
The Integrated mRNA and miRNA Approach Reveals Potential Regulators of Flowering Time in Arundina graminifolia

 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
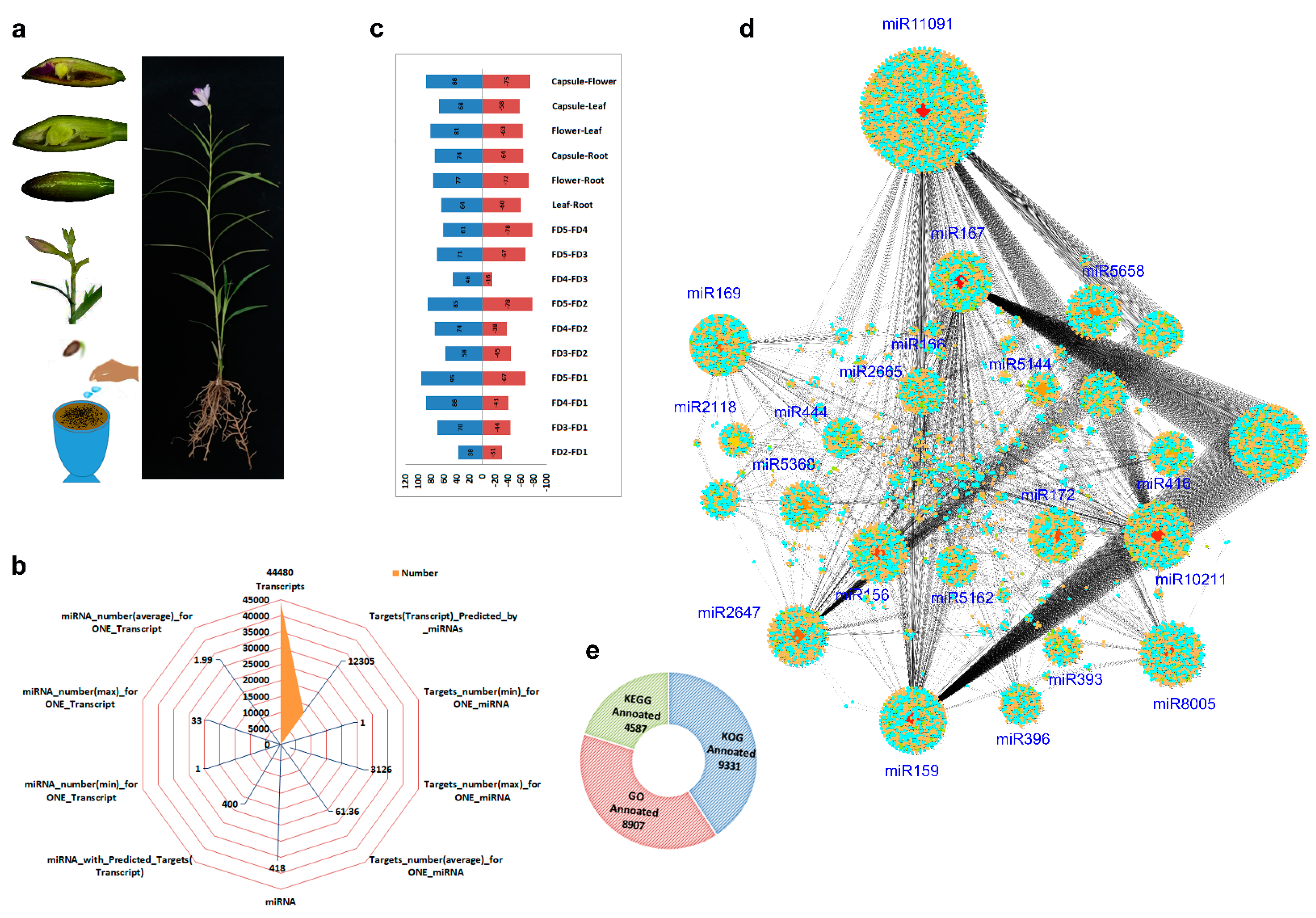
2.1. Continuous Flowering and Flower Characteristics of A. graminifolia
2.2. Transcriptome-miRNA Sequencing and Functional Annotation
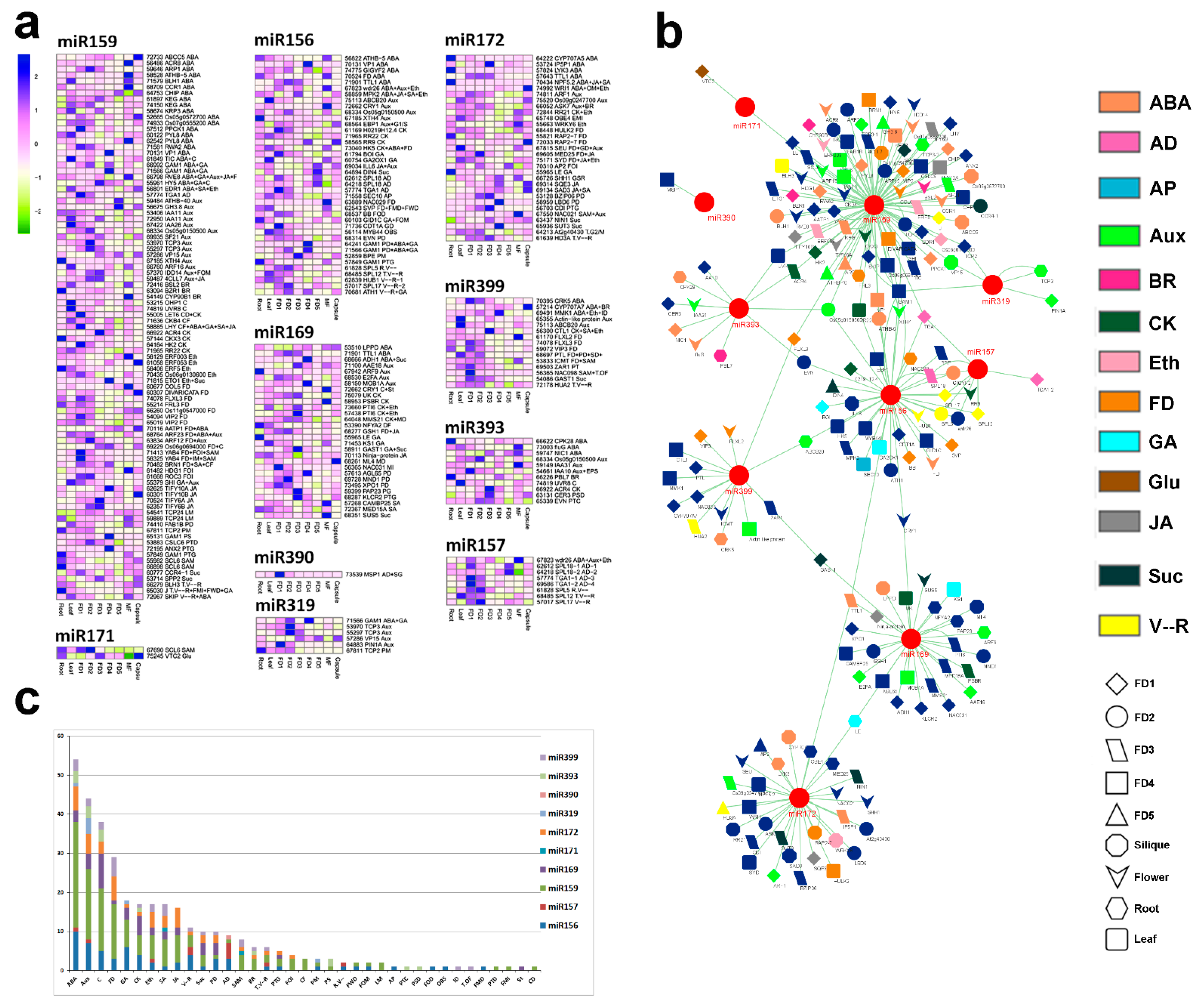
2.3. One miRNA with the Highest Targets Responds to Multi-Level Regulation
2.4. Major miRNA Classes Were Found for Flower Bud Control
2.5. Stage-Specific miRNAs during Bud Outgrowth
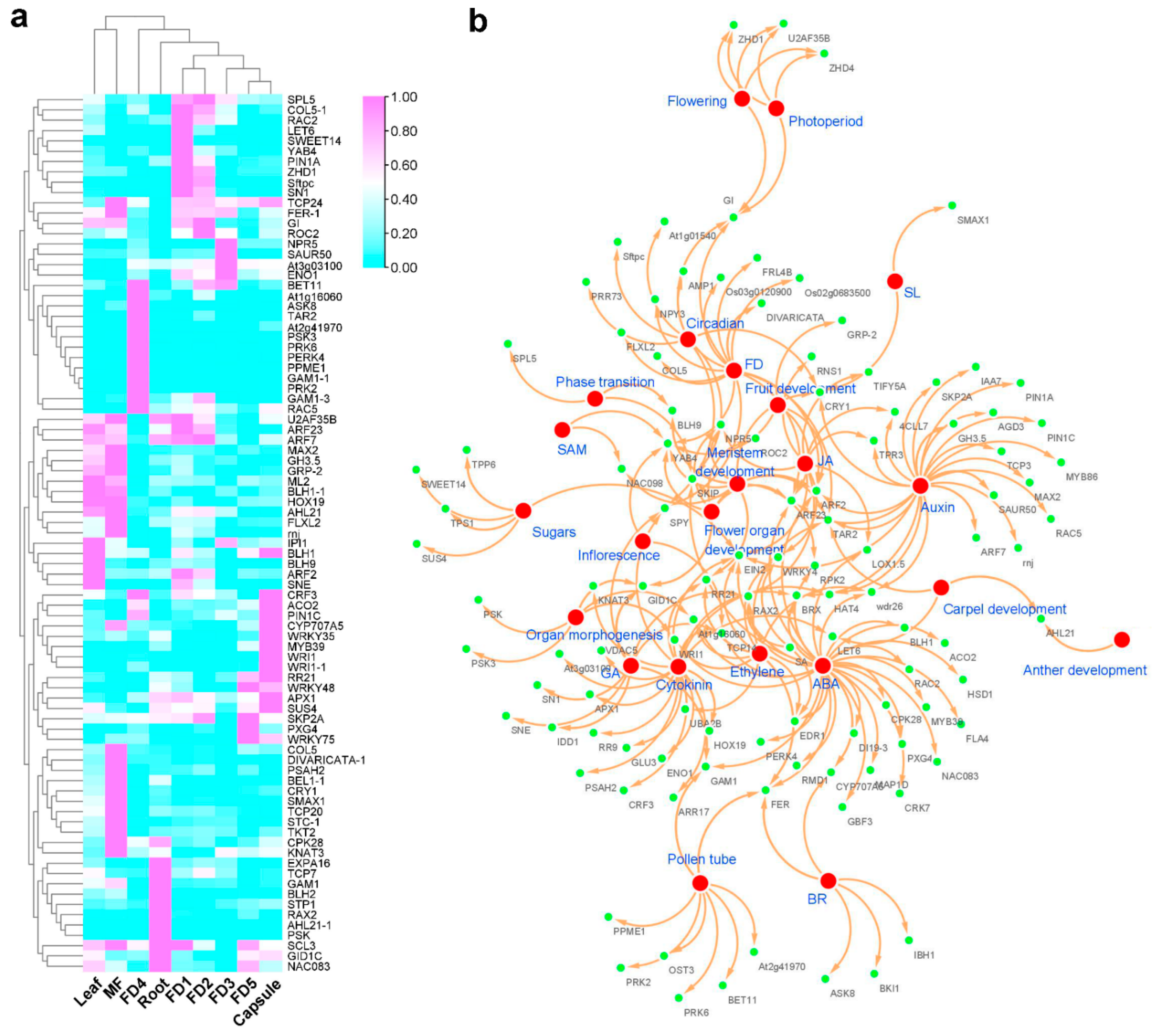
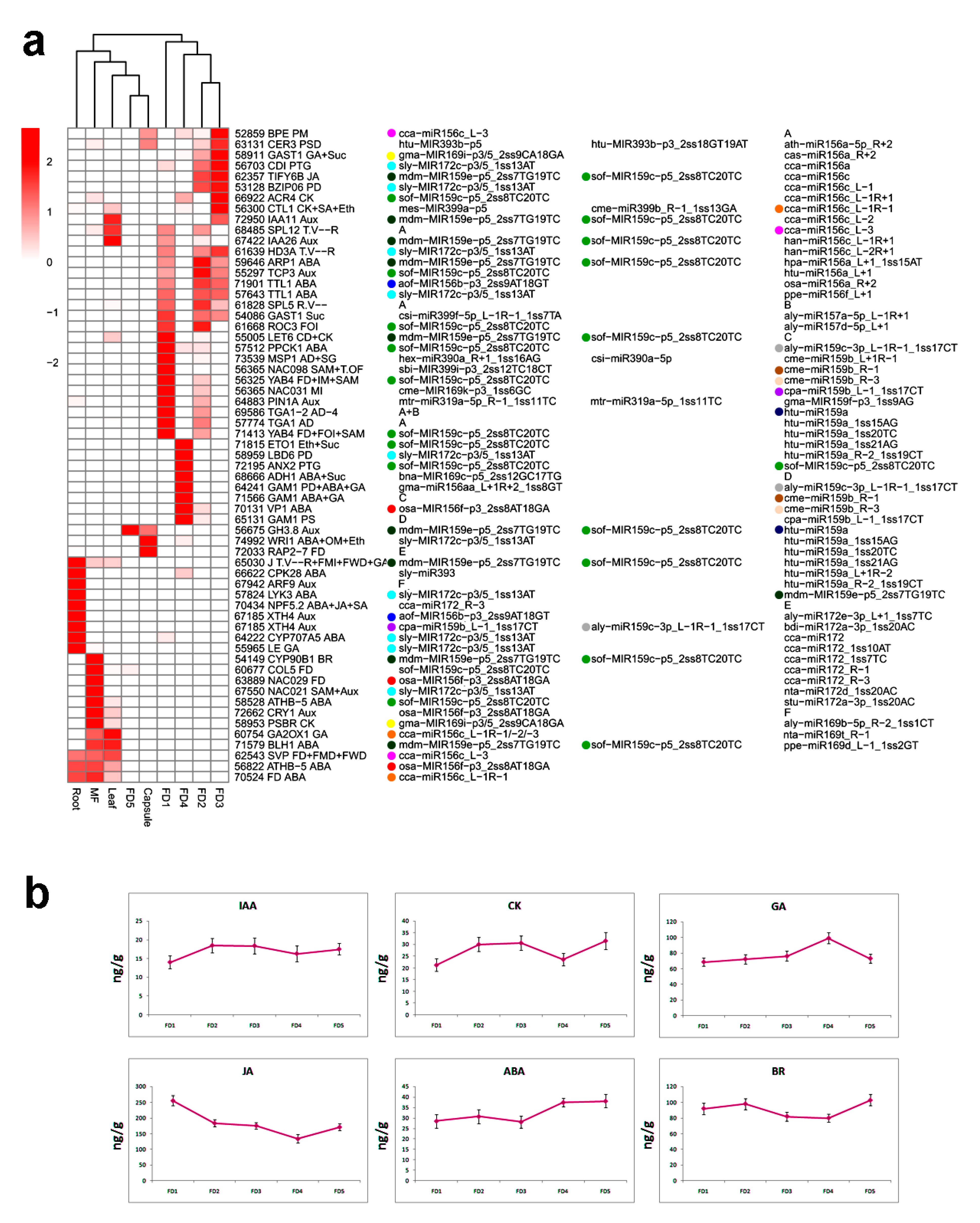
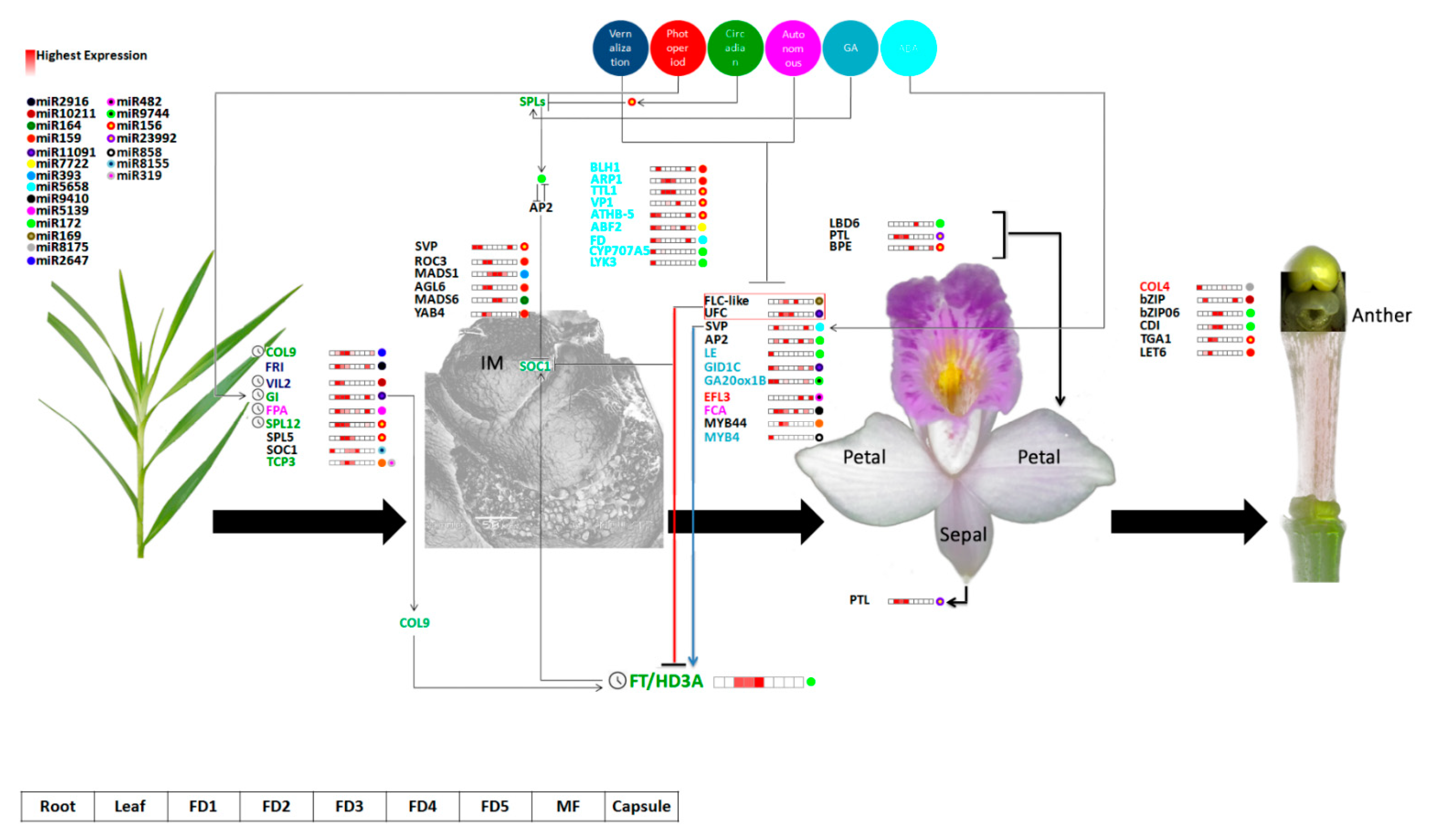
2.6. Hormonal Regulation Plays a Key Role in Flower Development
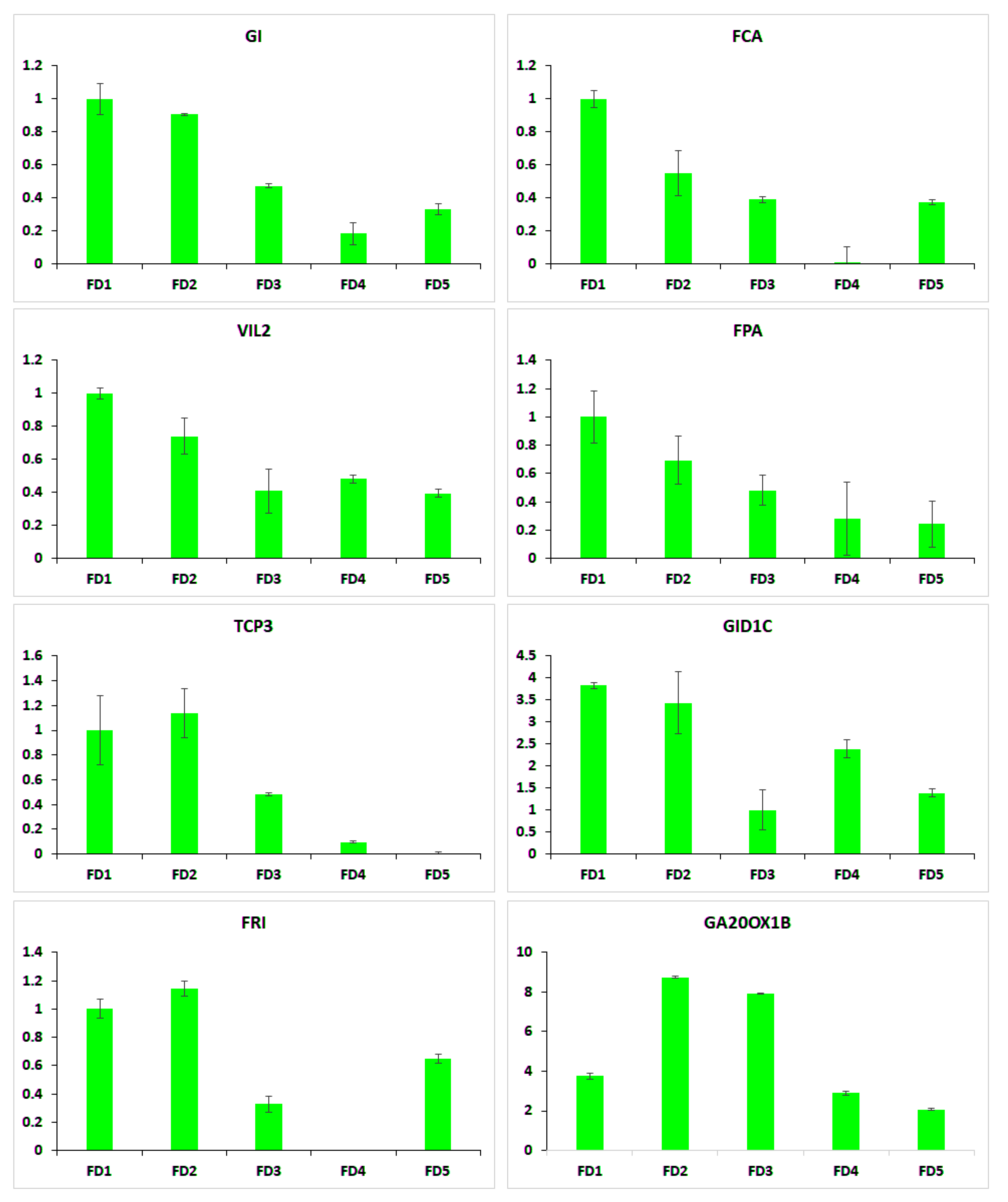
2.7. qRT-PCR of Selected Flowering Regulators
3. Discussion
4. Materials and Methods
4.1. Plant Preparation and Sampling
4.2. Transcriptome Sequencing
- RNA-seq library and sequencing
DEG Analysis
4.3. Micro RNA Sequencing
4.3.1. Conserved miRNA Alignment and Expression Analysis
4.3.2. Novel miRNA Prediction
4.4. Degradome Sequencing and Target Prediction
4.5. GO and Pathway Enrichment Analyses
4.6. Hormonal Analysis
4.7. Scanning Electron Microscopy
4.8. Quantitative Real Time PCR
4.9. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Cai, J.; Liu, X.; Vanneste, K.; Proost, S.; Tsai, W.-C.; Liu, K.-W.; Chen, L.-J.; He, Y.; Xu, Q.; Bian, C. The genome sequence of the orchid Phalaenopsis equestris. Nat. Genet. 2015, 47, 65. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wong, D.C.; Pichersky, E.; Peakall, R. The biosynthesis of unusual floral volatiles and blends involved in orchid pollination by deception: Current progress and future prospects. Front. Plant Sci. 2017, 8, 1955. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhan, Q.; Liang, Y.; Zhang, Z.; Liu, F.; Li, L.; Tang, X.; Liang, Z.; Chen, W.; Hu, M.; Tan, S. Geographic Patterns of the Richness and Density of Wild Orchids in Nature Reserves of Jiangxi, China. Diversity 2022, 14, 855. [Google Scholar] [CrossRef]
- Yang, F.; Zhu, G.; Wei, Y.; Gao, J.; Liang, G.; Peng, L.; Lu, C.; Jin, J. Low-temperature-induced changes in the transcriptome reveal a major role of CgSVP genes in regulating flowering of Cymbidium goeringii. BMC Genom. 2019, 20, 53. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seidenfaden, G.; Wood, J.J.; Holttum, R.E. The orchids of peninsular Malaysia and Singapore; Olsen & Olsen: Midvale, UT, USA, 1992. [Google Scholar]
- Hooker, J. Gnetaceae. Flora Br. India 1890, 5, 640–643. [Google Scholar]
- Auberon, F.; Olatunji, O.J.; Krisa, S.; Antheaume, C.; Herbette, G.; Bonté, F.; Mérillon, J.-M.; Lobstein, A. Two new stilbenoids from the aerial parts of Arundina graminifolia (Orchidaceae). Molecules 2016, 21, 1430. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, Z.Y.; Raven, P.H.; Hong, D.Y. Flora of China. Volume 25 (Orchidaceae); Science Press and Missouri Botanical Garden Press: Beijing, China, 2009. [Google Scholar]
