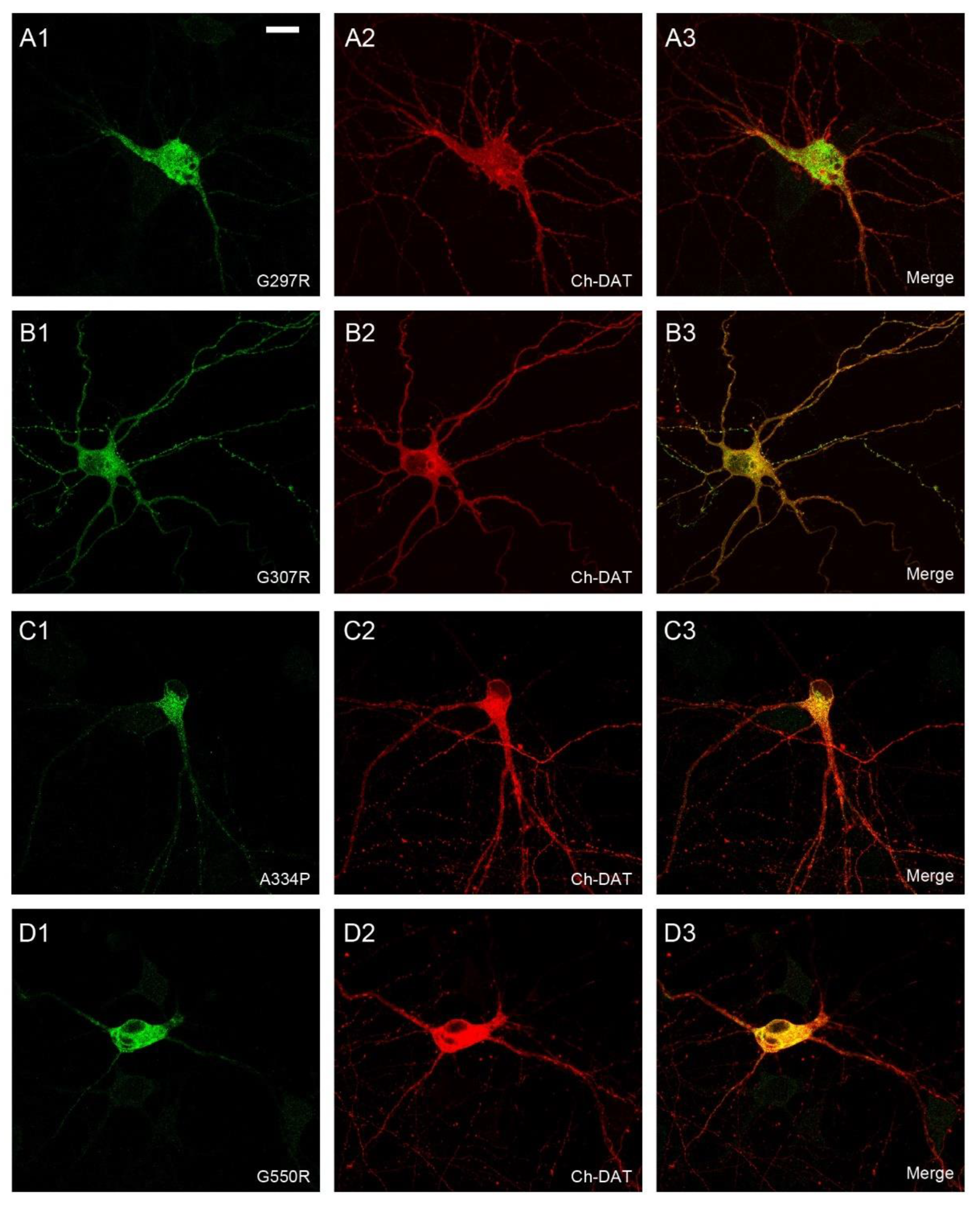
2.2. Effect of Various SLC6A1 Mutations on Transporter Function and Trafficking
2.3. Bioinformatic STUDIES on the Effect of Mutations in the SLC6A1 Gene
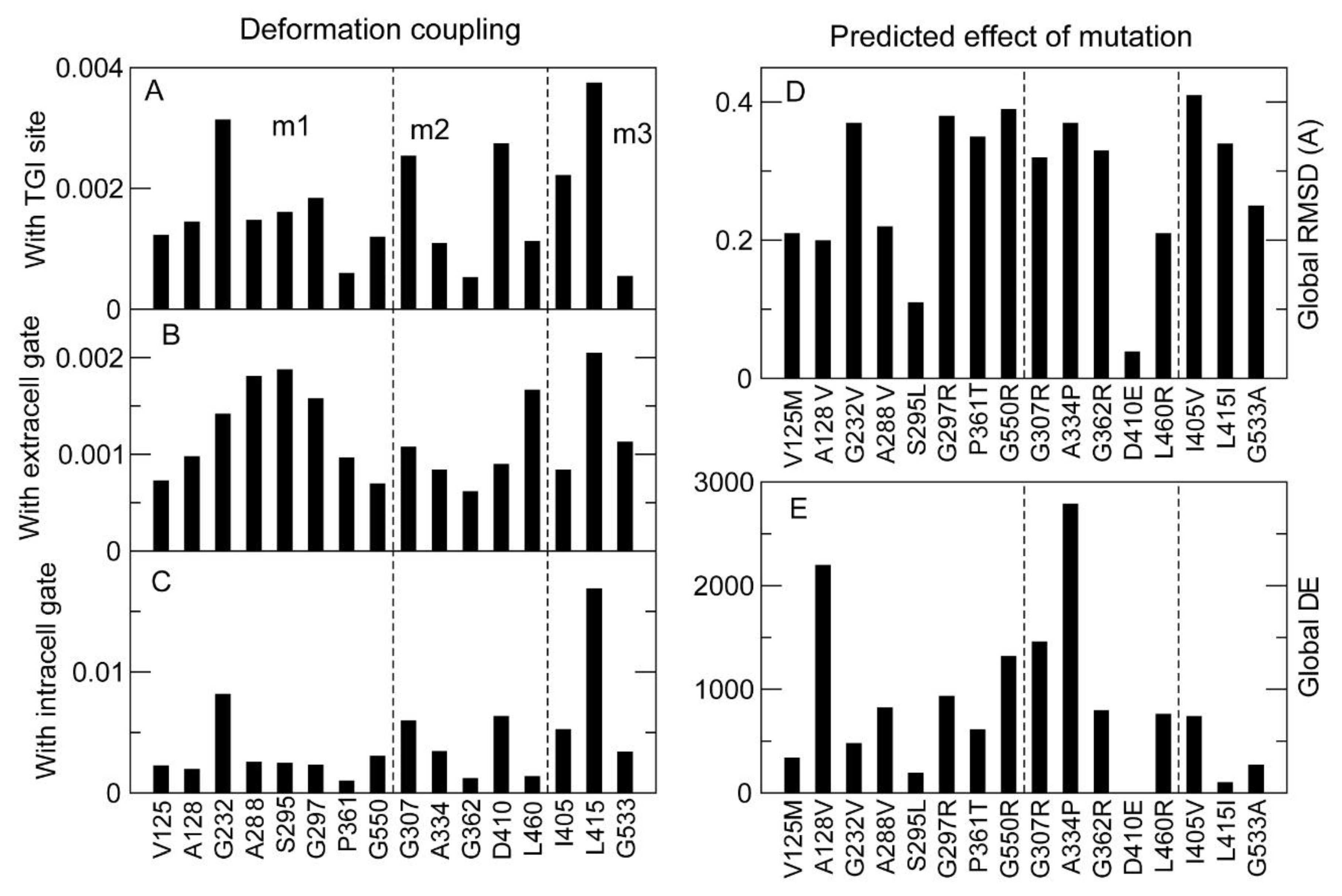
2.3.1. Prediction of Protein Stability Changes upon Mutation
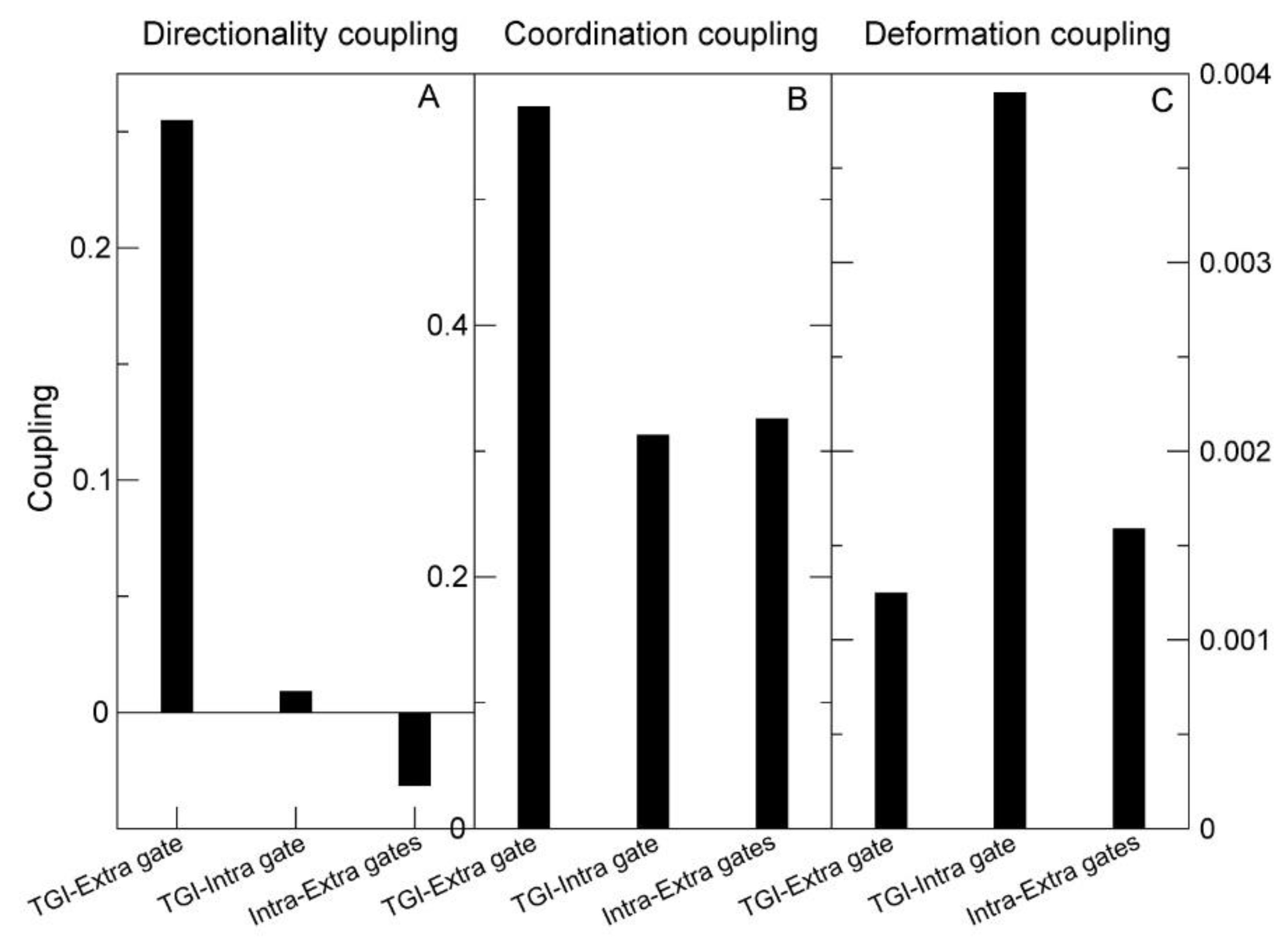
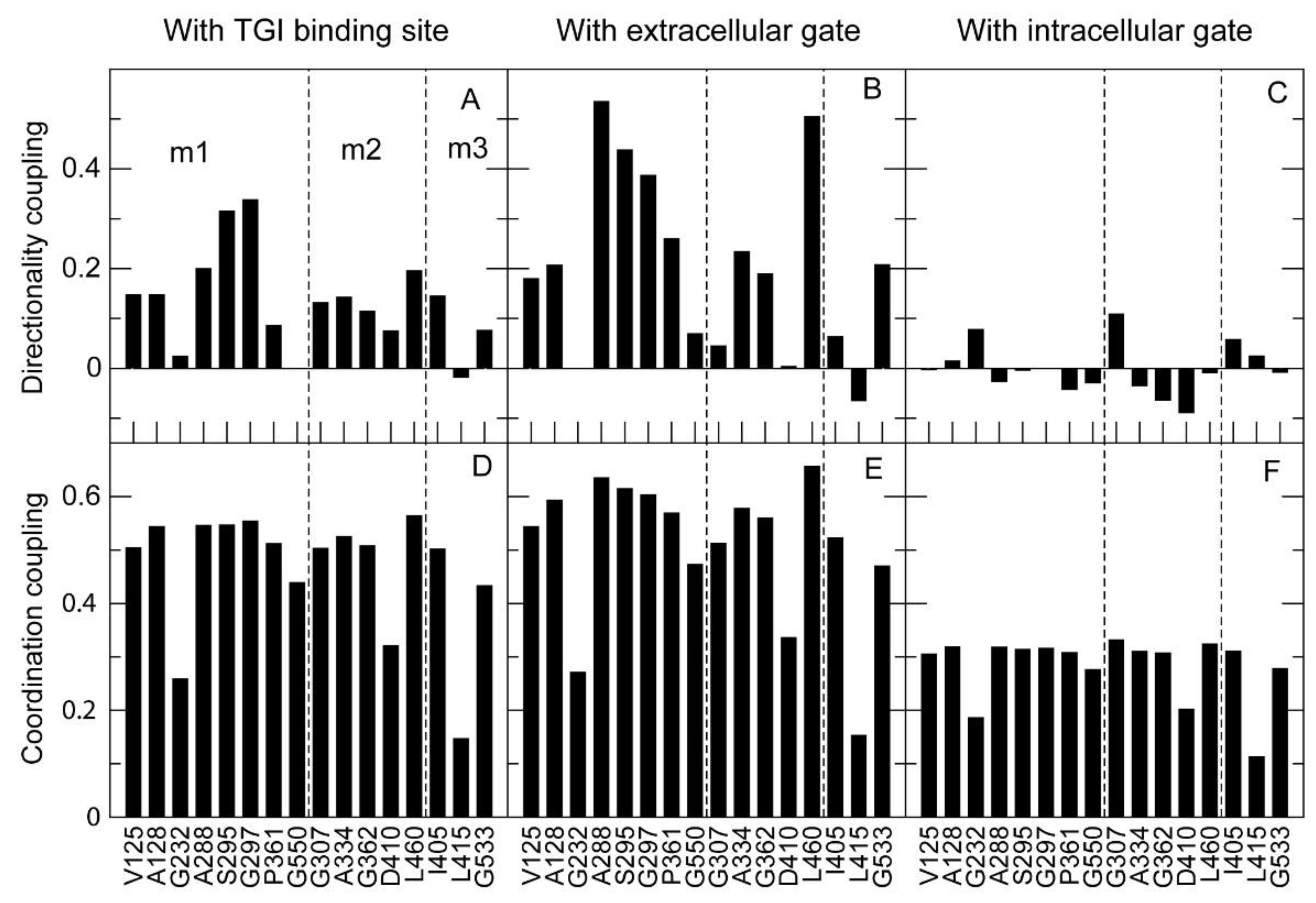
2.3.2. Analysis of the Structural Effects of Mutations with Normal Modes
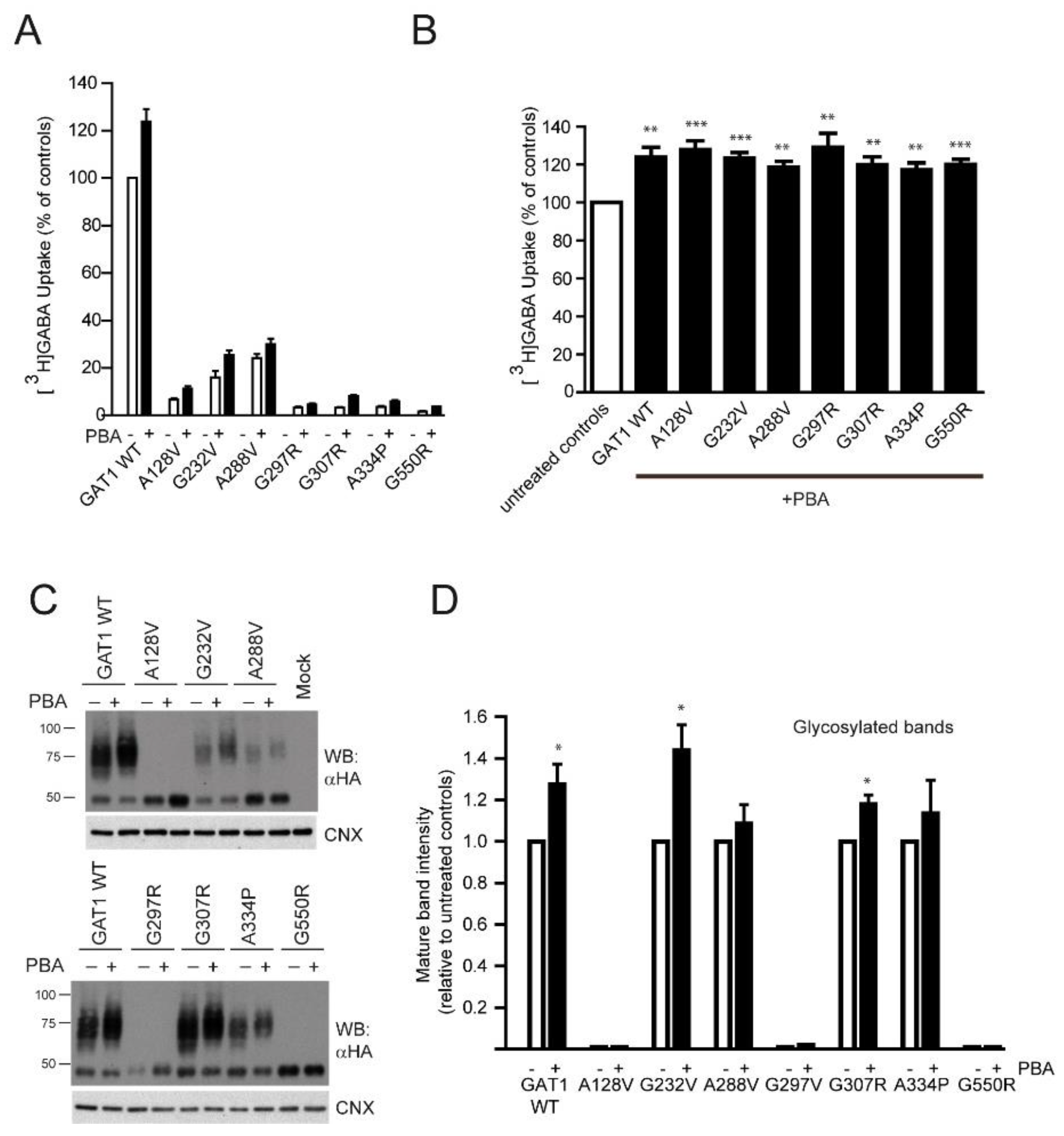
2.4. Rescue of GABA Uptake by the Chemical Chaperone 4-Phenylbutyrate
3. Discussion
4. Materials and Methods
4.1. Patients, Sample Preparation and DNA Analysis
4.2. Site-Directed Mutagenesis
4.3. Primary Cultures
4.4. Transfection
4.5. Immunofluorescence
4.6. Radioactive GABA Transport
4.7. Electrophoresis and Immunoblotting
4.8. Prediction of the Effect of Mutations on Stability and Structure
4.9. Analysis of Protein Dynamics with the Torsional Network Model (TNM)
4.10. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Carvill, G.L.; McMahon, J.M.; Schneider, A.; Zemel, M.; Myers, C.T.; Saykally, J.; Nguyen, J.; Robbiano, A.; Zara, F.; Specchio, N.; et al. Mutations in the GABA Transporter SLC6A1 Cause Epilepsy with Myoclonic-Atonic Seizures. Am. J. Hum. Genet. 2015, 96, 808–815. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johannesen, K.M.; Gardella, E.; Linnankivi, T.; Courage, C.; de Saint Martin, A.; Lehesjoki, A.-E.; Mignot, C.; Afenjar, A.; Lesca, G.; Abi-Warde, M.-T.; et al. Defining the Phenotypic Spectrum of SLC6A1 Mutations. Epilepsia 2018, 59, 389–402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fischer, F.P.; Kasture, A.S.; Hummel, T.; Sucic, S. Molecular and Clinical Repercussions of GABA Transporter 1 Variants Gone Amiss: Links to Epilepsy and Developmental Spectrum Disorders. Front. Mol. Biosci. 2022, 9, 834498. [Google Scholar] [CrossRef] [PubMed]
- Farhan, H.; Korkhov, V.M.; Paulitschke, V.; Dorostkar, M.M.; Scholze, P.; Kudlacek, O.; Freissmuth, M.; Sitte, H.H. Two Discontinuous Segments in the Carboxyl Terminus Are Required for Membrane Targeting of the Rat Gamma-Aminobutyric Acid Transporter-1 (GAT1). J. Biol. Chem. 2004, 279, 28553–28563. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Imoukhuede, P.I.; Moss, F.J.; Michael, D.J.; Chow, R.H.; Lester, H.A. Ezrin Mediates Tethering of the Gamma-Aminobutyric Acid Transporter GAT1 to Actin Filaments via a C-Terminal PDZ-Interacting Domain. Biophys. J. 2009, 96, 2949–2960. [Google Scholar] [CrossRef] [Green Version]
