
Synthesis and Antibacterial Evaluation of Ciprofloxacin Congeners with Spirocyclic Amine Periphery
 and
and
Abstract
:1. Introduction
2. Results and Discussion
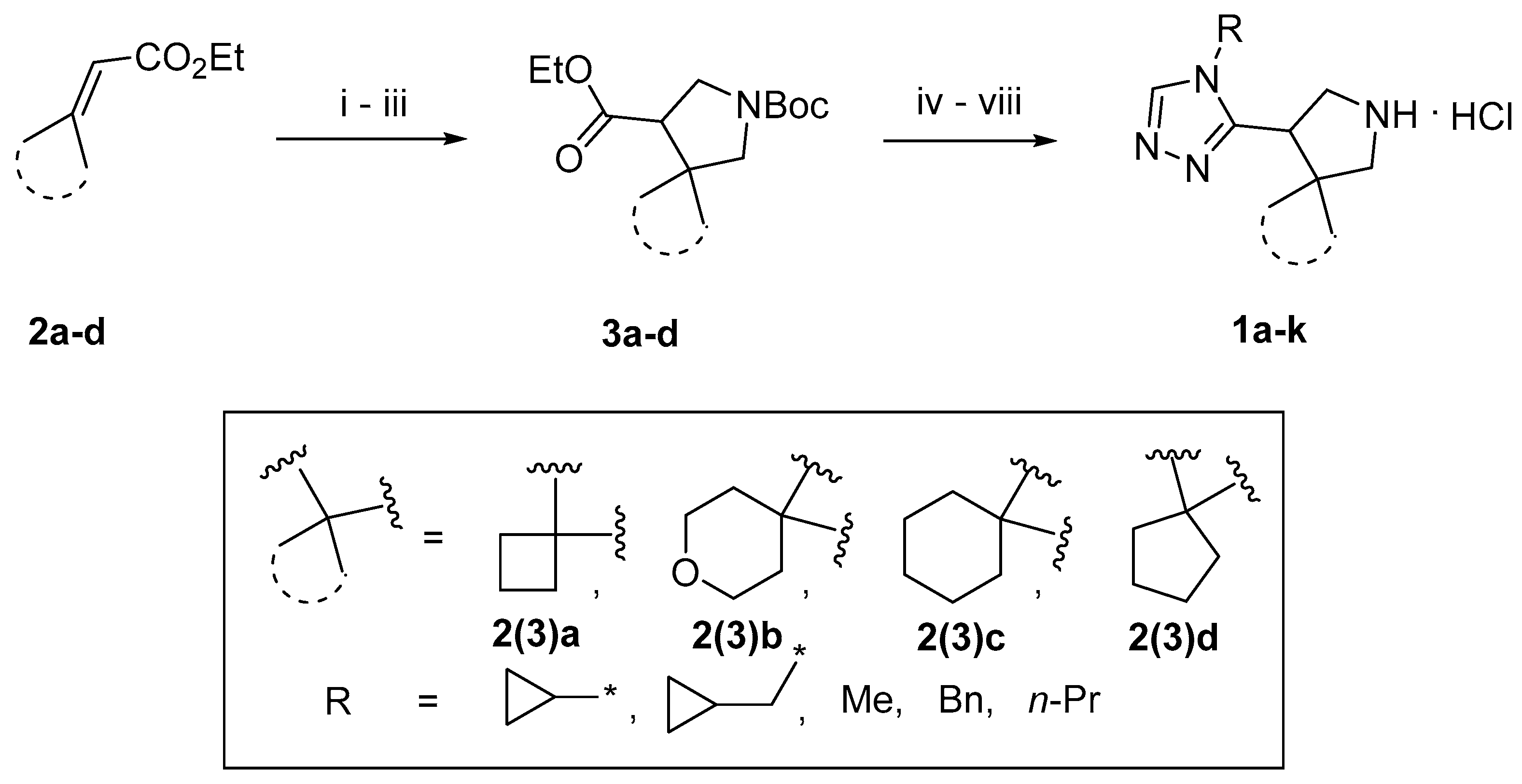
2.1. Compound Synthesis
2.2. Establishment of the Antibacterial Profile of Compounds 6a–k
3. Materials and Methods
3.1. Compound Synthesis
3.1.1. General
3.1.2. 6-tert-Butyl 8-ethyl 6-azaspiro[3.4]octane-6,8-dicarboxylate (3a)
3.1.3. 2-tert-Butyl 4-ethyl 8-oxa-2-azaspiro[4.5]decane-2,4-dicarboxylate (3b)
3.1.4. 2-tert-Butyl 4-ethyl 2-azaspiro[4.4]decane-2,4-dicarboxylate (3c)
3.1.5. 2-tert-Butyl 4-ethyl 2-azaspiro[4.4]nonane-2,4-dicarboxylate (3d)
3.1.6. 8-(4-Cyclopropyl-4H-1,2,4-triazol-3-yl)-6-azaspiro[3.4]octane hydrochloride (1a)
3.1.7. 8-(4-Propyl-4H-1,2,4-triazol-3-yl)-6-azaspiro[3.4]octane hydrochloride (1b)
3.1.8. 8-[4-(Cyclopropylmethyl)-4H-1,2,4-triazol-3-yl]-6-azaspiro[3.4]octane hydrochloride (1c)
3.1.9. 4-(4-Methyl-4H-1,2,4-triazol-3-yl)-2-azaspiro[4.4]nonane hydrochloride (1d)
3.1.10. 4-(4-Cyclopropyl-4H-1,2,4-triazol-3-yl)-2-azaspiro[4.4]nonane hydrochloride (1e)
3.1.11. 4-(4-Methyl-4H-1,2,4-triazol-3-yl)-2-azaspiro[4.5]decane hydrochloride (1f)
3.1.12. 4-[4-(Cyclopropylmethyl)-4H-1,2,4-triazol-3-yl]-2-azaspiro[4.5]decane hydrochloride (1g)
3.1.13. 4-(4-Benzyl-4H-1,2,4-triazol-3-yl)-2-azaspiro[4.5]decane hydrochloride (1h)
3.1.14. 4-(4-Methyl-4H-1,2,4-triazol-3-yl)-8-oxa-2-azaspiro[4.5]decane hydrochloride (1i)
3.1.15. 4-(4-Cyclopropyl-4H-1,2,4-triazol-3-yl)-8-oxa-2-azaspiro[4.5]decane hydrochloride (1j)
3.1.16. 4-[4-(Cyclopropylmethyl)-4H-1,2,4-triazol-3-yl]-8-oxa-2-azaspiro[4.5]decane hydrochloride (1k)
3.1.17. 1-Cyclopropyl-7-[8-(4-cyclopropyl-4H-1,2,4-triazol-3-yl)-6-azaspiro[3.4]oct-6-yl]-6-fluoro-4-oxo-1,4-dihydroquinoline-3-carboxylic acid (6a)
3.1.18. 1-Cyclopropyl-6-fluoro-4-oxo-7-[8-(4-propyl-4H-1,2,4-triazol-3-yl)-6-azaspiro[3.4]oct-6-yl]-1,4-dihydroquinoline-3-carboxylic acid (6b)
3.1.19. 1-Cyclopropyl-7-{8-[4-(cyclopropylmethyl)-4H-1,2,4-triazol-3-yl]-6-azaspiro[3.4]oct-6-yl}-6-fluoro-4-oxo-1,4-dihydroquinoline-3-carboxylic acid (6c)
3.1.20. 1-Cyclopropyl-6-fluoro-7-[4-(4-methyl-4H-1,2,4-triazol-3-yl)-2-azaspiro[4.4]non-2-yl]-4-oxo-1,4-dihydroquinoline-3-carboxylic acid (6d)
