
Alarmins and MicroRNAs, a New Axis in the Genesis of Respiratory Diseases: Possible Therapeutic Implications

 , , ,
, , ,
Abstract
:1. Introduction
1.1. Chronic Respiratory Diseases
1.2. General Considerations Regarding High Mobility Group Box 1, Interleukin-33 and microRNAs
1.2.1. High Mobility Group Box 1
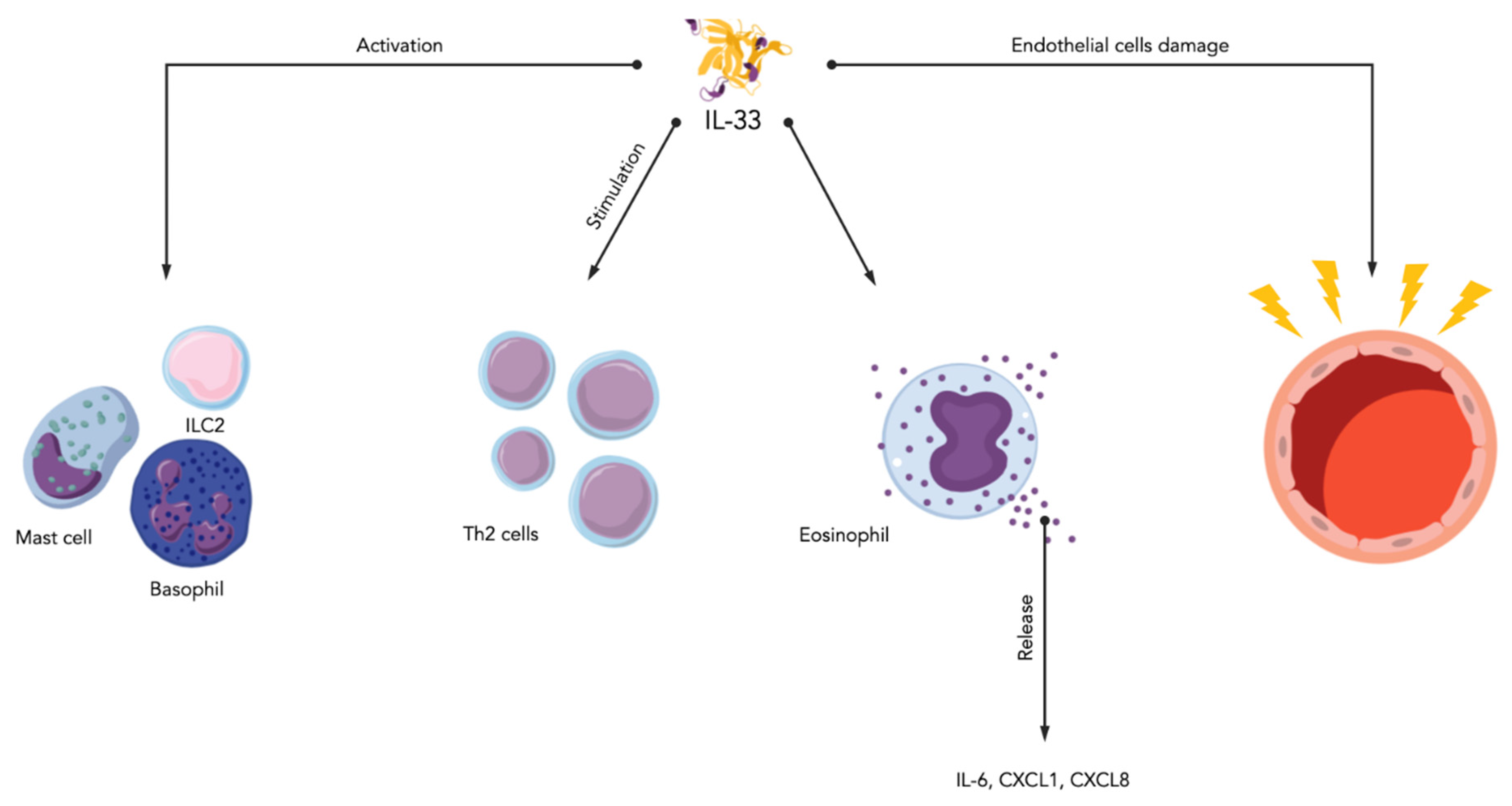
1.2.2. Interleukin-33
1.2.3. microRNAs
2. Alarmins and Chronic Respiratory Diseases
2.1. Asthma and Alarmins
2.2. Alarmins and Respiratory Syncytial Virus Infection
2.3. Alarmins and Cystic Fibrosis
3. MicroRNAs and Chronic Respiratory Diseases
3.1. MicroRNAs and Asthma
3.2. MicroRNAs and Acute Respiratory Distress Syndrome
4. Exosomal MicroRNA, Circular RNA, and Long Non-Coding RNA in Respiratory Diseases
5. Discussion
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- GBD Chronic Respiratory Disease Collaborators. Prevalence and attributable health burden of chronic respiratory diseases, 1990-2017: A systematic analysis for the Global Burden of Disease Study 2017. Lancet Respir. Med. 2020, 8, 585–596. [Google Scholar] [CrossRef] [PubMed]
- Asher, M.I.; García-Marcos, L.; Pearce, N.E.; Strachan, D.P. Trends in worldwide asthma prevalence. Eur. Respir. J. 2020, 56, 2002094. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Xu, J.; Yang, L.; Xu, Y.; Zhang, X.; Bai, C.; Kang, J.; Ran, P.; Shen, H.; Wen, F.; et al. China Pulmonary Health Study Group. Prevalence and risk factors of chronic obstructive pulmonary disease in China (the China Pulmonary Health [CPH] study): A national cross-sectional study. Lancet 2018, 391, 1706–1717. [Google Scholar] [CrossRef] [PubMed]
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA A Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef]
- Lynch, D.A.; Sverzellati, N.; Travis, W.D.; Brown, K.K.; Colby, T.V.; Galvin, J.R.; Goldin, J.G.; Hansell, D.M.; Inoue, Y.; Johkoh, T.; et al. Diagnostic criteria for idiopathic pulmonary fibrosis: A Fleischner Society White Paper. Lancet Respir. Med. 2018, 6, 138–153. [Google Scholar] [CrossRef]
- Wei, L.; Zhang, W.; Li, Y.; Zhai, J. The SIRT1-HMGB1 axis: Therapeutic potential to ameliorate inflammatory responses and tumor occurrence. Front. Cell Dev. Biol. 2022, 10, 986511. [Google Scholar] [CrossRef]
- Lotze, M.T.; Tracey, K.J. High-mobility group box 1 protein (HMGB1): Nuclear weapon in the immune arsenal. Nat. Rev. Immunol. 2005, 5, 331–342. [Google Scholar] [CrossRef]
- Lee, S.A.; Kwak, M.S.; Kim, S.; Shin, J.S. The role of high mobility group box 1 in innate immunity. Yonsei Med. J. 2014, 55, 1165–1176. [Google Scholar] [CrossRef] [Green Version]
- Murdaca, G.; Allegra, A.; Paladin, F.; Calapai, F.; Musolino, C.; Gangemi, S. Involvement of Alarmins in the Pathogenesis and Progression of Multiple Myeloma. Int. J. Mol. Sci. 2021, 22, 9039. [Google Scholar] [CrossRef]
- Li Pomi, F.; Borgia, F.; Custurone, P.; Vaccaro, M.; Pioggia, G.; Gangemi, S. Role of HMGB1 in Cutaneous Melanoma: State of the Art. Int. J. Mol. Sci. 2022, 23, 9327. [Google Scholar] [CrossRef]
- Tang, D.; Kang, R.; Livesey, K.M.; Cheh, C.W.; Farkas, A.; Loughran, P.; Hoppe, G.; Bianchi, M.E.; Tracey, K.J.; Zeh, H.J.; et al. Endogenous HMGB1 regulates autophagy. J. Cell Biol. 2010, 190, 881–892. [Google Scholar] [CrossRef] [PubMed]
- Ullah, M.A.; Loh, Z.; Gan, W.J.; Zhang, V.; Yang, H.; Li, J.H.; Yamamoto, Y.; Schmidt, A.M.; Armour, C.L.; Hughes, J.M.; et al. Receptor for advanced glycation end products and its ligand high-mobility group box-1 mediate allergic airway sensitization and airway inflammation. J. Allergy Clin. Immunol. 2014, 134, 440–450. [Google Scholar] [CrossRef] [PubMed]
- Gardella, S.; Andrei, C.; Ferrera, D.; Lotti, L.V.; Torrisi, M.R.; Bianchi, M.E.; Rubartelli, A. The nuclear protein HMGB1 is secreted by monocytes via a non-classical, vesicle-mediated secretory pathway. EMBO Rep. 2002, 3, 995–1001. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, J.S.; Gamboni-Robertson, F.; He, Q.; Svetkauskaite, D.; Kim, J.Y.; Strassheim, D.; Sohn, J.W.; Yamada, S.; Maruyama, I.; Banerjee, A.; et al. High mobility group box 1 protein interacts with multiple Toll-like receptors. Am. J. Physiol. Cell Physiol. 2006, 290, C917–C924. [Google Scholar] [CrossRef] [PubMed]
- Treutiger, C.J.; Mullins, G.E.; Johansson, A.S.; Rouhiainen, A.; Rauvala, H.M.; Erlandsson-Harris, H.; Andersson, U.; Yang, H.; Tracey, K.J.; Andersson, J.; et al. High mobility group 1 B-box mediates activation of human endothelium. J. Intern. Med. 2003, 254, 375–385. [Google Scholar] [CrossRef]
- Huebener, P.; Pradere, J.P.; Hernandez, C.; Gwak, G.Y.; Caviglia, J.M.; Mu, X.; Loike, J.D.; Jenkins, R.E.; Antoine, D.J.; Schwabe, R.F. The HMGB1/RAGE axis triggers neutrophil-mediated injury amplification following necrosis. J. Clin. Investig. 2019, 130, 1802. [Google Scholar] [CrossRef] [Green Version]
- Taverna, S.; Tonacci, A.; Ferraro, M.; Cammarata, G.; Cuttitta, G.; Bucchieri, S.; Pace, E.; Gangemi, S. High Mobility Group Box 1: Biological Functions and Relevance in Oxidative Stress Related Chronic Diseases. Cells 2022, 11, 849. [Google Scholar] [CrossRef]
- Stark, K.; Philippi, V.; Stockhausen, S.; Busse, J.; Antonelli, A.; Miller, M.; Schubert, I.; Hoseinpour, P.; Chandraratne, S.; von Brühl, M.L.; et al. Disulfide HMGB1 derived from platelets coordinates venous thrombosis in mice. Blood 2016, 128, 2435–2449. [Google Scholar] [CrossRef]
- Yu, Y.; Tang, D.; Kang, R. Oxidative stress-mediated HMGB1 biology. Front. Physiol. 2015, 6, 93. [Google Scholar] [CrossRef] [Green Version]
- Maugeri, N.; Franchini, S.; Campana, L.; Baldini, M.; Ramirez, G.A.; Sabbadini, M.G.; Rovere-Querini, P.; Manfredi, A.A. Circulating platelets as a source of the damage-associated molecular pattern HMGB1 in patients with systemic sclerosis. Autoimmunity 2012, 45, 584–587. [Google Scholar] [CrossRef]
- Ahrens, I.; Chen, Y.C.; Topcic, D.; Bode, M.; Haenel, D.; Hagemeyer, C.E.; Seeba, H.; Duerschmied, D.; Bassler, N.; Jandeleit-Dahm, K.A.; et al. HMGB1 binds to activated platelets via the receptor for advanced glycation end products and is present in platelet rich human coronary artery thrombi. Thromb. Haemost. 2015, 114, 994–1003. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carriere, V.; Roussel, L.; Ortega, N.; Lacorre, D.A.; Americh, L.; Aguilar, L.; Bouche, G.; Girard, J.P. IL-33, the IL-1-like cytokine ligand for ST2 receptor, is a chromatin-associated nuclear factor in vivo. Proc. Natl. Acad. Sci. USA 2007, 104, 282–287. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liew, F.Y.; Girard, J.P.; Turnquist, H.R. Interleukin-33 in health and disease. Nat. Rev. Immunol. 2016, 16, 676–689. [Google Scholar] [CrossRef] [PubMed]
- Qi, F.; Bai, S.; Wang, D.; Xu, L.; Hu, H.; Zeng, S.; Chai, R.; Liu, B. Macrophages produce IL-33 by activating MAPK signaling pathway during RSV infection. Mol. Immunol. 2017, 87, 284–292. [Google Scholar] [CrossRef]
- Murdaca, G.; Greco, M.; Tonacci, A.; Negrini, S.; Borro, M.; Puppo, F.; Gangemi, S. IL-33/IL-31 Axis in Immune-Mediated and Allergic Diseases. Int. J. Mol. Sci. 2019, 20, 5856. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Salvo, E.; Casciaro, M.; Gangemi, S. IL-33 genetics and epigenetics in immune-related diseases. Clin. Mol. Allergy 2021, 19, 18. [Google Scholar] [CrossRef]
- Yeoh, W.J.; Vu, V.P.; Krebs, P. IL-33 biology in cancer: An update and future perspectives. Cytokine 2022, 157, 155961. [Google Scholar] [CrossRef]
- Musolino, C.; Allegra, A.; Profita, M.; Alonci, A.; Saitta, S.; Russo, S.; Bonanno, A.; Innao, V.; Gangemi, S. Reduced IL-33 plasma levels in multiple myeloma patients are associated with more advanced stage of disease. Br. J. Haematol. 2013, 160, 709–710. [Google Scholar] [CrossRef]
- Allegra, A.; Alonci, A.; Campo, S.; Penna, G.; Petrungaro, A.; Gerace, D.; Musolino, C. Circulating microRNAs: New biomarkers in diagnosis, prognosis and treatment of cancer (review). Int. J. Oncol. 2012, 41, 1897–1912. [Google Scholar] [CrossRef] [Green Version]