- McHugh, E.M.; Zhu, W.; Milgram, S.; Mager, S. The GABA Transporter GAT1 and the MAGUK Protein Pals1: Interaction, Uptake Modulation, and Coexpression in the Brain. Mol. Cell. Neurosci. 2004, 26, 406–417. [Google Scholar] [CrossRef]
- Moss, F.J.; Imoukhuede, P.I.; Scott, K.; Hu, J.; Jankowsky, J.L.; Quick, M.W.; Lester, H.A. GABA Transporter Function, Oligomerization State, and Anchoring: Correlates with Subcellularly Resolved FRET. J. Gen. Physiol. 2009, 134, 489–521. [Google Scholar] [CrossRef] [Green Version]
- Ryan, R.M.; Ingram, S.L.; Scimemi, A. Regulation of Glutamate, GABA and Dopamine Transporter Uptake, Surface Mobility and Expression. Front. Cell. Neurosci. 2021, 15, 670346. [Google Scholar] [CrossRef]
- Fattorini, G.; Melone, M.; Conti, F. A Reappraisal of GAT-1 Localization in Neocortex. Front. Cell. Neurosci. 2020, 14, 9. [Google Scholar] [CrossRef]
- Motiwala, Z.; Aduri, N.G.; Shaye, H.; Han, G.W.; Lam, J.H.; Katritch, V.; Cherezov, V.; Gati, C. Structural Basis of GABA Reuptake Inhibition. Nature 2022, 606, 820–826. [Google Scholar] [CrossRef]
- Penmatsa, A.; Wang, K.H.; Gouaux, E. X-Ray Structure of Dopamine Transporter Elucidates Antidepressant Mechanism. Nature 2013, 503, 85–90. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coleman, J.A.; Green, E.M.; Gouaux, E. X-ray Structures and Mechanism of the Human Serotonin Transporter. Nature 2016, 532, 334–339. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shahsavar, A.; Stohler, P.; Bourenkov, G.; Zimmermann, I.; Siegrist, M.; Guba, W.; Pinard, E.; Sinning, S.; Seeger, M.A.; Schneider, T.R.; et al. Structural Insights into the Inhibition of Glycine Reuptake. Nature 2021, 591, 677–681. [Google Scholar] [CrossRef] [PubMed]
- Colas, C. Toward a Systematic Structural and Functional Annotation of Solute Carriers Transporters-Example of the SLC6 and SLC7 Families. Front. Pharmacol. 2020, 11, 1229. [Google Scholar] [CrossRef] [PubMed]
- Krishnamurthy, H.; Gouaux, E. X-ray Structures of LeuT in Substrate-Free Outward-Open and Apo Inward-Open States. Nature 2012, 481, 469–474. [Google Scholar] [CrossRef] [Green Version]
- Rudnick, G. Cytoplasmic Permeation Pathway of Neurotransmitter Transporters. Biochemistry 2011, 50, 7462–7475. [Google Scholar] [CrossRef] [Green Version]
- Yamashita, A.; Singh, S.K.; Kawate, T.; Jin, Y.; Gouaux, E. Crystal Structure of a Bacterial Homologue of Na+/Cl−-Dependent Neurotransmitter Transporters. Nature 2005, 437, 215–223. [Google Scholar] [CrossRef]