3.1.21. 1-Cyclopropyl-7-[4-(4-cyclopropyl-4H-1,2,4-triazol-3-yl)-2-azaspiro[4.4]non-2-yl]-6-fluoro-4-oxo-1,4-dihydroquinoline-3-carboxylic acid (6e)
3.1.22. 1-Cyclopropyl-6-fluoro-7-[4-(4-methyl-4H-1,2,4-triazol-3-yl)-2-azaspiro[4.5]dec-2-yl]-4-oxo-1,4-dihydroquinoline-3-carboxylic acid (6f)
3.1.23. 1-Cyclopropyl-7-{4-[4-(cyclopropylmethyl)-4H-1,2,4-triazol-3-yl]-2-azaspiro[4.5]dec-2-yl}-6-fluoro-4-oxo-1,4-dihydroquinoline-3-carboxylic acid (6g)
3.1.24. 7-[4-(4-Benzyl-4H-1,2,4-triazol-3-yl)-2-azaspiro[4.5]dec-2-yl]-1-cyclopropyl-6-fluoro-4-oxo-1,4-dihydroquinoline-3-carboxylic acid (6h)
3.1.25. 1-Cyclopropyl-6-fluoro-7-[4-(4-methyl-4H-1,2,4-triazol-3-yl)-8-oxa-2-azaspiro[4.5]dec-2-yl]-4-oxo-1,4-dihydroquinoline-3-carboxylic acid (6i)
3.1.26. 1-Cyclopropyl-7-[4-(4-cyclopropyl-4H-1,2,4-triazol-3-yl)-8-oxa-2-azaspiro[4.5]dec-2-yl]-6-fluoro-4-oxo-1,4-dihydroquinoline-3-carboxylic acid (6j)
3.1.27. 1-Cyclopropyl-7-{4-[4-(cyclopropylmethyl)-4H-1,2,4-triazol-3-yl]-8-oxa-2-azaspiro[4.5]dec-2-yl}-6-fluoro-4-oxo-1,4-dihydroquinoline-3-carboxylic acid (6k)
3.2. Biology Studies
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Zhanel, G.G.; Walkty, A.; Vercaigne, L.; Karlowsky, J.A.; Embil, J.; Gin, A.S.; Hoban, D.J. The new fluoroquinolones: A critical review. Can. J. Infect. Dis. 1999, 10, 207–238. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mitscher, L.A. Bacterial topoisomerase inhibitors: Quinolone and pyridone antibacterial agents. Chem. Rev. 2005, 105, 559–592. [Google Scholar] [CrossRef] [PubMed]
- Aldred, K.J.; Schwanz, H.A.; Li, G.; McPherson, S.A.; Turnbough, C.L.; Kerns, R.J.; Osheroff, N. Overcoming target-mediated quinolone resistance in topoisomerase IV by introducing metal-ion independent drug–enzyme interactions. ACS Chem. Biol. 2013, 12, 2660–2668. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dalhoff, A.; Schubert, S. The impact of protein binding on antibacterial activities of antibiotics is more than predicted by considering its numerical value alone: Impact of preparative and incubation methods on different pharmacodynamic endpoints of b-lactams, macrolides, or fluoroquinolones against gram-positive and gram-negative bacteria-Part I. J. Clin. Infect. Dis. Pract. 2016, 1, 110. [Google Scholar]