- Wang, S.; Lee, P.; Lee, Y.; Hsiao, Y.; Chen, Y.; Pan, Z. Duplicated C-Class MADS-Box genes reveal distinct roles in gynostemium development in Cymbidium ensifolium (Orchidaceae). Plant Cell Physiol. 2011, 52, 567–577. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiang, L.; Li, X.; Qin, D.; Guo, F.; Wu, C.; Miao, L. Functional analysis of FLOWERING LOCUS T orthologs from spring orchid (Cymbidium goeringii Rchb. f.) that regulates the vegetative to reproductive transition. Plant Physiol. Biochem. 2012, 58, 98–105. [Google Scholar] [CrossRef]
- Ahmad, S.; Peng, D.; Zhou, Y.; Zhao, K. The Genetic and Hormonal Inducers of Continuous Flowering in Orchids: An Emerging View. Cells 2022, 11, 657. [Google Scholar] [CrossRef]
- Bartel, B.; Bartel, D.P. MicroRNAs: At the root of plant development. Plant Physiol. 2003, 132, 709–717. [Google Scholar] [CrossRef] [Green Version]
- Tang, G.; Reinhart, B.J.; Bartel, D.P.; Zamore, P.D. A biochemical framework for RNA silencing in plants. Genes Dev. 2003, 17, 49–63. [Google Scholar] [CrossRef] [PubMed]
- Chen, X. Small RNAs and their roles in plant development. Annu. Rev. Cell Dev. Biol. 2009, 25, 21–44. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Voinnet, O. Origin, biogenesis, and activity of plant microRNAs. Cell 2009, 136, 669–687. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Y.T.; Wang, M.; Fu, S.X.; Yang, W.C.; Qi, C.K.; Wang, X.J. Small RNA profiling in two Brassica napus cultivars identifies microRNAs with oil production- and development-correlated expression and new small RNA classes. Plant Physiol. 2012, 158, 813–823. [Google Scholar] [CrossRef] [Green Version]
- Pei, H.; Ma, N.; Chen, J.; Zheng, Y.; Tian, J.; Li, J. Integrative analysis of miRNA and mRNA profiles in response to ethylene in rose petals during flower opening. PLoS ONE 2013, 8, e64290. [Google Scholar] [CrossRef] [PubMed]
- Vazquez, F.; Vaucheret, H.; Rajagopalan, R.; Lepers, C.; Gasciolli, V.; Mallory, A.C. Endogenous trans-acting siRNAs regulate the accumulation of Arabidopsis mRNAs. Mol. Cell 2004, 16, 69–79. [Google Scholar] [CrossRef]
- Allen, E.; Xie, Z.; Gustafson, A.M.; Carrington, J.C. microRNAdirected phasing during trans-acting siRNA biogenesis in plants. Cell 2005, 121, 207–221. [Google Scholar] [CrossRef] [Green Version]
- Schwab, R.; Palatnik, J.F.; Riester, M.; Schommer, C.; Schmid, M.; Weigel, D. Specific effects of microRNAs on the plant transcriptome. Dev. Cell 2005, 8, 517–527. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Palatnik, J.F.; Wollmann, H.; Schommer, C.; Schwab, R.; Boisbouvier, J. Sequence and expression differences underlie functional specialization of Arabidopsis microRNAs miR159 and miR319. Dev. Cell 2007, 13, 115–125. [Google Scholar] [CrossRef] [Green Version]