- Cheng, J.Z.; Carvill, G.L. Pathogenic Mechanisms Underlying SLC6A1 Variant-Mediated Neurodevelopmental Disorders. Brain 2021, 144, 2237–2239. [Google Scholar] [CrossRef]
- Mattison, K.A.; Butler, K.M.; Inglis, G.A.S.; Dayan, O.; Boussidan, H.; Bhambhani, V.; Philbrook, B.; da Silva, C.; Alexander, J.J.; Kanner, B.I.; et al. SLC6A1 Variants Identified in Epilepsy Patients Reduce γ-Aminobutyric Acid Transport. Epilepsia 2018, 59, e135–e141. [Google Scholar] [CrossRef] [Green Version]
- Cai, K.; Wang, J.; Eissman, J.; Wang, J.; Nwosu, G.; Shen, W.; Liang, H.-C.; Li, X.-J.; Zhu, H.-X.; Yi, Y.-H.; et al. A Missense Mutation in SLC6A1 Associated with Lennox-Gastaut Syndrome Impairs GABA Transporter 1 Protein Trafficking and Function. Exp. Neurol. 2019, 320, 112973. [Google Scholar] [CrossRef]
- Wang, J.; Poliquin, S.; Mermer, F.; Eissman, J.; Delpire, E.; Wang, J.; Shen, W.; Cai, K.; Li, B.-M.; Li, Z.-Y.; et al. Endoplasmic Reticulum Retention and Degradation of a Mutation in SLC6A1 Associated with Epilepsy and Autism. Mol. Brain 2020, 13, 76. [Google Scholar] [CrossRef]
- Nwosu, G.; Mermer, F.; Flamm, C.; Poliquin, S.; Shen, W.; Rigsby, K.; Kang, J.Q. 4-Phenylbutyrate Restored γ-Aminobutyric Acid Uptake and Reduced Seizures in SLC6A1 Patient Variant-Bearing Cell and Mouse Models. Brain Commun. 2022, 4, fcac144. [Google Scholar] [CrossRef]
- Mermer, F.; Poliquin, S.; Zhou, S.; Wang, X.; Ding, Y.; Yin, F.; Shen, W.; Wang, J.; Rigsby, K.; Xu, D.; et al. Astrocytic GABA Transporter 1 Deficit in Novel SLC6A1 Variants Mediated Epilepsy: Connected from Protein Destabilization to Seizures in Mice and Humans. Neurobiol. Dis. 2022, 172, 105810. [Google Scholar] [CrossRef]
- Mermer, F.; Poliquin, S.; Rigsby, K.; Rastogi, A.; Shen, W.; Romero-Morales, A.; Nwosu, G.; McGrath, P.; Demerast, S.; Aoto, J.; et al. Common Molecular Mechanisms of SLC6A1 Variant-Mediated Neurodevelopmental Disorders in Astrocytes and Neurons. Brain 2021, 144, 2499–2512. [Google Scholar] [CrossRef] [PubMed]
- El-Kasaby, A.; Kasture, A.; Koban, F.; Hotka, M.; Asjad, H.M.M.; Kubista, H.; Freissmuth, M.; Sucic, S. Rescue by 4-Phenylbutyrate of Several Misfolded Creatine Transporter-1 Variants Linked to the Creatine Transporter Deficiency Syndrome. Neuropharmacology 2019, 161, 107572. [Google Scholar] [CrossRef] [PubMed]