- Pranger, A.D.; van der Werf, T.S.; Kosterink, J.G.W.; Alffenaar, J.W.C. The Role of Fluoroquinolones in the Treatment of Tuberculosis in 2019. Drugs 2019, 79, 161–171. [Google Scholar] [CrossRef] [Green Version]
- Assar, S.; Nosratabadi, R.; Khorramdel Azad, H.; Masoumi, J.; Mohamadi, M.; Hassanshahi, G. A Review of Immunomodulatory Effects of Fluoroquinolones. Immunol. Invest. 2021, 50, 1007–1026. [Google Scholar] [CrossRef]
- Idowu, T.; Schweizer, F. Ubiquitous Nature of Fluoroquinolones: The Oscillation between Antibacterial and Anticancer Activities. Antibiotics 2017, 6, 26. [Google Scholar] [CrossRef] [Green Version]
- Sousa, J.; Alves, G.; Fortuna, A.; Falcão, A. Third and fourth generation fluoroquinolone antibacterials: A systematic review of safety and toxicity profiles. Curr. Drug Saf. 2014, 2, 89–105. [Google Scholar] [CrossRef]
- Federowicz, J.; Saczewski, J. Modifications of quinolones and fluoroquinolones: Hybrid compounds and dual-action molecules. Monatsh. Chem. 2018, 149, 1199–1245. [Google Scholar] [CrossRef] [Green Version]
- Naidoo, A.; Naidoo, K.; McIlleron, H.; Essack, S.; Padayatchi, N. A review of moxifloxacin for the treatment of drug-susceptible tuberculosis. J. Clin. Pharmacol. 2017, 57, 1369–1386. [Google Scholar] [CrossRef]
- Sharma, P.C.; Jain, A.; Jain, S.; Pahwa, R.; Yar, M.S. Ciprofloxacin: Review on developments in synthetic, analytical, and medicinal aspects. J. Enzyme Inhib. Med. Chem. 2010, 25, 577–589. [Google Scholar] [CrossRef] [PubMed]
- Schentag, J.J. Sparfloxacin: A review. Clin. Ther. 2000, 22, 372–387. [Google Scholar] [CrossRef] [PubMed]
- Totoli, E.G.; Saldago, H.R.N. Besifloxacin: A Critical Review of Its Characteristics, Properties, and Analytical Methods. Crit. Rev. Anal. Chem. 2018, 48, 132–142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sweeney, M.T.; Quesnell, R.; Tiwari, R.; LeMay, M.; Watts, J.L. In vitro activity and rodent efficacy of clinafloxacin for bovine and swine respiratory disease. Front. Microbiol. 2013, 4, 154. [Google Scholar] [CrossRef] [Green Version]
- McKeage, K. Finafloxacin: First global approval. Drugs 2015, 75, 687–693. [Google Scholar] [CrossRef]