- Jung, J.H.; Seo, P.J.; Ahn, J.H.; Park, C.M. Arabidopsis RNA-binding protein FCA regulates microRNA172 processing in thermosensory flowering. J. Biol. Chem. 2012, 287, 16007–16016. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Z.; Li, F.; Yang, S.; Dong, Y.; Yuan, Q.; Wang, F. Identification and functional analysis of flowering related microRNAs in common wild rice (Oryza rufipogon Griff.). PLoS ONE 2013, 8, e82844. [Google Scholar] [CrossRef]
- Chuck, G.; Meeley, R.; Irish, E.; Sakai, H.; Hake, S. The maize tasselseed4 microRNA controls sex determination and meristem cell fate by targeting Tasselseed6/indeterminate spikelet1. Nat. Genet. 2007, 39, 1517–1521. [Google Scholar] [CrossRef] [PubMed]
- Chen, X. A microRNA as a translational repressor of APETALA2 in Arabidopsis flower development. Science 2004, 303, 2022–2025. [Google Scholar] [CrossRef] [Green Version]
- Luo, Y.; Guo, Z.; Li, L. Evolutionary conservation of microRNA regulatory programs in plant flower development. Dev. Biol. 2013, 380, 133–144. [Google Scholar] [CrossRef] [Green Version]
- An, F.M.; Hsiao, S.R.; Chan, M.T. Sequencing-based approaches reveal low ambient temperature-responsive and tissue-specific microRNAs in phalaenopsis orchid. PLoS ONE 2011, 6, e18937. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aceto, S.; Sica, M.; Paolo, S.; D’Argenio, V.; Cantiello, P.; Salvatore, F. The analysis of the inflorescence miRNome of the orchid Orchis italica reveals a DEF-like MADS-box gene as a new miRNA target. PLoS ONE 2014, 9, e97839. [Google Scholar] [CrossRef]
- Lin, C.S.; Chen, J.J.; Huang, Y.T.; Hsu, C.T.; Lu, H.C.; Chou, M.L.; Chen, L.C.; Ou, C.I.; Liao, D.C.; Yeh, Y.Y. Catalog of Erycina pusilla miRNA and categorization of reproductive phase-related miRNAs and their target gene families. Plant Mol. Biol. 2013, 82, 193–204. [Google Scholar] [CrossRef]
- Curaba, J.; Singh, M.B.; Bhalla, P.L. miRNAs in the crosstalk between phytohormone signalling pathways. J. Exp. Bot. 2014, 65, 1425–1438. [Google Scholar] [CrossRef]
- Liu, H.-H.; Tian, X.; Li, Y.-J.; Wu, C.-A.; Zheng, C.-C. Microarray-based analysis of stress-regulated microRNAs in Arabidopsis thaliana. RNA 2008, 14, 836–843. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodriguez, R.E.; Mecchia, M.A.; Debernardi, J.M.; Schommer, C.; Weigel, D.; Palatnik, J.F. Control of cell proliferation in Arabidopsis thaliana by microRNA miR396. Development 2010, 137, 103–112. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Gu, X.; Xu, D.; Wang, W.; Wang, H.; Zeng, M.; Chang, Z.; Huang, H.; Cui, X. miR396-targeted AtGRF transcription factors are required for coordination of cell division and differentiation during leaf development in Arabidopsis. J. Exp. Bot. 2011, 62, 761–773. [Google Scholar] [CrossRef]
- Hewezi, T.; Maier, T.R.; Nettleton, D.; Baum, T.J. The Arabidopsis microRNA396-GRF1/GRF3 regulatory module acts as a developmental regulator in the reprogramming of root cells during cyst nematode infection. Plant Physiol. 2012, 159, 321–335. [Google Scholar] [CrossRef] [Green Version]