- Fujiwara, M.; Yamamoto, H.; Miyagi, T.; Seki, T.; Tanaka, S.; Hide, I.; Sakai, N. Effects of the Chemical Chaperone 4-Phenylbutylate on the Function of the Serotonin Transporter (SERT) Expressed in COS-7 Cells. J. Pharmacol. Sci. 2013, 122, 71–83. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arribas-González, E.; de Juan-Sanz, J.; Aragón, C.; López-Corcuera, B. Molecular Basis of the Dominant Negative Effect of a Glycine Transporter 2 Mutation Associated with Hyperekplexia. J. Biol. Chem. 2015, 290, 2150–2165. [Google Scholar] [CrossRef] [Green Version]
- Bhat, S.; El-Kasaby, A.; Freissmuth, M.; Sucic, S. Functional and Biochemical Consequences of Disease Variants in Neurotransmitter Transporters: A Special Emphasis on Folding and Trafficking Deficits. Pharmacol. Ther. 2021, 222, 107785. [Google Scholar] [CrossRef]
- Kahen, A.; Kavus, H.; Geltzeiler, A.; Kentros, C.; Taylor, C.; Brooks, E.; Green Snyder, L.; Chung, W. Neurodevelopmental Phenotypes Associated with Pathogenic Variants in SLC6A1. J. Med. Genet. 2022, 59, 536–543. [Google Scholar] [CrossRef]
- Cai, G.; Salonikidis, P.S.; Fei, J.; Schwarz, W.; Schülein, R.; Reutter, W.; Fan, H. The Role of N-Glycosylation in the Stability, Trafficking and GABA-Uptake of GABA-Transporter 1. Terminal N-Glycans Facilitate Efficient GABA-Uptake Activity of the GABA Transporter. FEBS J. 2005, 272, 1625–1638. [Google Scholar] [CrossRef]
- Echave, J. Evolutionary Divergence of Protein Structure: The Linearly Forced Elastic Network Model. Chem. Phys. Lett. 2008, 457, 413–416. [Google Scholar] [CrossRef]
- Mendez, R.; Bastolla, U. Torsional Network Model: Normal Modes in Torsion Angle Space Better Correlate with Conformation Changes in Proteins. Phys. Rev. Lett. 2010, 104, 228103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alfayate, A.; Rodriguez Caceres, C.; Gomes Dos Santos, H.; Bastolla, U. Predicted Dynamical Couplings of Protein Residues Characterize Catalysis, Transport and Allostery. Bioinformatics 2019, 35, 4971–4978. [Google Scholar] [CrossRef]
- Goodspeed, K.; Pérez-Palma, E.; Iqbal, S.; Cooper, D.; Scimemi, A.; Johannesen, K.M.; Stefanski, A.; Demarest, S.; Helbig, K.L.; Kang, J.; et al. Current Knowledge of SLC6A1-Related Neurodevelopmental Disorders. Brain Commun. 2020, 2, fcaa170. [Google Scholar] [CrossRef]
- Dayan-Alon, O.; Kanner, B.I. Internal Gate Mutants of the GABA Transporter GAT1 Are Capable of Substrate Exchange. Neuropharmacology 2019, 161, 107534. [Google Scholar] [CrossRef] [PubMed]