- Goulart, D.B.; Beyi, A.F.; Wu, Z.; Adiguzel, M.C.; Schroeder, A.; Singh, K.; Xu, C.; Ocal, M.M.; Dewell, R.; Dewell, G.A.; et al. Effect of Danofloxacin Treatment on the Development of Fluoroquinolone Resistance in Campylobacter jejuni in Calves. Antibiotics 2022, 11, 531. [Google Scholar] [CrossRef]
- Markham, A. Delafloxacin: First Global Approval. Drugs 2017, 77, 1481–1486. [Google Scholar] [CrossRef] [Green Version]
- Lukin, A.; Chudinov, M.; Rogacheva, E.; Kraeva, L.; Bakulina, O.; Krasavin, M. Exploration of spirocyclic derivatives of ciprofloxacin as antibacterial agents. Molecules 2022, 27, 4864. [Google Scholar] [CrossRef]
- Hiesinger, K.; Dar’in, D.; Proschak, E.; Krasavin, M. Spirocyclic Scaffolds in Medicinal Chemistry. J. Med. Chem. 2021, 64, 150–183. [Google Scholar] [CrossRef]
- Lovering, F.; Bikker, J.; Humblet, C. Escape from Flatland: Increasing Saturation as an Approach to Improving Clinical Success. J. Med. Chem. 2009, 52, 6752–6756. [Google Scholar] [CrossRef]
- Tratrat, C. 1,2,4-Triazole: A Privileged Scaffold for the Development of Potent Antifungal Agents—A Brief Review. Curr. Top. Med. Chem. 2020, 20, 2235–2258. [Google Scholar] [CrossRef] [PubMed]
- Mulani, M.S.; Kamble, E.E.; Kumkar, S.N.; Tawre, M.S.; Pardesi, K.R. Emerging Strategies to Combat ESKAPE Pathogens in the Era of Antimicrobial Resistance: A Review. Front. Microbiol. 2019, 10, 539. [Google Scholar] [CrossRef] [PubMed]
- Wiberg, K.B.; Hiatt, J.E. Solvolysis of 2-(1-cyclobuten-1-yl)ethyl tosylate. J. Am. Chem. Soc. 1968, 90, 6495–6500. [Google Scholar] [CrossRef]
- Koy, M.; Bellotti, P.; Katzenburg, F.; Daniliuc, C.G.; Glorius, F. Synthesis of All-Carbon Quaternary Centers by Palladium-Catalyzed Olefin Dicarbofunctionalization. Angew. Chem. Int. Ed. 2020, 59, 2375–2379. [Google Scholar] [CrossRef]
- Comito, R.J.; Finelli, F.G.; MacMillan, D.W.C. Enantioselective Intramolecular Aldehyde α-Alkylation with Simple Olefins: Direct Access to Homo-Ene Products. J. Am. Chem. Soc. 2013, 135, 9358–9361. [Google Scholar] [CrossRef] [Green Version]
- Cavero, M.; Motherwell, W.B.; Potier, P.; Weibel, J.-M. Thioepoxide formation by ring closure of allylthiyl radicals—A novel rearrangement of allylic thionitrites. Chem. Commun. 2002, 20, 2394–2395. [Google Scholar] [CrossRef]
- Bauer, A.W.; Kirby, W.M.; Sherris, J.C.; Turck, M. Antibiotic Susceptibility Testing by a Standardized Single Disk Method. Am. J. Clin. Pathol. 1966, 45, 493–496. [Google Scholar] [CrossRef]