- Mallory, A.C.; Bartel, D.P.; Bartel, B. MicroRNA-directed regulation of Arabidopsis AUXIN RESPONSE FACTOR 17 is essential for proper development and modulates expression of early auxin response genes. Plant Cell 2005, 17, 1360–1375. [Google Scholar] [CrossRef] [Green Version]
- Liu, P.P.; Montgomery, T.A.; Fahlgren, N.; Kasschau, K.D.; Nonogaki, H.; Carrington, J.C. Repression of AUXIN RESPONSE FACTOR10 by microRNA160 is critical for seed germination and post-germination stages. Plant J. 2007, 52, 133–146. [Google Scholar] [CrossRef]
- Yu, S.; Galvão, V.C.; Zhang, Y.-C.; Horrer, D.; Zhang, T.-Q.; Hao, Y.-H.; Feng, Y.-Q.; Wang, S.; Schmid, M.; Wang, J.-W. Gibberellin regulates the Arabidopsis floral transition through miR156-targeted SQUAMOSA PROMOTER BINDING–LIKE transcription factors. Plant Cell 2012, 24, 3320–3332. [Google Scholar] [CrossRef] [Green Version]
- Yu, N.; Cai, W.-J.; Wang, S.; Shan, C.-M.; Wang, L.-J.; Chen, X.-Y. Temporal control of trichome distribution by microRNA156-targeted SPL genes in Arabidopsis thaliana. Plant Cell 2010, 22, 2322–2335. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahmad, S.; Lu, C.; Gao, J.; Ren, R.; Wei, Y.; Wu, J.; Jin, J.; Zheng, C.; Zhu, G.; Yang, F. Genetic insights into the regulatory pathways for continuous flowering in a unique orchid Arundina graminifolia. BMC Plant Biol. 2021, 21, 587. [Google Scholar] [CrossRef] [PubMed]
- Kurokura, T.; Mimida, N.; Battey, N.H.; Hytönen, T. The regulation of seasonal flowering in the Rosaceae. J. Exp. Bot. 2013, 64, 4131–4141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choi, K.; Kim, J.; Hwang, H.-J.; Kim, S.; Park, C.; Kim, S.Y.; Lee, I. The FRIGIDA complex activates transcription of FLC, a strong flowering repressor in Arabidopsis, by recruiting chromatin modification factors. Plant Cell 2011, 23, 289–303. [Google Scholar] [CrossRef] [Green Version]
- Reinhart, B.J.; Weinstein, E.G.; Rhoades, M.W.; Bartel, B.; Bartel, D.P. MicroRNAs in plants. Genes Dev. 2002, 16, 1616–1626. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Liu, H.; Li, D.; Chen, H. Identification and characterization of maize microRNAs involved in the very early stage of seed germination. BMC Genom. 2011, 12, 154. [Google Scholar] [CrossRef]
- Achard, P.; Herr, A.; Baulcombe, D.C.; Harberd, N.P. Modulation of floral development by a gibberellin-regulated microRNA. Development 2004, 131, 3357–3365. [Google Scholar] [CrossRef] [Green Version]
- Quesada, V.; Dean, C.; Simpson, G.G. Regulated RNA processing in the control of Arabidopsis flowering. Int. J. Dev. Biol. 2005, 49, 773–780. [Google Scholar] [CrossRef] [PubMed]
- Xie, K.; Wu, C.; Xiong, L. Genomic organization, differential expression, and interaction of SQUAMOSA promoter-binding-like transcription factors and microRNA156 in rice. Plant Physiol. 2006, 142, 280–293. [Google Scholar] [CrossRef] [Green Version]