- Ben-Yona, A.; Kanner, B.I. Functional Defects in the External and Internal Thin Gates of the γ-Aminobutyric Acid (GABA) Transporter GAT-1 Can Compensate Each Other. J. Biol. Chem. 2013, 288, 4549–4556. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kasture, A.; El-Kasaby, A.; Szöllősi, D.; Asjad, H.M.M.; Grimm, A.; Stockner, T.; Hummel, T.; Freissmuth, M.; Sucic, S. Functional Rescue of a Misfolded Drosophila Melanogaster Dopamine Transporter Mutant Associated with a Sleepless Phenotype by Pharmacological Chaperones. J. Biol. Chem. 2016, 291, 20876–20890. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rees, E.; Han, J.; Morgan, J.; Carrera, N.; Escott-Price, V.; Pocklington, A.J.; Duffield, M.; Hall, L.S.; Legge, S.E.; Pardiñas, A.F.; et al. De Novo Mutations Identified by Exome Sequencing Implicate Rare Missense Variants in SLC6A1 in Schizophrenia. Nat. Neurosci. 2020, 23, 179–184. [Google Scholar] [CrossRef] [PubMed]
- Islam, M.P.; Herman, G.E.; de los Reyes, E.C. Language Regression in an Atypical SLC6A1 Mutation. Semin. Pediatr. Neurol. 2018, 26, 25–27. [Google Scholar] [CrossRef]
- Devries, S.; Mulder, M.; Charron, J.G.; Prokop, J.W.; Mark, P.R. SLC6A1 G443D Associated with Developmental Delay and Epilepsy. Cold Spring Harb. Mol. Case Stud. 2020, 6, a005371. [Google Scholar] [CrossRef]
- Posar, A.; Visconti, P. Mild Phenotype Associated with SLC6A1 Gene Mutation: A Case Report with Literature Review. J. Pediatr. Neurosci. 2019, 14, 100–102. [Google Scholar] [CrossRef] [PubMed]
- Chollet, M.E.; Skarpen, E.; Iversen, N.; Sandset, P.M.; Skretting, G. The Chemical Chaperone Sodium 4-Phenylbutyrate Improves the Secretion of the Protein CA267T Mutant in CHO-K1 Cells Trough the GRASP55 Pathway. Cell Biosci. 2015, 5, 57. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lucariello, M.; Vidal, E.; Vidal, S.; Saez, M.; Roa, L.; Huertas, D.; Pineda, M.; Dalfó, E.; Dopazo, J.; Jurado, P.; et al. Whole Exome Sequencing of Rett Syndrome-like Patients Reveals the Mutational Diversity of the Clinical Phenotype. Hum. Genet. 2016, 135, 1343–1354. [Google Scholar] [CrossRef] [Green Version]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and Guidelines for the Interpretation of Sequence Variants: A Joint Consensus Recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef] [Green Version]
- Kopanos, C.; Tsiolkas, V.; Kouris, A.; Chapple, C.E.; Albarca Aguilera, M.; Meyer, R.; Massouras, A. VarSome: The Human Genomic Variant Search Engine. Bioinformatics 2019, 35, 1978–1980. [Google Scholar] [CrossRef] [Green Version]