- Musolino, C.; Oteri, G.; Allegra, A.; Mania, M.; D’Ascola, A.; Avenoso, A.; Innao, V.; Allegra, A.G.; Campo, S. Altered microRNA expression profile in the peripheral lymphoid compartment of multiple myeloma patients with bisphosphonate-induced osteonecrosis of the jaw. Ann. Hematol. 2018, 97, 1259–1269. [Google Scholar] [CrossRef]
- Mott, J.L.; Mohr, A.M. Overview of MicroRNA biology. Semin. Liver Dis. 2015, 35, 3–11. [Google Scholar]
- Chevillet, J.R.; Lee, I.; Briggs, H.A.; He, Y.; Wang, K. Issues and prospects of microRNA-based biomarkers in blood and other body fluids. Molecules 2014, 19, 6080–6105. [Google Scholar] [CrossRef] [PubMed]
- Tomasello, L.; Distefano, R.; Nigita, G.; Croce, C.M. The MicroRNA family gets wider: The IsomiRs classifcation and role. Front. Cell Dev. Biol. 2021, 9, 668648. [Google Scholar] [CrossRef] [PubMed]
- O’Brien, J.; Hayder, H.; Zayed, Y.; Peng, C. Overview of MicroRNA biogenesis, mechanisms of actions, and circulation. Front. Endocrinol. 2018, 9, 402. [Google Scholar] [CrossRef] [Green Version]
- Papi, A.; Brightling, C.; Pedersen, S.E.; Reddel, H.K. Asthma. Lancet 2018, 391, 783–800. [Google Scholar] [CrossRef]
- Bloom, C.; de Preux, L.; Sheikh, A.; Quint, J. Health and cost impact of stepping down asthma medication for UK patients, 2001–2017: A population-based observational study. PLoS Med. 2020, 17, e1003145. [Google Scholar] [CrossRef]
- Hoshi, M.; Matsunaga, M.; Nogami, K.; Hamada, K.; Kobori, T.; Kainuma, K.; Nagao, M.; Fujisawa, T. Three cases of severe adolescent asthma treated with mepolizumab: Lung function trajectories. Asia Pac. Allergy 2020, 10, e13. [Google Scholar] [CrossRef] [Green Version]
- Fahy, J.V. Type 2 inflammation in asthma–present in most, absent in many. Nat. Rev. Immunol. 2015, 15, 57–65. [Google Scholar] [CrossRef] [Green Version]
- Kubo, M. Innate and adaptive type 2 immunity in lung allergic inflammation. Immunol. Rev. 2017, 278, 162–172. [Google Scholar] [CrossRef]
- Kim, H.Y.; De Kruyff, R.H.; Umetsu, D.T. The many paths to asthma: Phenotype shaped by innate and adaptive immunity. Nat. Immunol. 2010, 11, 577–584. [Google Scholar] [CrossRef] [Green Version]
- Pulendran, B.; Artis, D. New paradigms in type 2 immunity. Science 2012, 337, 431–435. [Google Scholar] [CrossRef] [PubMed]
- Halim, T.Y.F.; McKenzie, A.N.J. New kids on the block: Group 2 innate lymphoid cells and type 2 inflammation in the lung. Chest 2013, 144, 1681–1686. [Google Scholar] [CrossRef] [PubMed]
- Neill, D.R.; Wong, S.H.; Bellosi, A.; Flynn, R.J.; Daly, M.; Langford, T.K.; Bucks, C.; Kane, C.M.; Fallon, P.G.; Pannell, R.; et al. Nuocytes represent a new innate effector leukocyte that mediates type-2 immunity. Nature 2010, 464, 1367–1370. [Google Scholar] [CrossRef] [Green Version]
- Price, A.E.; Liang, H.E.; Sullivan, B.M.; Reinhardt, R.L.; Eisley, C.J.; Erle, D.J.; Locksley, R.M. Systemically dispersed innate IL-13-expressing cells in type 2 immunity. Proc. Natl. Acad. Sci. USA 2010, 107, 11489–11494. [Google Scholar] [CrossRef] [Green Version]
- Fallon, P.G.; Ballantyne, S.J.; Mangan, N.E.; Barlow, J.L.; Dasvarma, A.; Hewett, D.R.; McIlgorm, A.; Jolin, H.E.; McKenzie, A.N. Identification of an interleukin (IL)-25-dependent cell population that provides IL-4, IL-5, and IL-13 at the onset of helminth expulsion. J. Exp. Med. 2006, 203, 1105–1116. [Google Scholar] [CrossRef] [PubMed]
- Herbert, D.R.; Douglas, B.; Zullo, K. Group 2 Innate Lymphoid Cells (ILC2): Type 2 immunity and Helminth immunity. Int. J. Mol. Sci. 2019, 20, 2276. [Google Scholar] [CrossRef] [Green Version]
- Halim, T.Y.; Krauss, R.H.; Sun, A.C.; Takei, F. Lung natural helper cells are a critical source of Th2 cell-type cytokines in protease allergen-induced airway inflammation. Immunity 2012, 36, 451–463. [Google Scholar] [CrossRef] [Green Version]
- Mjösberg, J.M.; Trifari, S.; Crellin, N.K.; Peters, C.P.; van Drunen, C.M.; Piet, B.; Fokkens, W.J.; Cupedo, T.; Spits, H. Human IL-25- and IL-33-responsive type 2 innate lymphoid cells are defined by expression of CRTH2 and CD161. Nat. Immunol. 2011, 12, 1055–1062. [Google Scholar] [CrossRef]
- Cayrol, C.; Girard, J.P. The IL-1-like cytokine IL-33 is inactivated after maturation by caspase-1. Proc. Natl. Acad. Sci. USA 2009, 106, 9021902–9021906. [Google Scholar] [CrossRef] [Green Version]
- Kulkarni, N.S.; Hollins, F.; Sutcliffe, A.; Saunders, R.; Shah, S.; Siddiqui, S.; Gupta, S.; Haldar, P.; Green, R.; Pavord, I.; et al. Eosinophil protein in airway macrophages: A novel biomarker of eosinophilic inflammation in patients with asthma. J. Allergy Clin. Immunol. 2010, 126, 61–69.e3. [Google Scholar] [CrossRef] [Green Version]
- Suzukawa, M.; Morita, H.; Nambu, A.; Arae, K.; Shimura, E.; Shibui, A.; Yamaguchi, S.; Suzukawa, K.; Nakanishi, W.; Oboki, K.; et al. Epithelial cell-derived IL-25, but not Th17 cell-derived IL-17 or IL-17F, is crucial for murine asthma. J. Immunol. 2012, 189, 3641–3652. [Google Scholar] [CrossRef] [PubMed]
- Soumelis, V.; Reche, P.A.; Kanzler, H.; Yuan, W.; Edward, G.; Homey, B.; Gilliet, M.; Ho, S.; Antonenko, S.; Lauerma, A.; et al. Human epithelial cells trigger dendritic cell mediated allergic inflammation by producing TSLP. Nat. Immunol. 2002, 3, 673–680. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lai, Y.; Altemeier, W.A.; Vandree, J.; Piliponsky, A.M.; Johnson, B.; Appel, C.L.; Frevert, C.W.; Hyde, D.M.; Ziegler, S.F.; Smith, D.E.; et al. Increased density of intraepithelial mast cells in patients with exercise-induced bronchoconstriction regulated through epithelially derived thymic stromal lymphopoietin and IL-33. J. Allergy Clin. Immunol. 2014, 133, 1448–1455. [Google Scholar] [CrossRef] [Green Version]
- Wenzel, S.E. Asthma Phenotypes: The Evolution from Clinical to Molecular Approaches. Nat. Med. 2012, 18, 716–725. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.W.; Baek, M.G.; Choi, S.; Ahn, Y.H.; Bang, J.Y.; Sohn, K.H.; Kang, M.G.; Jung, J.W.; Choi, J.H.; Cho, S.H.; et al. Peripheral blood transcriptomic clusters uncovered immune phenotypes of asthma. Respir. Res. 2022, 23, 237. [Google Scholar] [CrossRef] [PubMed]
- Chia, N.; Kumar, R.K.; Foster, P.S.; Herbert, C. Enhanced Pro-Inflammatory Response of Macrophages to Interleukin-33 in an Allergic Environment. Int. Arch. Allergy Immunol. 2018, 176, 74–82. [Google Scholar] [CrossRef]
- Ding, W.; Zou, G.L.; Zhang, W.; Lai, X.N.; Chen, H.W.; Xiong, L.X. Interleukin-33: Its emerging role in allergic diseases. Molecules 2018, 23, 1665. [Google Scholar] [CrossRef] [Green Version]
- Kearley, J.; Barker, J.E.; Robinson, D.S.; Lloyd, C.M. Resolution of airway inflammation and hyperreactivity after in vivo transfer of CD4+CD25+ regulatory T cells is interleukin 10 dependent. J. Exp. Med. 2005, 202, 1539–1547. [Google Scholar] [CrossRef] [Green Version]
- Oczypok, E.A.; Milutinovic, P.S.; Alcorn, J.F.; Khare, A.; Crum, L.T.; Manni, M.L.; Epperly, M.W.; Pawluk, A.M.; Ray, A.; Oury, T.D. Pulmonary receptor for advanced glycation end-products promotes asthma pathogenesis through IL-33 and accumulation of group 2 innate lymphoid cells. J. Allergy Clin. Immunol. 2015, 136, 747–756.e4. [Google Scholar] [CrossRef] [Green Version]
- Watanabe, T.; Asai, K.; Fujimoto, H.; Tanaka, H.; Kanazawa, H.; Hirata, K. Increased levels of HMGB-1 and endogenous secretory RAGE in induced sputum from asthmatic patients. Respir. Med. 2011, 105, 519–525. [Google Scholar] [CrossRef] [Green Version]
- Shim, E.J.; Chun, E.; Lee, H.S.; Bang, B.R.; Kim, T.W.; Cho, S.H.; Min, K.U.; Park, H.W. The role of high-mobility group box-1 (HMGB1) in the pathogenesis of asthma. Clin. Exp. Allergy 2012, 42, 958–965. [Google Scholar] [CrossRef] [PubMed]
- Cuppari, C.; Manti, S.; Chirico, V.; Caruso, R.; Salpietro, V.; Giacchi, V.; Laganà, F.; Arrigo, T.; Salpietro, C.; Leonardi, S. Sputum high mobility group box-1 in asthmatic children: A noninvasive sensitive biomarker reflecting disease status. Ann. Allergy Asthma Immunol. 2015, 115, 103–107. [Google Scholar] [CrossRef] [PubMed]
- Hong, S.M.; Cho, J.S.; Um, J.Y.; Shin, J.M.; Park, I.H.; Lee, S.H.; Lee, S.H.; Lee, H.M. Increased expression of high-mobility group protein B1 in chronic rhinosinusitis. Am. J. Rhinol. Allergy 2013, 27, 278–282. [Google Scholar] [CrossRef] [PubMed]