- Gandikota, M.; Birkenbihl, R.P.; Höhmann, S.; Cardon, G.H.; Saedler, H.; Huijser, P. The miRNA156/157 recognition element in the 3′ UTR of the Arabidopsis SBP box gene SPL3 prevents early flowering by translational inhibition in seedlings. Plant J. 2007, 49, 683–693. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.W.; Czech, B.; Weigel, D. miR156-regulated SPL transcription factors define an endogenous flowering pathway in Arabidopsis thaliana. Cell 2009, 138, 738–749. [Google Scholar] [CrossRef] [Green Version]
- Lu, S.; Sun, Y.H.; Shi, R.; Clark, C.; Li, L.; Chiang, V.L. Novel and mechanical stress-responsive microRNAs in Populus trichocarpa that are absent from Arabidopsis. Plant Cell 2005, 17, 2186–2203. [Google Scholar] [CrossRef] [Green Version]
- Aukerman, M.J.; Sakai, H. Regulation of flowering time and floral organ identity by a MicroRNA and its APETALA2-like target genes. Plant Cell 2003, 15, 2730–2741. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jung, J.H.; Seo, Y.H.; Seo, P.J.; Reyes, J.L.; Yun, J.; Chua, N.H. The GIGANTEA-regulated microRNA172 mediates photoperiodic flowering independent of CONSTANS in Arabidopsis. Plant Cell 2007, 19, 2736–2748. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.J.; Huang, J.Q.; Huang, Y.J.; Li, Z.; Zheng, B.S. Discovery and profiling of novel and conserved microRNAs during flower development in Carya cathayensis via deep sequencing. Planta 2012, 236, 613–621. [Google Scholar] [CrossRef] [PubMed]
- Wu, G.; Park, M.Y.; Conway, S.R.; Wang, J.W.; Weigel, D.; Poethig, R.S. The sequential action of miR156 and miR172 regulates developmental timing in Arabidopsis. Cell 2009, 138, 750–759. [Google Scholar] [CrossRef] [PubMed]
- Wu, G.; Poethig, R.S. Temporal regulation of shoot development in Arabidopsis thaliana by miR156 and its target SPL3. Development 2006, 133, 3539–3547. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mizoguchi, T.; Wright, L.; Fujiwara, S.; Cremer, F.; Lee, K.; Onouchi, H.; Mouradov, A.; Fowler, S.; Kamada, H.; Putterill, J. Distinct roles of GIGANTEA in promoting flowering and regulating circadian rhythms in Arabidopsis. Plant Cell 2005, 17, 2255–2270. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Teotia, S.; Tang, G. To bloom or not to bloom: Role of microRNAs in plant flowering. Mol. Plant 2015, 8, 359–377. [Google Scholar] [CrossRef] [Green Version]
- Xu, M.Y.; Zhang, L.; Li, W.W.; Hu, X.L.; Wang, M.-B.; Fan, Y.L.; Zhang, C.Y.; Wang, L. Stress-induced early flowering is mediated by miR169 in Arabidopsis thaliana. J. Exp. Bot. 2014, 65, 89–101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johanson, U.; West, J.; Lister, C.; Michaels, S.; Amasino, R.; Dean, C. Molecular analysis of FRIGIDA, a major determinant of natural variation in Arabidopsis flowering time. Science 2000, 290, 344–347. [Google Scholar] [CrossRef] [PubMed]
- Sunkar, R.; Zhou, X.; Zheng, Y.; Zhang, W.; Zhu, J.-K. Identification of novel and candidate miRNAs in rice by high throughput sequencing. BMC Plant Biol. 2008, 8, 25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Palatnik, J.F.; Allen, E.; Wu, X.; Schommer, C.; Schwab, R.; Carrington, J.C.; Weigel, D. Control of leaf morphogenesis by microRNAs. Nature 2003, 425, 257–263. [Google Scholar] [CrossRef] [Green Version]