- Ibáñez, I.; Bartolomé-Martín, D.; Piniella, D.; Giménez, C.; Zafra, F. Activity Dependent Internalization of the Glutamate Transporter GLT-1 Requires Calcium Entry through the NCX Sodium/Calcium Exchanger. Neurochem. Int. 2019, 123, 125–132. [Google Scholar] [CrossRef] [PubMed]
- Piniella, D.; Martínez-Blanco, E.; Ibáñez, I.; Bartolomé-Martín, D.; Porlan, E.; Díez-Guerra, J.; Giménez, C.; Zafra, F. Identification of Novel Regulatory Partners of the Glutamate Transporter GLT-1. Glia 2018, 66, 2737–2755. [Google Scholar] [CrossRef]
- Pandurangan, A.P.; Ochoa-Montaño, B.; Ascher, D.B.; Blundell, T.L. SDM: A Server for Predicting Effects of Mutations on Protein Stability. Nucleic Acids Res. 2017, 45, W229–W235. [Google Scholar] [CrossRef] [Green Version]
- Pires, D.E.V.; Rodrigues, C.H.M.; Ascher, D.B. MCSM-Membrane: Predicting the Effects of Mutations on Transmembrane Proteins. Nucleic Acids Res. 2020, 48, W147–W153. [Google Scholar] [CrossRef]
- Pires, D.E.V.; Ascher, D.B.; Blundell, T.L. DUET: A Server for Predicting Effects of Mutations on Protein Stability Using an Integrated Computational Approach. Nucleic Acids Res. 2014, 42, W314–W319. [Google Scholar] [CrossRef] [Green Version]
- Frappier, V.; Chartier, M.; Najmanovich, R.J. ENCoM Server: Exploring Protein Conformational Space and the Effect of Mutations on Protein Function and Stability. Nucleic Acids Res. 2015, 43, W395–W400. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodrigues, C.H.; Pires, D.E.; Ascher, D.B. DynaMut: Predicting the Impact of Mutations on Protein Conformation, Flexibility and Stability. Nucleic Acids Res. 2018, 46, W350–W355. [Google Scholar] [CrossRef] [PubMed]
- Rodrigues, C.H.M.; Pires, D.E.V.; Ascher, D.B. DynaMut2: Assessing Changes in Stability and Flexibility upon Single and Multiple Point Missense Mutations. Protein Sci. 2021, 30, 60–69. [Google Scholar] [CrossRef] [PubMed]
- Laimer, J.; Hiebl-Flach, J.; Lengauer, D.; Lackner, P. MAESTROweb: A Web Server for Structure-Based Protein Stability Prediction. Bioinformatics 2016, 32, 1414–1416. [Google Scholar] [CrossRef]
- Savojardo, C.; Fariselli, P.; Martelli, P.L.; Casadio, R. INPS-MD: A Web Server to Predict Stability of Protein Variants from Sequence and Structure. Bioinformatics 2016, 32, 2542–2544. [Google Scholar] [CrossRef] [PubMed]
- Dehouck, Y.; Kwasigroch, J.M.; Gilis, D.; Rooman, M. PoPMuSiC 2.1: A Web Server for the Estimation of Protein Stability Changes upon Mutation and Sequence Optimality. BMC Bioinform. 2011, 12, 151. [Google Scholar] [CrossRef] [PubMed]