- Dzaman, K.; Szczepanski, M.J.; Molinska-Glura, M.; Krzeski, A.; Zagor, M. Expression of the receptor for advanced glycation end products, a target for high mobility group box 1 protein, and its role in chronic recalcitrant rhinosinusitis with nasal polyps. Arch. Immunol. Ther. Exp. 2015, 63, 223–230. [Google Scholar] [CrossRef] [Green Version]
- Werder, R.B.; Ullah, M.A.; Rahman, M.M.; Simpson, J.; Lynch, J.P.; Collinson, N.; Rittchen, S.; Rashid, R.B.; Sikder, M.A.A.; Handoko, H.Y.; et al. Targeting the P2Y13 Receptor Suppresses IL-33 and HMGB1 Release and Ameliorates Experimental Asthma. Am. J. Respir. Crit. Care Med. 2022, 205, 300–312. [Google Scholar] [CrossRef]
- Tiotiu, A.; Badi, Y.; Kermani, N.Z.; Sanak, M.; Kolmert, J.; Wheelock, C.E.; Hansbro, P.M.; Dahlén, S.-E.; Sterk, P.J.; Djukanovic, R.; et al. Association of Differential Mast Cell Activation with Granulocytic Inflammation in Severe Asthma. Am. J. Respir. Crit. Care Med. 2022, 205, 397–411. [Google Scholar] [CrossRef]
- Yao, X.; Xie, R.; Cao, Y.; Tang, J.; Men, Y.; Peng, H.; Yang, W. Simvastatin induced ferroptosis for triple-negative breast cancer therapy. J. Nanobiotechnol. 2021, 19, 311. [Google Scholar] [CrossRef]
- Dixon, S.J.; Lemberg, K.M.; Lamprecht, M.R.; Skouta, R.; Zaitsev, E.M.; Gleason, C.E.; Patel, D.N.; Bauer, A.J.; Cantley, A.M.; Yang, W.S.; et al. Ferroptosis: An iron-dependent form of nonapoptotic cell death. Cell 2012, 149, 1060–1072. [Google Scholar] [CrossRef] [Green Version]
- Alvarez, S.W.; Sviderskiy, V.O.; Terzi, E.M.; Papagiannakopoulos, T.; Moreira, A.L.; Adams, S.; Sabatini, D.M.; Birsoy, K.; Possemato, R. NFS1 undergoes positive selection in lung tumours and protects cells from ferroptosis. Nature 2017, 551, 639–643. [Google Scholar] [CrossRef] [Green Version]
- Zhang, W.; Sun, Y.; Bai, L.; Zhi, L.; Yang, Y.; Zhao, Q.; Chen, C.; Qi, Y.; Gao, W.; He, W.; et al. RBMS1 regulates lung cancer ferroptosis through translational control of SLC7A11. J. Clin. Investig. 2021, 131, e152067. [Google Scholar] [CrossRef]
- Liu, P.; Feng, Y.; Li, H.; Chen, X.; Wang, G.; Xu, S.; Li, Y.; Zhao, L. Ferrostatin-1 alleviates lipopolysaccharide-induced acute lung injury via inhibiting ferroptosis. Cell Mol. Biol. Lett. 2020, 25, 10. [Google Scholar] [CrossRef]
- Qiu, Y.B.; Wan, B.B.; Liu, G.; Wu, Y.X.; Chen, D.; Lu, M.D.; Chen, J.L.; Yu, R.Q.; Chen, D.Z.; Pang, Q.F. Nrf2 protects against seawater drowning-induced acute lung injury via inhibiting ferroptosis. Respir. Res. 2020, 21, 232. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Zhuang, X.; Qiao, T. Role of ferroptosis in the process of acute radiation-induced lung injury in mice. Biochem. Biophys. Res. Commun. 2019, 519, 240–245. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, M.; Minagawa, S.; Araya, J.; Sakamoto, T.; Hara, H.; Tsubouchi, K.; Hosaka, Y.; Ichikawa, A.; Saito, N.; Kadota, T.; et al. Involvement of cigarette smoke-induced epithelial cell ferroptosis in COPD pathogenesis. Nat. Commun. 2019, 10, 3145. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, Y.; Chen, H.; Xuan, N.; Zhou, L.; Wu, Y.; Zhu, C.; Li, M.; Weng, Q.; Shen, J.; Zhang, H.; et al. Induction of ferroptosis-like cell death of eosinophils exerts synergistic effects with glucocorticoids in allergic airway inflammation. Thorax 2020, 75, 918–927. [Google Scholar] [CrossRef] [PubMed]
- Stockwell, B.R.; Friedmann Angeli, J.P.; Bayir, H.; Bush, A.I.; Conrad, M.; Dixon, S.J.; Fulda, S.; Gascón, S.; Hatzios, S.K.; Kagan, V.E.; et al. Ferroptosis: A Regulated Cell Death Nexus Linking Metabolism, Redox Biology, and Disease. Cell 2017, 171, 273–285. [Google Scholar] [CrossRef] [Green Version]
- Allinne, J.; Scott, G.; Lim, W.K.; Birchard, D.; Erjefält, J.S.; Sandén, C.; Ben, L.H.; Agrawal, A.; Kaur, N.; Kim, J.H.; et al. IL-33 blockade affects mediators of persistence and exacerbation in a model of chronic airway inflammation. J. Allergy Clin. Immunol. 2019, 144, 1624–1637.e10. [Google Scholar] [CrossRef] [Green Version]
- Bao, C.; Liu, C.; Liu, Q.; Hua, L.; Hu, J.; Li, Z.; Xu, S. Liproxstatin-1 alleviates LPS/IL-13-induced bronchial epithelial cell injury and neutrophilic asthma in mice by inhibiting ferroptosis. Int. Immunopharmacol. 2022, 109, 108770. [Google Scholar] [CrossRef]
- Liu, T.; Barrett, N.A.; Kanaoka, Y.; Buchheit, K.; Laidlaw, T.M.; Garofalo, D.; Lai, J.; Katz, H.R.; Feng, C.; Boyce, J.A. Cysteinyl leukotriene receptor 2 drives lung immunopathology through a platelet and high mobility box 1-dependent mechanism. Mucosal Immunol. 2019, 12, 679–690. [Google Scholar] [CrossRef]
- Smith, D.K.; Seales, S.; Budzik, C. Respiratory syncytial virus bronchiolitis in children. Am. Fam. Physician 2017, 95, 94–99. [Google Scholar]
- Walsh, E.E. Respiratory Syncytial Virus Infection: An Illness for All Ages. Clin. Chest Med. 2017, 38, 29–36. [Google Scholar] [CrossRef]
- Johnson, J.E.; Gonzales, R.A.; Olson, S.J.; Wright, P.F.; Graham, B.S. The histopathology of fatal untreated human respiratory syncytial virus infection. Mod. Pathol. 2007, 20, 108–119. [Google Scholar] [CrossRef] [PubMed]
- Piedimonte, G.; Perez, M.K. Respiratory syncytial virus infection and bronchiolitis. Pediatr. Rev. 2014, 35, 519–530. [Google Scholar] [CrossRef] [PubMed]
- Plesca, D.A.; Cora, F.; Buzoianu, E.; Moiceanu, M.; Hurduc, V. Risk factors for severe bronchiolitis—A retrospective study. Eur. Respir. J. 2012, 40, 4660. [Google Scholar]
- Borchers, A.T.; Chang, C.; Gershwin, M.E.; Gershwin, L.J. Respiratory syncytial virus—A comprehensive review. Clin. Rev. Allergy Immunol. 2013, 45, 331–379. [Google Scholar] [CrossRef] [PubMed]
- Hashem, M.; Hall, C.B. Respiratory syncytial virus in healthy adults: The cost of a cold. J. Clin. Virol. 2003, 27, 14–21. [Google Scholar] [CrossRef]
- Camelo, A.; Rosignoli, G.; Ohne, Y.; Stewart, R.A.; Overed-Sayer, C.; Sleeman, M.A.; May, R.D. IL-33, IL-25, and TSLP induce a distinct phenotypic and activation profile in human type 2 innate lymphoid cells. Blood Adv. 2017, 1, 577–589. [Google Scholar] [CrossRef] [Green Version]
- Hinde, H.L.; Wu, Q.; Bentley, J.K.; Han, M.B.M.; Rajput, C.; Hong, J.Y.; Lei, J. The Innate Cytokines IL-25, IL-33, and TSLP Cooperate in the Induction of Type 2 Innate Lymphoid Cell Expansion and Mucous Metaplasia in Rhinovirus-Infected Immature Mice. J. Immunol. 2017, 199, 1308–1318. [Google Scholar]
- Zhang, K.; Jin, Y.; Lai, D.; Wang, J.; Wang, Y.; Wu, X.; Scott, M.; Li, Y.; Hou, J.; Billiar, T.; et al. RAGE-induced ilc2 expansion in acute lung injury due to haemorrhagic shock. Thorax 2020, 75, 209–219. [Google Scholar] [CrossRef] [Green Version]
- Saravia, J.; You, D.; Shrestha, B.; Jaligama, S.; Siefker, D.; Lee, G.I.; Harding, J.N.; Jones, T.L.; Rovnaghi, C.; Bagga, B.; et al. Respiratory Syncytial Virus Disease Is Mediated by Age-Variable IL-33. PLoS Pathog. 2015, 11, e1005217. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.; Wu, J.; Qi, F.; Zeng, S.; Xu, L.; Hu, H.; Wang, D.; Liu, B. Natural helper cells contribute to pulmonary eosinophilia by producing IL-13 via IL-33/ST2 pathway in a murine model of respiratory syncytial virus infection. Int. Immunopharmacol. 2015, 28, 337–343. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.H.; Lai, A.C.Y.; Chi, P.Y.; Thio, C.L.P.; Chen, W.Y.; Tsai, C.H.; Lee, Y.L.; Lukacs, N.W.; Chang, Y.J. Pulmonary IL-33 orchestrates innate immune cells to mediate RSV-evoked airway hyperreactivity and eosinophilia. Allergy 2020, 75, 818–830. [Google Scholar] [CrossRef] [PubMed]
- Fonseca, W.; Malinczak, C.-A.; Schuler, C.F.; Best, S.K.K.; Rasky, A.J.; Morris, S.B.; Cui, T.X.; Popova, A.P.; Lukacs, N.W. Uric acid pathway activation during respiratory virus infection promotes Th2 immune response via innate cytokine production and ILC2 accumulation. Mucosal Immunol. 2020, 13, 691–701. [Google Scholar] [CrossRef] [PubMed]
- Gause, W.C.; Wynn, T.A.; Allen, J.E. Type 2 immunity and wound healing: Evolutionary refinement of adaptive immunity by helminths. Nat. Rev. Immunol. 2013, 13, 607–614. [Google Scholar] [CrossRef] [PubMed]