- Schommer, C.; Palatnik, J.F.; Aggarwal, P.; Chételat, A.; Cubas, P.; Farmer, E.E.; Nath, U.; Weigel, D. Control of jasmonate biosynthesis and senescence by miR319 targets. PLoS Biol. 2008, 6, e230. [Google Scholar] [CrossRef] [Green Version]
- Sarvepalli, K.; Nath, U. Hyper-activation of the TCP4 transcription factor in Arabidopsis thaliana accelerates multiple aspects of plant maturation. Plant J. 2011, 67, 595–607. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peng, J. Gibberellin and jasmonate crosstalk during stamen development. J. Integr. Plant Biol. 2009, 51, 1064–1070. [Google Scholar] [CrossRef] [PubMed]
- Barbier, F.F.; Lunn, J.E.; Beveridge, C.A. Ready, steady, go! A sugar hit starts the race to shoot branching. Curr. Opin. Plant Biol. 2015, 25, 39–45. [Google Scholar] [CrossRef] [PubMed]
- Beveridge, C.A.; Dun, E.A.; Rameau, C. Pea has its tendrils in branching discoveries spanning a century from auxin to strigolactones. Plant Physiol. 2009, 151, 985–990. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Müller, D.; Leyser, O. Auxin, cytokinin and the control of shoot branching. Ann. Bot. 2011, 107, 1203–1212. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kebrom, T.H.; Spielmeyer, W.; Finnegan, E.J. Grasses provide new insights into regulation of shoot branching. Trends Plant Sci. 2013, 18, 41–48. [Google Scholar] [CrossRef]
- Wang, H.; Wang, H. Phytochrome signaling: Time to tighten up the loose ends. Mol. Plant 2015, 8, 540–551. [Google Scholar] [CrossRef] [Green Version]
- Yuan, C.; Xi, L.; Kou, Y.; Zhao, Y.; Zhao, L. Current perspectives on shoot branching regulation. Front. Agric. Sci. Eng. 2015, 2, 38–52. [Google Scholar] [CrossRef] [Green Version]
- Ruttink, T. A molecular timetable for apical bud formation and dormancy induction in poplar. Plant Cell 2007, 19, 2370–2390. [Google Scholar] [CrossRef] [Green Version]
- Tylewicz, S. Photoperiodic control of seasonal growth is mediated by ABA acting on cell-cell communication. Science 2018, 360, 212–215. [Google Scholar] [CrossRef] [Green Version]
- Rohde, A. PtABI3 impinges on the growth and differentiation of embryonic leaves during bud set in poplar. (vol 14, pg 1885, 2002). Plant Cell 2002, 14, 1885–1901. [Google Scholar] [CrossRef] [Green Version]
- Singh, R.K.; Miskolczi, P.; Maurya, J.P.; Bhalerao, R.P. A tree ortholog of SHORT VEGETATIVE PHASE floral repressor mediates photoperiodic control of bud dormancy. Curr. Biol. 2019, 29, 128–133.e122. [Google Scholar] [CrossRef]
- Tian, Q.; Uhlir, N.J.; Reed, J.W. Arabidopsis SHY2/IAA3 inhibits auxin-regulated gene expression. Plant Cell 2002, 14, 301–319. [Google Scholar] [CrossRef] [Green Version]
- Kant, S.; Bi, Y.-M.; Zhu, T.; Rothstein, S.J. SAUR39, a small auxin-up RNA gene, acts as a negative regulator of auxin synthesis and transport in rice. Plant Physiol. 2009, 151, 691–701. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ori, N.; Cohen, A.R.; Etzioni, A.; Brand, A.; Yanai, O.; Shleizer, S.; Menda, N.; Amsellem, Z.; Efroni, I.; Pekker, I. Regulation of LANCEOLATE by miR319 is required for compound-leaf development in tomato. Nat. Genet. 2007, 39, 787–791. [Google Scholar] [CrossRef]