- Bastolla, U. Detecting Selection on Protein Stability through Statistical Mechanical Models of Folding and Evolution. Biomolecules 2014, 4, 291–314. [Google Scholar] [CrossRef] [Green Version]
- Minning, J.; Porto, M.; Bastolla, U. Detecting Selection for Negative Design in Proteins through an Improved Model of the Misfolded State. Proteins 2013, 81, 1102–1112. [Google Scholar] [CrossRef] [Green Version]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A Visualization System for Exploratory Research and Analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [Green Version]
- Tirion, M.M. Large Amplitude Elastic Motions in Proteins from a Single-Parameter, Atomic Analysis. Phys. Rev. Lett. 1996, 77, 1905–1908. [Google Scholar] [CrossRef]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Patient no. | Gender/ Age at Inclusion | Family History | Cognition before Epilepsy Onset | Age at Epilepsy Onset | Seizure Type | EEG | Cognition after Seizure Onset | Behavioral Problems | Neurological Findings | Effective AED | Mutation |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | F/47 y | None | Moderate ID | 5 y | Absences atonic, eyelid myoclonia | Generalized spike and wave discharges at 3 Hz | Moderate ID | Autism Spectrum Disorders | Hypotonia | Sz free on VPA + CLZ | NM_003042.3: c.919G>A (NP_003033.3: p.Gly307Arg) de novo |
| 2 | M/2.5 y | None | Moderate ID | 15 m | Absences, eyelid myoclonia | Slow background rhythm, occipital spike-wave activity. | Moderate ID | Autism Spectrum Disorders | Hypotonia, Midline stereotypies | Sz free on VPA + CLZ | NM_003042.3: c.919G>A (NP_003033.3: p.Gly307Arg) de novo |
| Mutation | Membr Fraction (%) | Dynamut2 | Dynamut | ENCoM | mCSM | SDM | DUET | Maestro Web | Pop_ Music | INPS _3D | Delta GREM | -TNM _RMSD | -TNM_DE | Accessi-bility | Hum_Div | Hum_Var |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| V125M | 25 | −0.39 | 0.68 | −0.166 | −0.292 | −0.63 | −0.2 | −0.318 | −1.19 | −1.164 | −0.905 | −0.21 | −339 | 0.5 | Pr-Da | Pr-Da |
| A128V | 2 | −0.25 | −0.298 | −0.386 | −0.469 | −1.03 | −0.33 | −0.463 | −0.73 | −0.509 | −0.914 | −0.2 | −2200 | 0 | Pr-Da | Pr-Da |
| G232V | 20 | −0.58 | −0.67 | 0.071 | −0.057 | 0.09 | 0.311 | 0.154 | −0.76 | −1.391 | −0.121 | −0.37 | −480 | 40.23 | Pr-Da | Pr-Da |
| A288V | 10 | −0.51 | −0.092 | −0.581 | −0.169 | −1.03 | −0.007 | −0.515 | −0.59 | −1.424 | −0.381 | −0.22 | −823 | 0 | Pr-Da | Pr-Da |
| S295L | 2 | −0.65 | −0.023 | 0.073 | −0.286 | 2.07 | 0.542 | −0.55 | −0.67 | −0.703 | 0.04 | −0.11 | −195 | 15.76 | Pr-Da | Pr-Da |