- Phipps, S.; Ying, S.; Wangoo, A.; Ong, Y.E.; Levi-Schaffer, F.; Kay, A.B. The Relationship Between Allergen-Induced Tissue Eosinophilia and Markers of Repair and Remodeling in Human Atopic Skin. J. Immunol. 2002, 169, 4604–4612. [Google Scholar] [CrossRef] [Green Version]
- Monticelli, L.A.; Sonnenberg, G.F.; Abt, M.C.; Alenghat, T.; Ziegler, C.G.; Doering, T.A.; Angelosanto, J.M.; Laidlaw, B.J.; Yang, C.Y.; Sathaliyawala, T.; et al. Innate lymphoid cells promote lung-tissue homeostasis after infection with influenza virus. Nat. Immunol. 2011, 12, 1045–1054. [Google Scholar] [CrossRef]
- Sonnenberg, G.F.; Artis, D. Innate lymphoid cells in the initiation, regulation and resolution of inflammation. Nat. Med. 2015, 21, 698–708. [Google Scholar] [CrossRef] [Green Version]
- Kaiko, G.E.; Loh, Z.; Spann, K.; Lynch, J.P.; Lalwani, A.; Zheng, Z.; Davidson, S.; Uematsu, S.; Akira, S.; Hayball, J.; et al. Toll-like receptor 7 gene deficiency and early-life Pneumovirus infection interact to predispose toward the development of asthma-like pathology in mice. J. Allergy Clin. Immunol. 2013, 131, 1331–1339. [Google Scholar] [CrossRef]
- Manti, S.; Harford, T.J.; Salpietro, C.; Rezaee, F.; Piedimonte, G. Induction of high-mobility group Box-1 in vitro and in vivo by respiratory syncytial virus. Pediatr. Res. 2018, 83, 1049–1056. [Google Scholar] [CrossRef]
- Hosakote, Y.M.; Brasier, A.R.; Casola, A.; Garofalo, R.P.; Kurosky, A. Respiratory Syncytial Virus Infection Triggers Epithelial HMGB1 Release as a Damage-Associated Molecular Pattern Promoting a Monocytic Inflammatory Response. J. Virol. 2016, 90, 9618–9631. [Google Scholar] [CrossRef] [Green Version]
- Rayavara, K.; Kurosky, A.; Sta_ord, S.J.; Garg, N.J.; Brasier, A.R.; Garofalo, R.P.; Hosakote, Y.M. Proinflammatory Effects of Respiratory Syncytial Virus–Induced Epithelial HMGB1 on Human Innate Immune Cell Activation. J. Immunol. 2018, 201, 2753–2766. [Google Scholar] [CrossRef] [Green Version]
- Norlander, A.E.; Peebles, R.S., Jr. Innate Type 2 Responses to Respiratory Syncytial Virus Infection. Viruses 2020, 12, 521. [Google Scholar] [CrossRef] [PubMed]
- Hirsiger, S.; Simmen, H.P.; Werner, C.M.; Wanner, G.A.; Rittirsch, D. Danger signals activating the immune response after trauma. Mediat. Inflamm. 2012, 2012, 315941. [Google Scholar] [CrossRef] [Green Version]
- Akdis, M.; Burgler, S.; Crameri, R.; Eiwegger, T.; Fujita, H.; Gomez, E.; Klunker, S.; Meyer, N.; O’Mahony, L.; Palomares, O.; et al. Interleukins, from 1 to 37, and interferon-γ: Receptors, functions, and roles in diseases. J. Allergy Clin. Immunol. 2011, 127, e1–e70. [Google Scholar] [CrossRef]
- Roussel, L.; Farias, R.; Rousseau, S. IL-33 is expressed in epithelia from patients with cystic fibrosis and potentiates neutrophil recruitment. J. Allergy Clin. Immunol. 2013, 131, 913–916. [Google Scholar] [CrossRef] [PubMed]
- Schmitz, J.; Owyang, A.; Oldham, E.; Song, Y.; Murphy, E.; McClanahan, T.K.; Zurawski, G.; Moshrefi, M.; Qin, J.; Li, X.; et al. IL-33, an interleukin-1-like cytokine that signals via the IL-1 receptor-related protein ST2 and induces T helper type 2-associated cytokines. Immunity 2005, 23, 479–490. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yanai, H.; Ban, T.; Taniguchi, T. High-mobility group box family of proteins: Ligand and sensor for innate immunity. Trends Immunol. 2012, 33, 633–640. [Google Scholar] [CrossRef]
- Rowe, S.M.; Jackson, P.L.; Liu, G.; Hardison, M.; Livraghi, A.; Solomon, G.M.; McQuaid, D.B.; Noerager, B.D.; Gaggar, A.; Clancy, J.P.; et al. Potential role of high-mobility group box 1 in cystic fibrosis airway disease. Am. J. Respir. Crit. Care Med. 2008, 178, 822–831. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Liao, H.; Ochani, M.; Justiniani, M.; Lin, X.; Yang, L.; Al-Abed, Y.; Wang, H.; Metz, C.; Miller, E.J.; et al. Cholinergic agonists inhibit HMGB1 release and improve survival in experimental sepsis. Nat. Med. 2004, 10, 1216–1221. [Google Scholar] [CrossRef]
- Brazova, J.; Sediva, A.; Pospisilova, D.; Vavrova, V.; Pohunek, P.; Macek, M., Jr.; Bartunkova, J.; Lauschmann, H. Differential cytokine profile in children with cystic fibrosis. Clin. Immunol. 2005, 115, 210–215. [Google Scholar] [CrossRef]
- Hartl, D.; Griese, M.; Kappler, M.; Zissel, G.; Reinhardt, D.; Rebhan, C.; Schendel, D.J.; Krauss-Etschmann, S. Pulmonary T(H)2 response in Pseudomonas aeruginosa-infected patients with cystic fibrosis. J. Allergy Clin. Immunol. 2006, 117, 204–211. [Google Scholar] [CrossRef] [PubMed]
- Tiringer, K.; Treis, A.; Kanolzer, S.; Witt, C.; Ghanim, B.; Gruber, S.; Schmidthaler, K.; Renner, S.; Dehlink, E.; Nachbaur, E.; et al. Differential expression of IL-33 and HMGB1 in the lungs of stable cystic fibrosis patients. Eur. Respir. J. 2014, 44, 802–805. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heffler, E.; Allegra, A.; Pioggia, G.; Picardi, G.; Musolino, C.; Gangemi, S. MicroRNA Profiling in Asthma: Potential Biomarkers and Therapeutic Targets. Am. J. Respir. Cell Mol. Biol. 2017, 57, 642–650. [Google Scholar] [CrossRef] [PubMed]
- Bartel, S.; Carraro, G.; Alessandrini, F.; Krauss-Etschmann, S.; Ricciardolo, F.L.M.; Bellusci, S. miR-142-3p is associated with aberrant WNT signaling during airway remodeling in asthma. Am. J. Physiol. Lung Cell Mol. Physiol. 2018, 315, 328–333. [Google Scholar] [CrossRef]
- Pua, H.H.; Happ, H.C.; Gray, C.J.; Mar, D.J.; Chiou, N.T.; Hesse, L.E.; Ansel, K.M. Increased hematopoietic extracellular RNAs and vesicles in the lung during allergic airway responses. Cell Rep. 2019, 26, 933–944.e934. [Google Scholar] [CrossRef] [Green Version]
- Zhang, K.; Liang, Y.; Feng, Y.; Wu, W.; Zhang, H.; He, J.; Hu, Q.; Zhao, J.; Xu, Y.; Liu, Z.; et al. Decreased epithelial and sputum miR-221-3p associates with airway eosinophilic inflammation and CXCL17 expression in asthma. Am. J. Physiol. Lung Cell Mol. Physiol. 2018, 315, 253–264. [Google Scholar] [CrossRef]
- Liang, Y.; Feng, Y.; Wu, W.; Chang, C.; Chen, D.; Chen, S.; Zhen, G. microRNA-218-5p plays a protective role in eosinophilic airway inflammation via targeting δ-catenin, a novel catenin in asthma. Clin. Exp. Allergy 2020, 50, 29–40. [Google Scholar] [CrossRef]
- Huo, X.; Zhang, K.; Yi, L.; Mo, Y.; Liang, Y.; Zhao, J.; Zhang, Z.; Xu, Y.; Zhen, G. Decreased epithelial and plasma miR-181b-5p expression associates with airway eosinophilic inflammation in asthma. Clin. Exp. Allergy 2016, 46, 1281–1290. [Google Scholar] [CrossRef]
- Niu, Z.-R.; Han, T.; Sun, X.-L.; Luan, L.-X.; Gou, W.-L.; Zhu, X.-M. MicroRNA-30a-3p is overexpressed in the placentas of patients with preeclampsia and affects trophoblast invasion and apoptosis by its effects on IGF-1. Am. J. Obstet. Gynecol. 2018, 218, 249.e1–249.e12. [Google Scholar] [CrossRef]
- Wang, Y.; Wang, F.; He, J.; Du, J.; Zhang, H.; Shi, H.; Chen, Y.; Wei, Y.; Xue, W.; Yan, J.; et al. miR-30a-3p targets MAD2L1 and regulates proliferation of gastric cancer cells. OncoTargets Ther. 2019, 12, 11313–11324. [Google Scholar] [CrossRef] [Green Version]
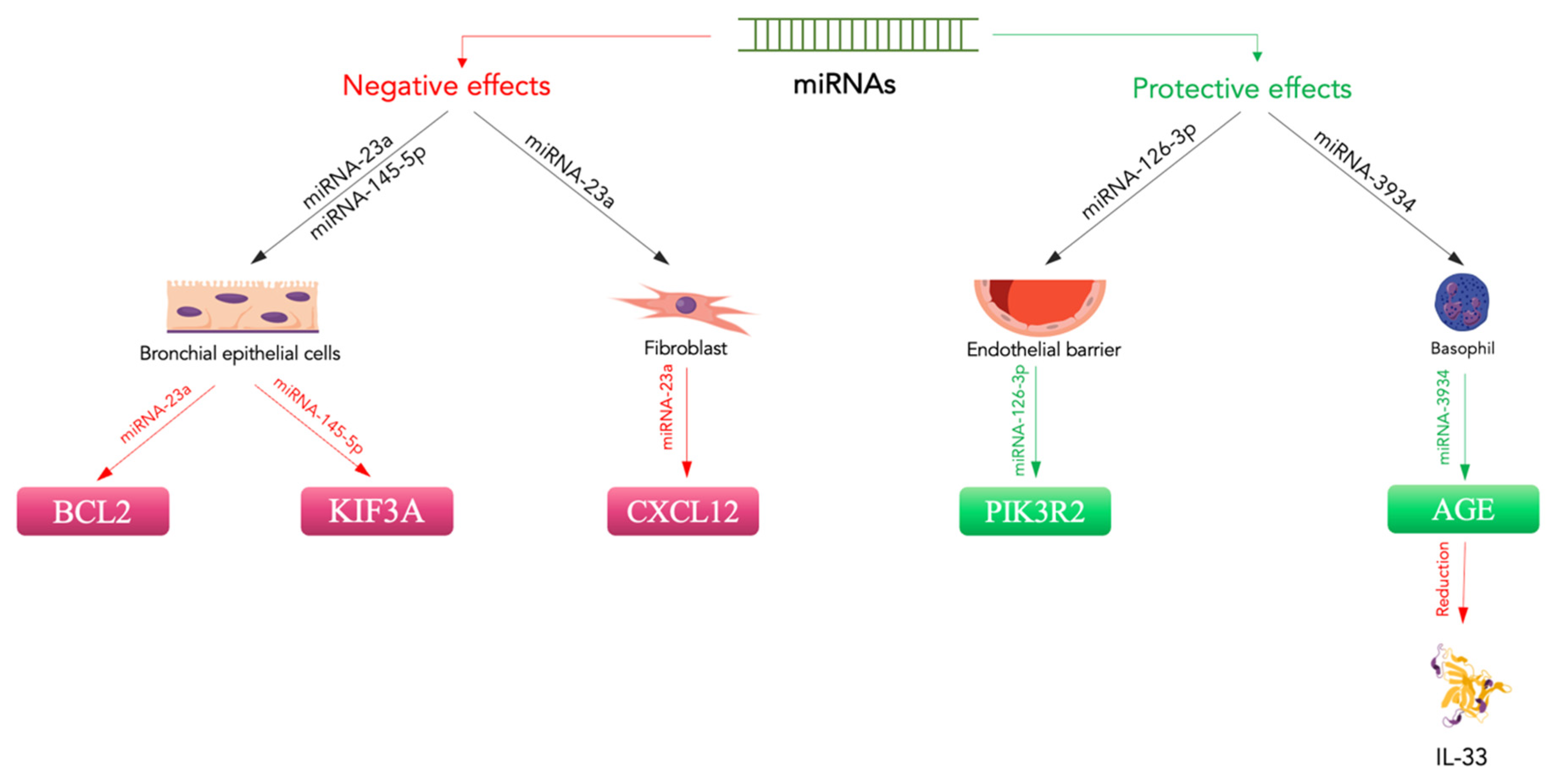
- Xiong, T.; Du, Y.; Fu, Z.; Geng, G. MicroRNA-145-5p promotes asthma pathogenesis by inhibiting kinesin family member 3A expression in mouse airway epithelial cells. J. Int. Med. Res. 2019, 47, 3307–3319. [Google Scholar] [CrossRef] [PubMed]
- Jin, A.; Bao, R.; Roth, M.; Liu, L.; Yang, X.; Tang, X.; Yang, X.; Sun, Q.; Lu, S. microRNA-23a contributes to asthma by targeting BCL2 in airway epithelial cells and CXCL12 in fibroblasts. J. Cell Physiol. 2019, 234, 21153–21165. [Google Scholar] [CrossRef] [PubMed]
- Dou, L.; Wang, W.; Wang, J.; Zhang, X.; Hu, X.; Zheng, W.; Han, K.; Wang, G. miR-3934 regulates the apoptosis and secretion of inflammatory cytokines of basophils via targeting RAGE in asthma. Allergy Asthma Clin. Immunol. 2022, 18, 66. [Google Scholar] [CrossRef] [PubMed]