- EUCAST SOP 9.2; Procedure for Establishing Zone Diameter Breakpoints and Quality Control Criteria. EUCAST: Växjö, Sweden, 2020. Available online: https://www.eucast.org/fileadmin/src/media/PDFs/EUCAST_files/EUCAST_SOPs/2020/EUCAST_SOP_9.2_Disk_diffusion_breakpoints_and_QC_ranges_final.pdf (accessed on 15 December 2022).
- Wiegand, I.; Hilpert, K.; Hancock, R.E. Agar and broth dilution methods to determine the minimal inhibitory concentration (MIC) of antimicrobial substances. Nat. Protoc. 2008, 3, 163–175. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Structure | MIC (μg/mL) | |||||
|---|---|---|---|---|---|---|---|
| E1 | S | K | A | P | E2 | ||
| 6a |  | 3.0 | 1.5 | 2.5 | 6.0 | 750 | 3.0 |
| 6b |  | 6.0 | 1.5 | >750 | 100 | >750 | 6.0 |
| 6c |  | 3.0 | 1.5 | 12.0 | 12.0 | 750 | 3.0 |
| 6d |  | 3.0 | 1.5 | 190 | 12.0 | >750 | 1.5 |
| 6e |  | 3.0 | 1.5 | 190 | 6.0 | 750 | 1.5 |
| 6f |  | NT | NT | NT | NT | NT | NT |
| 6g |  | NT | NT | NT | NT | NT | NT |
| 6h |  | NT | NT | NT | NT | NT | NT |
| 6i |  | NT | NT | NT | NT | NT | NT |
| 6j |  | NT | NT | NT | NT | NT | NT |
| 6k |  | NT | NT | NT | NT | NT | NT |
| 3w [18] |  | NT | inactive | inactive | 0.15 | inactive | NT |
| ciprofloxacin |  | 1.25 | 1.25 | 0.6 | 2.5 | 0.6 | 0.3 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lukin, A.; Komarova, K.; Vinogradova, L.; Rogacheva, E.; Kraeva, L.; Krasavin, M. Synthesis and Antibacterial Evaluation of Ciprofloxacin Congeners with Spirocyclic Amine Periphery. Int. J. Mol. Sci. 2023, 24, 954. https://doi.org/10.3390/ijms24020954
Lukin A, Komarova K, Vinogradova L, Rogacheva E, Kraeva L, Krasavin M. Synthesis and Antibacterial Evaluation of Ciprofloxacin Congeners with Spirocyclic Amine Periphery. International Journal of Molecular Sciences. 2023; 24(2):954. https://doi.org/10.3390/ijms24020954
Chicago/Turabian StyleLukin, Alexei, Kristina Komarova, Lyubov Vinogradova, Elizaveta Rogacheva, Lyudmila Kraeva, and Mikhail Krasavin. 2023. "Synthesis and Antibacterial Evaluation of Ciprofloxacin Congeners with Spirocyclic Amine Periphery" International Journal of Molecular Sciences 24, no. 2: 954. https://doi.org/10.3390/ijms24020954