| G297R | 0 | −0.19 | 0.069 | −0.744 | −0.545 | −3.1 | −0.865 | 0.0178 | −2.23 | −0.622 | −0.85 | −0.38 | −935 | 7.06 | Pr-Da | Pr-Da |
| G307R | 80 | −0.37 | −0.763 | −0.8 | −0.763 | −1.89 | −0.72 | −0.395 | −0.16 | −0.778 | −0.518 | −0.32 | −1460 | 1.91 | Ps-Da | Beni |
| A334P | 35 | 0.59 | 0.383 | −0.188 | 0.035 | −4.45 | −0.643 | −0.0535 | −1.87 | −1.971 | −2.444 | −0.37 | −2790 | 2.42 | Pr-Da | Pr-Da |
| P361T | 5 | −1.17 | −0.116 | −0.209 | −1.284 | 0.37 | −0.815 | −0.483 | −0.63 | −0.9492 | 0.5486 | −0.35 | −613 | 0.86 | Pr-Da | Pr-Da |
| G362R | 50 | −0.86 | −0.548 | −0.641 | −0.938 | −1.188 | −0.862 | 0.096 | −1.32 | −0.636 | 0.491 | −0.33 | −797 | 3.5 | Pr-Da | Ps-Da |
| D410E | 75 | −0.53 | 0.076 | −0.459 | −0.594 | 0.03 | −0.351 | −0.5 | −0.11 | −0.279 | −1.245 | −0.039 | −5.29 | 65.67 | Pr-Da | Ps-Da |
| L460R | 75 | −1.38 | −1.097 | 0.109 | −1.427 | −3.02 | −1.524 | 0.278 | −1.53 | −1.769 | 0 | −0.21 | −763 | 0.58 | Pr-Da | Pr-Da |
| G550R | 2 | −0.86 | 0.738 | −0.067 | −0.897 | 0.86 | −0.444 | −0.366 | −0.25 | −0.313 | 0.551 | −0.39 | −1320 | 41.07 | Ps-Da | Beni |
| Correlation with membrane fraction | 1 | −0.119 | −0.494 | −0.19 | −0.315 | −0.349 | −0.466 | 0.279 | 0.116 | −0.145 | −0.173 | 0.26 | 0.082 | 0.134 | n.a. | n.a. |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Piniella, D.; Canseco, A.; Vidal, S.; Xiol, C.; Díaz de Bustamante, A.; Martí-Carrera, I.; Armstrong, J.; Bastolla, U.; Zafra, F. Experimental and Bioinformatic Insights into the Effects of Epileptogenic Variants on the Function and Trafficking of the GABA Transporter GAT-1. Int. J. Mol. Sci. 2023, 24, 955. https://doi.org/10.3390/ijms24020955
Piniella D, Canseco A, Vidal S, Xiol C, Díaz de Bustamante A, Martí-Carrera I, Armstrong J, Bastolla U, Zafra F. Experimental and Bioinformatic Insights into the Effects of Epileptogenic Variants on the Function and Trafficking of the GABA Transporter GAT-1. International Journal of Molecular Sciences. 2023; 24(2):955. https://doi.org/10.3390/ijms24020955
Chicago/Turabian StylePiniella, Dolores, Ania Canseco, Silvia Vidal, Clara Xiol, Aránzazu Díaz de Bustamante, Itxaso Martí-Carrera, Judith Armstrong, Ugo Bastolla, and Francisco Zafra. 2023. "Experimental and Bioinformatic Insights into the Effects of Epileptogenic Variants on the Function and Trafficking of the GABA Transporter GAT-1" International Journal of Molecular Sciences 24, no. 2: 955. https://doi.org/10.3390/ijms24020955