- Chu, M.; Qin, S.; Wu, R.; Zhou, X.; Tang, X.; Zhang, S.; Zhao, Q.; Wang, H.; Liu, Y.; Han, X.; et al. Role of miR-126a-3p in endothelial injury in endotoxic mice. Crit Care Med. 2016, 44, e639–e650. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.; Wang, X.; Chen, Q.; Chen, M.; Cheng, L.; Dai, L.; Jiang, H.; Sun, Z. The effect of early goal-directed therapy on mortality in patients with severe sepsis and septic shock: A meta-analysis. J. Surg. Res. 2016, 202, 389–397. [Google Scholar] [CrossRef]
- Zhuang, Y.; Peng, H.; Mastej, V.; Chen, W. MicroRNA regulation of endothelial junction proteins and clinical consequence. Mediat. Inflamm. 2016, 2016, 5078627. [Google Scholar] [CrossRef] [Green Version]
- Xi, T.; Jin, F.; Zhu, Y.; Wang, J.; Tang, L.; Wang, Y.; Liebeskind, D.S.; He, Z. MicroRNA-126-3p attenuates blood-brain barrier disruption, cerebral edema and neuronal injury following intracerebral hemorrhage by regulating PIK3R2 and Akt. Biochem Biophys. Res. Commun. 2017, 494, 144–151. [Google Scholar] [CrossRef]
- Wang, Z.; Zhang, J.; Feng, T.; Zhang, D.; Pan, Y.; Liu, X.; Xu, J.; Qiao, X.; Cui, W.; Dong, L. Construction of lncRNA-Mediated Competing Endogenous RNA Networks Correlated With T2 Asthma. Front. Genet. 2022, 13, 872499. [Google Scholar] [CrossRef]
- Mogilyansky, E.; Rigoutsos, I. The miR-17/92 cluster: A comprehensive update on its genomics, genetics, functions and increasingly important and numerous roles in health and disease. Cell Death Differ. 2013, 20, 1603–1614. [Google Scholar] [CrossRef]
- Simpson, L.J.; Patel, S.; Bhakta, N.R.; Choy, D.F.; Brightbill, H.D.; Ren, X.; Wang, Y.; Pua, H.H.; Baumjohann, D.; Montoya, M. M et al.. A microRNA upregulated in asthma airway T cells promotes TH2 cytokine production. Nat. Immunol. 2014, 15, 1162–1170. [Google Scholar] [CrossRef]
- Singh, P.B.; Pua, H.H.; Happ, H.C.; Schneider, C.; von Moltke, J.; Locksley, R.M.; Baumjohann, D.; Ansel, K.M. MicroRNA regulation of type 2 innate lymphoid cell homeostasis and function in allergic inflammation. J. Exp. Med. 2017, 214, 3627–3643. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haley, K.J.; Lasky-Su, J.; Manoli, S.E.; Smith, L.A.; Shahsafaei, A.; Weiss, S.T.; Tantisira, K. RUNX transcription factors: Association with pediatric asthma and modulated by maternal smoking. Am. J. Physiol. Lung Cell Mol. Physiol. 2011, 301, 693–701. [Google Scholar] [CrossRef] [Green Version]
- Shi, N.; Zhang, J.; Chen, S.-Y. Runx2, a novel regulator for goblet cell differentiation and asthma development. FASEB J. 2017, 31, 412–420. [Google Scholar] [CrossRef] [Green Version]
- Van der Deen, M.; Akech, J.; Lapointe, D.; Gupta, S.; Young, D.W.; Montecino, M.A.; Galindo, M.; Lian, J.B.; Stein, J.L.; Stein, G.S.; et al. Genomic promoter occupancy of runt-related transcription factor RUNX2 in Osteosarcoma cells identifies genes involved in cell adhesion and motility. J. Biol. Chem. 2012, 287, 4503–4517. [Google Scholar] [CrossRef] [PubMed]
- Wu, W.; Gao, J.; Chen, D.; Chen, G.; Feng, Y.; Chang, C.; Chen, S.; Yi, L.; Zhen, G. Epithelial microRNA-30a-3p targets RUNX2/HMGB1 axis to suppress airway eosinophilic inflammation in asthma. Respir. Res. 2022, 23, 17. [Google Scholar] [CrossRef] [PubMed]
- Weinberger, S.E.; Cockrill, B.A.; Mandel, J. Acute respiratory distress syndrome. In Principles of Pulmonary Medicine; Elsevier: Amsterdam, The Netherlands, 2019; pp. 357–369. [Google Scholar]
- Xu, B.; Chen, S.S.; Liu, M.Z.; Gan, C.X.; Li, J.Q.; Guo, G.H. Stem cell derived exosomes-based therapy for acute lung injury and acute respiratory distress syndrome: A novel therapeutic strategy. Life Sci. 2020, 254, 117766. [Google Scholar] [CrossRef]
- Thompson, B.T.; Chambers, R.C.; Liu, K.D. Acute respiratory distress syndrome. N. Engl. J. Med. 2017, 377, 562–572. [Google Scholar] [CrossRef] [PubMed]
- Rawal, G.; Yadav, S.; Kumar, R. Acute respiratory distress syndrome: An update and review. J. Transl. Int. Med. 2018, 6, 74–77. [Google Scholar] [CrossRef] [Green Version]
- Pais, F.M.; Sinha, P.; Liu, K.D.; Matthay, M.A. Influence of clinical factors and exclusion criteria on mortality in ARDS observational studies and randomized controlled trials. Respir. Care 2018, 63, 1060–1069. [Google Scholar] [CrossRef] [Green Version]
- Bellani, G.; Laffey, J.G.; Pham, T.; Fan, E.; Brochard, L.; Esteban, A.; Gattinoni, L.; van Haren, F.; Larsson, A.; McAuley, D.F.; et al. Epidemiology. Patterns of care, and mortality for patients with acute respiratory distress syndrome in intensive care units in 50 countries. JAMA 2016, 315, 788–800. [Google Scholar] [CrossRef]
- Sun, X.; Icli, B.; Wara, A.K.; Belkin, N.; He, S.; Kobzik, L.; Hunninghake, G.M.; Vera, M.P.; MICU Registry. MicroRNA-181b regulates NF-kB-mediated vascular inflammation. J. Clin. Investig. 2012, 122, 1973–1990. [Google Scholar] [PubMed]
- Rao, R.; Rieder, S.A.; Nagarkatti, P.; Nagarkatti, M. Staphylococcal enterotoxin B-induced microRNA-155 targets SOCS1 to promote acute inflammatory lung injury. Infect. Immun. 2014, 82, 2971–2979. [Google Scholar] [CrossRef] [Green Version]
- He, B.; Zhou, W.; Rui, Y.; Liu, L.; Chen, B.; Su, X. MicroRNA-574-5p Attenuates Acute Respiratory Distress Syndrome by Targeting HMGB1. Am. J. Respir. Cell Mol. Biol. 2021, 64, 196–207. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.W.; Zhu, Y.; Matthay, M.A. Cell-based therapy for acute lung injury: Are we there yet? Anesthesiology 2012, 116, 1189–1191. [Google Scholar] [CrossRef] [PubMed]
- Kawasaki, T.; Nishiwaki, T.; Sekine, A.; Nishimura, R.; Suda, R.; Urushibara, T.; Suzuki, T.; Takayanagi, S.; Terada, J.; Sakao, S.; et al. Vascular repair by tissue resident endothelial progenitor cells in endotoxin-induced lung injury. Am. J. Respir. Cell Mol. Biol. 2015, 53, 500–512. [Google Scholar] [CrossRef] [Green Version]
- Cao, J.P.; He, X.Y.; Xu, H.T.; Zou, Z.; Shi, X.Y. Autologous transplantation of peripheral blood-derived circulating endothelial progenitor cells attenuates endotoxin-induced acute lung injury in rabbits by direct endothelial repair and indirect immunomodulation. Anesthesiology 2012, 116, 1278–1287. [Google Scholar] [CrossRef] [Green Version]
- Lam, C.F.; Liu, Y.C.; Hsu, J.K.; Yeh, P.A.; Su, T.Y.; Huang, C.C.; Lin, M.W.; Wu, P.C.; Chang, P.J.; Tsai, Y.C. Autologous transplantation of endothelial progenitor cells attenuates acute lung injury in rabbits. Anesthesiology 2008, 108, 392–401. [Google Scholar] [CrossRef] [Green Version]
- Mao, M.; Wang, S.N.; Lv, X.J.; Wang, Y.; Xu, J.C. Intravenous delivery of bone marrow-derived endothelial progenitor cells improves survival and attenuates lipopolysaccharide-induced lung injury in rats. Shock 2010, 34, 196–204. [Google Scholar] [CrossRef]
- Fan, H.; Goodwin, A.J.; Chang, E.; Zingarelli, B.; Borg, K.; Guan, S.; Halushka, P.V.; Cook, J.A. Endothelial progenitor cells and a SDF-1alpha analogue synergistically improve survival in sepsis. Am. J. Respir. Crit. Care Med. 2014, 189, 1509–1519. [Google Scholar] [CrossRef] [Green Version]
- Valadi, H.; Ekstrom, K.; Bossios, A.; Sjostrand, M.; Lee, J.J.; Lotvall, J.O. Exosome mediated transfer of mRNAs and microRNAs is a novel mechanism of genetic exchange between cells. Nat. Cell Biol. 2007, 9, 654–659. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Y.; Li, P.; Goodwin, A.J.; Cook, J.A.; Halushka, P.V.; Chang, E.; Fan, H. Exosomes from endothelial progenitor cells improve the outcome of a murine model of sepsis. Mol. Ther. 2018, 26, 1375–1384. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Conlan, R.S.; Pisano, S.; Oliveira, M.I.; Ferrari, M.; Mendes Pinto, I. Exosomes as reconfigurable therapeutic systems. Trends Mol. Med. 2017, 23, 636–650. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Li, P.; Goodwin, A.J.; Cook, J.A.; Halushka, P.V.; Chang, E.; Zingarelli, B.; Fan, H. Exosomes from endothelial progenitor cells improve outcomes of the lipopolysaccharide-induced acute lung injury. Crit. Care 2019, 23, 44. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.; Sun, D.; Pu, W.C.; Wang, J.; Peng, Y. Circular RNAs in cancer: Biogenesis, function, and clinical significance. Trends Cancer 2020, 6, 319–336. [Google Scholar] [CrossRef] [PubMed]