- Yanai, O.; Shani, E.; Russ, D.; Ori, N. Gibberellin partly mediates LANCEOLATE activity in tomato. Plant J. 2011, 68, 571–582. [Google Scholar] [CrossRef]
- Liu, Q.; Zhang, Y.-C.; Wang, C.-Y.; Luo, Y.-C.; Huang, Q.-J.; Chen, S.-Y.; Zhou, H.; Qu, L.-H.; Chen, Y.-Q. Expression analysis of phytohormone-regulated microRNAs in rice, implying their regulation roles in plant hormone signaling. FEBS Lett. 2009, 583, 723–728. [Google Scholar] [CrossRef] [Green Version]
- Srivastava, S.; Srivastava, A.K.; Suprasanna, P.; D’souza, S. Identification and profiling of arsenic stress-induced microRNAs in Brassica juncea. J. Exp. Bot. 2013, 64, 303–315. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sunkar, R.; Zhu, J.-K. Novel and stress-regulated microRNAs and other small RNAs from Arabidopsis. Plant Cell 2004, 16, 2001–2019. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahmad, S.; Lu, C.; Wei, Y.; Gao, J.; Jin, J.; Zheng, C.; Zhu, G.; Yang, F. The de novo transcriptome identifies important zinc finger signatures associated with flowering in the orchid Arundina graminifolia. Sci. Hortic. 2022, 291, 110572. [Google Scholar] [CrossRef]
- Yang, F.; Lu, C.; Wei, Y.; Wu, J.; Ren, R.; Gao, J.; Ahmad, S.; Jin, J.; Xv, Y.; Liang, G. Organ-Specific Gene Expression Reveals the Role of the Cymbidium ensifolium-miR396/Growth-Regulating Factors Module in Flower Development of the Orchid Plant Cymbidium ensifolium. Front. Plant Sci. 2021, 12, 799778. [Google Scholar] [CrossRef]
- Ahmad, S.; Lu, C.; Wu, J.; Wei, Y.; Gao, J.; Jin, J.; Zheng, C.; Zhu, G.; Yang, F. Transcriptional cascade in the regulation of flowering in the Bamboo Orchid Arundina graminifolia. Biomolecules 2021, 11, 771. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, S.; Gao, J.; Wei, Y.; Lu, C.; Zhu, G.; Yang, F. The transcriptome profiling of flavonoids and bibenzyls reveals medicinal importance of rare orchid Arundina graminifolia. Front. Plant Sci. 2022, 13, 1934. [Google Scholar] [CrossRef] [PubMed]
- Nishihara, M.; Hikage, T.; Yamada, E.; Nakatsuka, T. A single-base substitution suppresses flower color mutation caused by a novel miniature inverted-repeat transposable element in gentian. Mol Genet Genom. 2011, 286, 371–382. [Google Scholar] [CrossRef] [PubMed]
- Grabherr, M.G.; Haas, B.J.; Yassour, M.; Levin, J.Z.; Thompson, D.A.; Amit, I.; Adiconis, X.; Fan, L.; Raychowdhury, R.; Zeng, Q. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol. 2011, 29, 644. [Google Scholar] [CrossRef] [Green Version]