- Vo, J.N.; Cieslik, M.; Zhang, Y.; Shukla, S.; Xiao, L.; Zhang, Y.; Wu, Y.M.; Dhanasekaran, S.M.; Engelke, C.G.; Cao, X.; et al. The Landscape of Circular RNA in Cancer. Cell 2019, 176, 869–881.e13. [Google Scholar] [CrossRef] [Green Version]
- Allegra, A.; Cicero, N.; Tonacci, A.; Musolino, C.; Gangemi, S. Circular RNA as a Novel Biomarker for Diagnosis and Prognosis and Potential Therapeutic Targets in Multiple Myeloma. Cancers 2022, 14, 1700. [Google Scholar] [CrossRef]
- Ye, Y.; Zhao, L.; Li, Q.; Xi, C.; Li, Y.; Li, Z. circ_0007385 served as competing endogenous RNA for miR-519d-3p to suppress malignant behaviors and cisplatin resistance of non-small cell lung cancer cells. Thorac. Cancer 2020, 11, 2196–2208. [Google Scholar] [CrossRef]
- Gál, Z.; Gézsi, A.; Semsei, Á.F.; Nagy, A.; Sultész, M.; Csoma, Z.; Tamási, L.; Gálffy, G.; Szalai, C. Investigation of circulating lncRNAs as potential biomarkers in chronic respiratory diseases. J. Transl. Med. 2020, 18, 422. [Google Scholar] [CrossRef]
- Menk, M.; Estenssoro, E.; Sahetya, S.K.; Neto, A.S.; Sinha, P.; Slutsky, A.S.; Summers, C.; Yoshida, T.; Bein, T.; Ferguson, N.D. Current and evolving standards of care for patients with ARDS. Intensive Care Med. 2020, 46, 2157–2167. [Google Scholar] [CrossRef]
- Aranda-Valderrama, P.; Kaynar, A.M. The basic science and molecular mechanisms of lung injury and acute respiratory distress syndrome. Int. Anesthesiol. Clin. 2018, 56, 1–25. [Google Scholar] [CrossRef]
- Patel, V.J.; Biswas Roy, S.; Mehta, H.J.; Joo, M.; Sadikot, R.T. Alternative and natural therapies for acute lung injury and acute respiratory distress syndrome. Biomed. Res. Int. 2018, 2018, 2476824. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jagrosse, M.L.; Dean, D.A.; Rahman, A.; Nilsson, B.L. RNAi therapeutic strategies for acute respiratory distress syndrome. Transl. Res. 2019, 214, 30–49. [Google Scholar] [CrossRef] [PubMed]
- Zoulikha, M.; Xiao, Q.; Boafo, G.F.; Sallam, M.A.; Chen, Z.; He, W. Pulmonary delivery of siRNA against acute lung injury/acute respiratory distress syndrome. Acta Pharm. Sin. B 2022, 12, 600–620. [Google Scholar] [CrossRef] [PubMed]
- Aldakheel, F.M.; Thomas, P.S.; Bourke, J.E.; Matheson, M.C.; Dharmage, S.C.; Lowe, A.J. Relationships between adult asthma and oxidative stress markers and pH in exhaled breath condensate: A systematic review. Allergy 2016, 71, 741–757. [Google Scholar] [CrossRef] [PubMed]
- Sahiner, U.M.; Birben, E.; Erzurum, S.; Sackesen, C.; Kalayci, Ö. Oxidative stress in asthma: Part of the puzzle. Pediatr. Allergy Immunol. 2018, 29, 789–800. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Feng, X.; Fan, Y.; Zhu, G.; Bai, C. Molecular hydrogen alleviates asthma through inhibiting IL-33/ILC2 axis. Inflamm. Res. 2021, 70, 569–579. [Google Scholar] [CrossRef]
- Ohsawa, I.; Ishikawa, M.; Takahashi, K.; Watanabe, M.; Nishimaki, K.; Yamagata, K.; Katsura, K.; Katayama, Y.; Asoh, S.; Ohta, S. Hydrogen acts as a therapeutic antioxidant by selectively reducing cytotoxic oxygen radicals. Nat. Med. 2007, 13, 688–694. [Google Scholar] [CrossRef]
- Kura, B.; Bagchi, A.K.; Singal, P.K.; Barancik, M.; LeBaron, T.W.; Valachova, K.; Šoltés, L.; Slezák, J. Molecular hydrogen: Potential in mitigating oxidative-stress-induced radiation injury. Can. J. Physiol. Pharmacol. 2019, 97, 287–292. [Google Scholar] [CrossRef]
- Magat, J.M.; Thomas, J.L.; Dumouchel, J.P.; Murray, F.; Li, W.X.; Li, J. Endogenous IL-33 and its autoamplification of IL-33/ST2 pathway play an important role in asthma. J. Immunol. 2020, 204, 1592–1597. [Google Scholar] [CrossRef] [Green Version]
- Solberg, O.D.; Ostrin, E.J.; Love, M.I.; Peng, J.C.; Bhakta, N.R.; Hou, L.; Nguyen, C.; Solon, M.; Nguyen, C.; Barczak, A.J.; et al. Airway epithelial miRNA expression is altered in asthma. Am. J. Respir. Crit. Care Med. 2012, 186, 965–974. [Google Scholar] [CrossRef] [Green Version]
- Kim, R.Y.; Horvat, J.C.; Pinkerton, J.W.; Starkey, M.R.; Essilfie, A.T.; Mayall, J.R.; Nair, P.M.; Hansbro, N.G.; Jones, B.; Haw, T.J.; et al. MicroRNA-21 drives severe, steroid-insensitive experimental asthma by amplifying phosphoinositide 3-kinase-mediated suppression of histone deacetylase 2. J. Allergy Clin. Immunol. 2017, 139, 519–532. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ungvari, I.; Hadadi, E.; Virag, V.; Bikov, A.; Nagy, A.; Semsei, A.F.; Galffy, G.; Tamasi, L.; Horvath, I.; Szalai, C. Implication of BIRC5 in asthma pathogenesis. Int. Immunol. 2012, 24, 293–301. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yin, S.C.; Liao, E.C.; Ye, C.X.; Chang, C.Y.; Tsai, J.J. Effect of mite allergenic components on innate immune response: Synergy of protease (Group 1 & 3) and non-protease (Group 2 & 7) allergens. Immunobiology 2018, 223, 443–448. [Google Scholar] [CrossRef] [PubMed]
- Gour, N.; Lajoie, S. Epithelial Cell Regulation of Allergic Diseases. Curr. Allergy Asthma Rep. 2016, 16, 65. [Google Scholar] [CrossRef] [PubMed]
- Gruzieva, O.; Merid, S.K.; Melén, E. An update on epigenetics and childhood respiratory diseases. Paediatr. Respir. Rev. 2014, 15, 348–354. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.; Liu, J.; Ma, R.; Hao, J.; Liang, Y.; Zhao, J.; Zhang, A.; Meng, H.; Lu, J. Interleukin-33: Metabolic checkpoints, metabolic processes, and epigenetic regulation in immune cells. Front. Immunol. 2022, 13, 900826. [Google Scholar] [CrossRef]
- Kozlova, A.L.; Valieva, M.E.; Maluchenko, N.V.; Studitsky, V.M. HMGB Proteins as DNA Chaperones That Modulate Chromatin Activity. Mol. Biol. 2018, 52, 737–749. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Authors | Chronic Respiratory Disease | Alarmins | Purpose of the Study | Model of Study | Results |
|---|---|---|---|---|---|
| Chia et al. [56] (2018) | Asthma | IL-33 | To demonstrate that IL-33-activated macrophages contribute to the worsening of airway inflammation in exacerbations of allergic asthma | In vitro | IL-33-stimulated cells led to an increase in pro-inflammatory cytokines potentially involved in allergic asthma exacerbations. |
| Watanabe et al. [60] (2011) | Asthma | HMGB1 | To demonstrate the correlation between HMGB1, neutrophilic inflammation and disease severity in asthma | Human | HMGB1 levels in induced sputum were significantly higher in asthmatic patients than in healthy controls and were inversely correlated with %FEV1 and FEV1/FVC. HMGB1 levels were directly proportional to disease severity and the percentage of neutrophils in induced sputum. |
| Shim et al. [61] (2012) | Asthma | HMGB1 | To evaluate the role of HMGB1 in the pathogenesis of eosinophilic asthma | Human and mouse | Sputum HMGB1 levels were increased in asthmatics compared with controls and positively correlated with the number of sputum eosinophils and the expression of TNF-α, IL-5, and IL-13. In a mouse model of asthma, eosinophilic airway inflammation was attenuated by the inhibition of HMGB1. |
| Cuppari et al. [62] (2015) | Asthma | HMGB1 | To evaluate HMGB1 levels in sputum in children with stable allergic asthma and their correlation with lung function | Human | Sputum HMGB1 levels were increased in children with asthma compared with healthy controls. HMGB1 correlated positively with total IgE levels and negatively with lung function. |
| Hong et al. [63] (2013) | Chronic rhinosinusitis | HMGB1 | To measure the expression of HMGB1 in the paranasal sinus mucosa and the difference in expression between CRS patients and healthy controls | Human | By both RT-PCR and real-time PCR, HMGB1 mRNA expression was significantly increased in the tissues of CRS patients compared with controls. HMGB1 protein level was significantly increased in CRS tissues by Western blot. Immunohistochemistry demonstrated that HMGB1 was mainly expressed in CRS. |
| Dzaman et al. [64] (2015) | CRSwNP | HMGB1 | To evaluate the expression of the RAGE and HMGB1 in CRSwNP and their correlation with the risk of relapse and disease severity | Human | The expression levels of the RAGE and HMGB1 in tissues correlated with disease severity. Elevated RAGE expression was associated with increased disease severity, allergy, and AERD in patients with CRSwNP. |
| Werder et al. [65] (2022) | Asthma | HMGB1 and IL-33 | To evaluate whether P2Y13-R (P2Y13 receptor) regulates the release of IL-33 and HMGB1 | Mouse (in vivo and in vitro) | In vitro exposure to aeroallergens or viruses induced the release of IL-33 and HMGB1. This response was blocked by the genetic deletion or pharmacological antagonism of P2Y13. In mouse models, the prophylactic or therapeutic isolation of P2Y13-R attenuated the onset of asthma and reduced the severity of rhinovirus-associated exacerbation. |
| Allinne et al. [77] (2019) | Asthma | IL-33 | To demonstrate that increasing IL-33 levels maintains pulmonary inflammation, promoting remodelling and exacerbation | Mouse | IL-33 drives inflammation and bronchial remodelling. The use of an IL-33 neutralizing antibody reduced inflammation and improved the remodelling of both lung epithelium and lung parenchyma. |
| Liu et al. [79] (2019) | AERD (aspirin-exacerbated respiratory disease) | HMGB1 | To demonstrate that platelets activated through CysLT2R facilitate the genesis of IL-33-dependent eosinophilic inflammation through the action of HMGB1 and the RAGE | Mouse | Mouse models subjected to platelet depletion, HMGB1 neutralisation and RAGE isolation did not respond to lysine-aspirin (Lys-ASA) inhalation. They also did not record an increase in IL-33, mast cell activation and changes in airway resistance. |
| Saravia et al. [90] (2015) | Respiratory syncytial virus infection | IL-33 | To demonstrate that IL-33 release in the lungs during RSV infection is age-dependent | Mouse | RSV infection induced IL-33 expression and ILC2 increase in the lungs of neonatal mice but not adults. Using anti-IL-33 antibodies or an IL-33 receptor knockout mouse during infection inhibited the immunopathogenesis of RSV. |
| Liu et al. [91] (2015) | Respiratory syncytial virus infection | IL-33 | To investigate the role of natural lung helper cells in the infiltration of eosinophils into the lung using BALB/c mice infected with RSV | Mouse | RSV infection induced an increase in the absolute number of natural helper cells in the lungs of mice and an increase in IL-13, suggesting that these cells may be the source of IL-13. Lung natural helper cells produced IL-13 stimulated by IL-33, which was especially increased in the lungs of mice after intranasal RSV infection. |
| Wu et al. [92] (2020) | Respiratory syncytial virus infection | IL-33 | To study the role in the pathophysiology triggered by RSV infection of each innate immune cell activated by IL-33 (including ILC2s and ST2+ myeloid cells) | Mouse | IL-33-activated ILC2s led to the development of airway hyperresponsiveness (AHR) and inflammation during RSV infection. Myeloid cell-derived IL-33 was required for airway inflammation, suggesting the importance of IL-33 signalling. |
| Fonseca et al. [93] (2020) | Respiratory syncytial virus infection | IL-33 | To evaluate the role of the uric acid pathway during RSV infection | Mouse (in vivo and in vitro) | The inhibition of uric acid pathway activation during infection reduced the expression of IL-33, TSLP and CCL2 in airway epithelial cells and IL-1β in bone marrow-derived macrophages. A reduction in ILC2s, macrophages, and IL-33 was observed in mice treated with XOI or interleukin-1 receptor antagonist during infection. |
| Manti et al. [99] (2018) | Respiratory syncytial virus infection | HMGB1 | To evaluate the role of HMGB1 in RSV infection | Mouse (in vivo and in vitro) | RSV infection strongly induced HMGB1 expression both in vitro and in vivo. |
| Hosakote et al. [100] (2016) | Respiratory syncytial virus infection | HMGB1 | To study the mechanism of action of HMGB1 released extracellularly in airway epithelial cells to establish its role in RSV infection | In vitro | RSV infection of human airway epithelial cells induced a significant release of HMGB1. Treatment with antioxidants considerably inhibited the extracellular release of HMGB1. HMGB1 appears to function as a paracrine factor by stimulating epithelial cells and monocytes. |
| Rayavara et al. [101] (2018) | Respiratory syncytial virus infection | HMGB1 | To study the mechanism of action of HMGB1 released extracellularly in airway epithelial cells to establish its role in RSV infection | In vitro | HMGB1 determined the phosphorylation of NF-κB and P38 MAPK but did not stimulate cytokine release from airway epithelial cells. HMGB1 induced the release of cytokines from immune cells. |
| Norlander et al. [102] (2020) | Respiratory syncytial virus infection | HMGB1 and IL-33 | To describe the contributions of ILC2s and alarmins in RSV infection | Review | From the literature review, it emerged that ILC2s and alarmins are key mediators in the early phase of the type 2 response to RSV infection. |
| Roussel et al. [105] (2013) | Cystic fibrosis | IL-33 | To demonstrate the correlation between IL-33, CFTR mutations and CF-related lung diseases | In vitro | IL-33 was upregulated in CF-related lung disease. IL-33 was present in the AEC nuclei of patients with CF and was released after tissue injury. IL-33 bioactivity was increased by neutrophil elastase. |
| Rowe et al. [108] (2008) | Cystic fibrosis | HMGB1 | To determine whether HMGB1 contributes to lung inflammation in cystic fibrosis and regulates neutrophil chemotaxis and lung matrix degradation | Human and mouse | Sputum HMGB1 levels were increased in subjects with CF. The chemotaxis of human neutrophils stimulated by purified HMGB1 was partially dependent on CXC chemokine receptors. The intratracheal administration of purified HMGB1 induced the recruitment of neutrophils to the airways of mice. |
| Tiringer et al. [112] (2014) | Cystic fibrosis | HMGB1 and IL-33 | To evaluate the expression of IL-33 and HMGB1 in the lungs of patients with stable cystic fibrosis | Human | IL-33 levels were significantly higher in clinically stable CF patients than in healthy subjects. HMGB1 levels were higher in non-CF controls with recurrent infections. In clinically stable CF patients who were able to perform pulmonary function tests, IL-33 levels were negatively correlated with forced vital capacity. |
| Zhang et al. [167] (2021) | Asthma | IL-33 | To evaluate the effects of molecular hydrogen on the pathogenesis of asthma | Mouse (in vivo and in vitro) | The serum and BALF levels of IL-33 were significantly increased by OVA and inhibited by H2 in mice. H2 reduced the HDM-induced apoptosis of 16HBE cells and upregulation of IL-33. |
| Magat et al. [170] (2020) | Asthma | IL-33 | To evaluate the role of IL-33 and its self-amplification of the IL-33/ST2 pathway in Ag-dependent and Ag-independent asthma-like models | Mouse | IL-33 auto-amplified itself and ST2 protein expression in airway epithelial cells. IL-33/ST2 pathway auto-amplification was not present in IL-33 knockout mice. In ST2 knockout mice, IL-33-induced eosinophilic airway inflammation was completely decreased. |
| Authors | Chronic Respiratory Disease | MiRNA | Purpose of the Study | Model of Study | Results |
|---|---|---|---|---|---|
| Heffler et al. [113] (2017) | Asthma | Main miRNAs involved in the pathology | To evaluate the main miRNAs involved in asthma and their potential roles as biomarkers or therapeutic targets | Review | Several miRNAs are involved in asthma and could be disease biomarkers or therapeutic targets. |
| Bartel et al. [114] (2018) | Asthma | MiR-142-3p | To study the connection between miR-142-3p and the proliferation of airway smooth muscle (ASM) precursor cells in asthma | Human and mouse | MiR-142-3p was increased in hyperproliferative regions of the lung in asthma. Bronchial biopsies of patients with early or late-onset severe asthma showed differential expression of miR-142-3p. |
| Pua et al. [115] (2019) | Asthma | MiR-223, miR-142a | To study the ex-miRNA release during inflammation | Mouse | The extracellular microRNAs (ex-miRNAs) present in the lung had a composition related to the epithelial lining of the airways. |