- Garg, R.; Singh, V.K.; Rajkumar, M.S.; Kumar, V.; Jain, M. Global transcriptome and coexpression network analyses reveal cultivar-specific molecular signatures associated with seed development and seed size/weight determination in chickpea. Plant J. 2017, 91, 1088–1107. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Huang, H.; Ma, Y.P.; Fu, J.X.; Wang, L.L.; Dai, S.L. Construction and de novo characterization of a transcriptome of Chrysanthemum lavandulifolium: Analysis of gene expression patterns in floral bud emergence. Plant Cell Tiss. Org. 2014, 116, 297–309. [Google Scholar] [CrossRef]
- Conesa, A.; Gotz, S.; Garcia-Gomez, J.M.; Terol, J.; Talon, M.; Robles, M. Blast2GO: A universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics 2005, 21, 3674–3676. [Google Scholar] [CrossRef] [Green Version]
- Kanehisa, M.; Goto, S. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000, 28, 27–30. [Google Scholar] [CrossRef]
- Audic, S.; Claverie, J.-M. The significance of digital gene expression profiles. Genome Res. 1997, 7, 986–995. [Google Scholar] [CrossRef]
- Yang, F.; Zhu, G. MicroRNA transcriptome variations in the multi-tepal mutant provide insights into the floral patterning of the orchid Cymbidium goeringii. In Proceedings of the II Asian Horticultural Congress, Chengdu, China, 26–28 September 2016; Volume 1208, pp. 85–96. [Google Scholar]
- Addo-Quaye, C.; Miller, W.; Axtell, M.J. CleaveLand: A pipeline for using degradome data to find cleaved small RNA targets. Bioinformatics 2009, 25, 130–131. [Google Scholar] [CrossRef] [Green Version]
- Fahlgren, N.; Howell, M.D.; Kasschau, K.D.; Chapman, E.J.; Sullivan, C.M.; Cumbie, J.S.; Givan, S.A.; Law, T.F.; Grant, S.R.; Dangl, J.L. High-throughput sequencing of Arabidopsis microRNAs: Evidence for frequent birth and death of MIRNA genes. PLoS ONE 2007, 2, e219. [Google Scholar] [CrossRef] [PubMed]
- Maere, S.; Heymans, K.; Kuiper, M. BiNGO: A Cytoscape plugin to assess overrepresentation of gene ontology categories in biological networks. Bioinformatics 2005, 21, 3448–3449. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pan, X.; Welti, R.; Wang, X. Quantitative analysis of major plant hormones in crude plant extracts by high-performance liquid chromatography–mass spectrometry. Nat. Protoc. 2010, 5, 986. [Google Scholar] [CrossRef] [PubMed]






Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ahmad, S.; Lu, C.; Gao, J.; Wei, Y.; Xie, Q.; Jin, J.; Zhu, G.; Yang, F. The Integrated mRNA and miRNA Approach Reveals Potential Regulators of Flowering Time in Arundina graminifolia. Int. J. Mol. Sci. 2023, 24, 1699. https://doi.org/10.3390/ijms24021699
Ahmad S, Lu C, Gao J, Wei Y, Xie Q, Jin J, Zhu G, Yang F. The Integrated mRNA and miRNA Approach Reveals Potential Regulators of Flowering Time in Arundina graminifolia. International Journal of Molecular Sciences. 2023; 24(2):1699. https://doi.org/10.3390/ijms24021699
Chicago/Turabian StyleAhmad, Sagheer, Chuqiao Lu, Jie Gao, Yonglu Wei, Qi Xie, Jianpeng Jin, Genfa Zhu, and Fengxi Yang. 2023. "The Integrated mRNA and miRNA Approach Reveals Potential Regulators of Flowering Time in Arundina graminifolia" International Journal of Molecular Sciences 24, no. 2: 1699. https://doi.org/10.3390/ijms24021699