| Zhang et al. [116] (2018) | Asthma | MiR-221-3p | To study the correlation between epithelial and sputum miR-221-3p and eosinophilic inflammation in asthma and to evaluate miR-221-3p as a new biomarker | Human | After induction of allergic airway inflammation, epithelial and sputum miR-221-3p were found to be excellent biomarkers of eosinophilic airway inflammation in asthma. Decreased epithelial miR-221-3p led to the upregulation of the anti-inflammatory chemokine CXCL17. |
| Liang et al. [117] (2020) | Asthma | MiR-218-5p | To determine the role of epithelial miR-218-5p and its target gene in eosinophilic airway inflammation | Human and mouse | The epithelial expression of miR-218-5p was significantly reduced in asthmatic patients and negatively correlated with eosinophils and other type 2 biomarkers. CTNND2 (encoding δ-catenin) was a target of miR-218-5p and its epithelial expression was positively correlated with airway eosinophilia. |
| Huo et al. [118] (2016) | Asthma | MiR-181b-5p | To evaluate the association between miR-181b-5p and eosinophilic airway inflammation and the possible mechanism by which miR-181b-5p participates in eosinophilic inflammation | Human (in vivo and in vitro) | The epithelial expression of miR-181b-5p was reduced in subjects with asthma and inversely correlated with sputum and bronchial submucosal eosinophilia. Plasma miR-181b-5p increased after treatment with inhaled corticosteroids. In human bronchial epithelial cells, miR-181b-5p regulated IL-13-induced IL-1β and CCL11 expression by targeting SPP1. |
| Xiong et al. [121] (2019) | Asthma | MiR-145-5p | To study the mechanisms by which miR-145-5p can induce asthma | Mouse | In the airway epithelial cells of asthmatic mice exposed to house dust mites, KIF3A was increased, while miR-145-5p was decreased. The use of miR-145-5p antagonists significantly improved the symptoms. |
| Jin et al. [122] (2019) | Asthma | MiR-23a | To evaluate the effect of miR-23a on BCL2 and CXCL12 in asthma | Mouse | MiR-23a was upregulated in lung tissues after exposure to the antigen. BCL2 in the epithelium and CXCL12 in fibroblasts were downregulated. The use of a miR-23a mimic or inhibitor changed the expression of BCL2 and CXCL12. |
| Dou et al. [123] (2022) | Asthma | MiR-3934 | To study the role of miR-3934 in the pathogenesis of asthmatic disease | Human (in vivo and in vitro) | MiR-3934 was downregulated in the basophils of asthmatic patients. The use of miR-3934 mimics resulted in a reduction in the expression of the IL-6, IL-8, and IL-33 pro-inflammatory cytokines. MiR-3934 proved to be a good biomarker. |
| Wang et al. [128] (2022) | Asthma | Main miRNAs involved in the pathology | To identify the competing endogenous RNA network mechanism underlying T2 asthma | In vitro | A total of 30 lncRNAs, 22 miRNAs and 202 mRNAs were differentially expressed in airway biopsies from patients with T2 asthma. |
| Simpson et al. [130] (2014) | Asthma | MiR-19a | To study miRNAs and their pathways that control the responses of type 2 helper T cells (Th2 cells) involved in the pathogenesis of asthma | Human and mouse | Elevated expression of miR-19a was revealed in human airway-infiltrating T cells from patients with asthma. Modulation of miR-19 activity weakened Th2 cytokine production in both human and mouse T cells. |
| Singh et al. [131] (2017) | Asthma | MiR-19a | To evaluate the role of miRNAs in the regulation of ILC2 biological activity | Mouse (in vivo and in vitro) | Papain-exposed mice without the miR-17~92 cluster showed reduced airway inflammation. The miR-17~92 cluster member miR-19a induced IL-13 and IL-5 production through the inhibition of SOCS1 and A20. |
| Wu et al. [135] (2022) | Asthma | MiR-30a-3p | To study the correlation between miR-30a-3p, RUNX2, and HMGB1 in allergic airway inflammation | Human and mouse | The expression of miR-30a-3p was significantly reduced in asthmatic patients compared with control subjects. The epithelial expression of miR-30a-3p was negatively correlated with eosinophils in induced sputum and bronchial biopsies and the fraction of exhaled nitric oxide in patients with asthma. RUNX2 is a target of miR-30a-3p, and the airway overexpression of mmu-miR-30a-3p suppressed the expression of RUNX2 and HMGB1, relieving airway eosinophilia. |
| Sun et al. [142] (2012) | ARDS | MiR-181b | To evaluate the role of miR-181b in NF-κB-mediated EC activation and vascular inflammation in response to pro-inflammatory stimuli | In vivo and in vitro | MiR-181b inhibited the expression of importin-α3- and NF-κB-sensitive genes, such as adhesion molecules VCAM-1 and E-selectin. The inhibition of miR-181b exacerbated endotoxin-induced NF-κB activity, leucocyte influx and lung injury. Critical patients with sepsis showed reduced levels of miR-181b compared with control ICU subjects. |
| Rao et al. [143] (2014) | ARDS | MiR-155 | To study miRNAs overexpressed after exposure to staphylococcal enterotoxin B (SEB) and the consequent development of acute inflammatory lung injury | Mouse | The most expressed miRNA was miR-155; miR-155(−/−) mice were protected from SEB-mediated inflammation and lung injury. There was a functional link between SEB-induced miR-155 and IFN-γ. MiR-155(−/−) mice showed increased expression of SOCS1, and miR-155 overexpression led to its suppression and a reduction in IFN-γ. |
| He et al. [144] (2021) | ARDS | MiR-574-5p | To study the correlation between miR-574-5p and HMGB1 and possible new therapeutic strategies for the treatment of ARDS | Human and mouse (in vivo and in vitro) | The expression of miR-574-5p was upregulated in ARDS patients and after LPS stimulation in vitro and in vivo; miR-574-5p suppresses inflammatory responses and NLRP3 inflammasome activation through the inhibition of HMGB1. |
| Zhou et al. [154] (2019) | ARDS | MiR-126 | To study the effects of endothelial progenitor cell (EPC) exosomes and miR-126 in LPS-induced acute lung injury (ALI) | Mouse | The intratracheal administration of EPC exosomes reduced lung injury after LPS-induced ALI. Furthermore, EPC exosomes reduced myeloperoxidase (MPO) activity, lung injury score and pulmonary oedema. |
| Solberg et al. [171] (2012) | Asthma | MiR-34/449 family | To determine if airway epithelial miRNA expression is impaired in asthma and identify IL-13-regulated miRNAs | Human | Compared with control subjects, 217 miRNAs were differentially expressed in subjects with steroid-naïve asthma and 200 miRNAs in subjects with steroid-using asthma. Treatment with inhaled corticosteroids induced a statistically significant change for only nine miRNAs. |
| Kim et al. [172] (2017) | Asthma | MiR-21 | To study mouse models with severe and steroid-insensitive asthma to identify pathogenic mechanisms and study new therapeutic approaches | Mouse | Infection induced increases in miR-21 expression levels in the lung during SSIAAD, while expression of the miR-21 target phosphatase and tensin homolog was decreased. Treatment with a miR-21-specific antagomir (Ant-21) increased tensin and phosphatase homolog levels. This led to the suppression of airway hyperresponsiveness and the restoration of steroid sensitivity to allergic airway disease. |
| Key Points |
|---|
|
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Allegra, A.; Murdaca, G.; Gammeri, L.; Ettari, R.; Gangemi, S. Alarmins and MicroRNAs, a New Axis in the Genesis of Respiratory Diseases: Possible Therapeutic Implications. Int. J. Mol. Sci. 2023, 24, 1783. https://doi.org/10.3390/ijms24021783
Allegra A, Murdaca G, Gammeri L, Ettari R, Gangemi S. Alarmins and MicroRNAs, a New Axis in the Genesis of Respiratory Diseases: Possible Therapeutic Implications. International Journal of Molecular Sciences. 2023; 24(2):1783. https://doi.org/10.3390/ijms24021783
Chicago/Turabian StyleAllegra, Alessandro, Giuseppe Murdaca, Luca Gammeri, Roberta Ettari, and Sebastiano Gangemi. 2023. "Alarmins and MicroRNAs, a New Axis in the Genesis of Respiratory Diseases: Possible Therapeutic Implications" International Journal of Molecular Sciences 24, no. 2: 1783. https://doi.org/10.3390/ijms24021783